

Abstract
AIMS
To identify prospectively a safe therapeutic window for administration of a novel oral transforming growth factor β (TGF-β) inhibitor, LY2157299 monohydrate, based on a pharmacokinetic/pharmacodynamic (PK/PD) model. Simulations of population plasma exposures and biomarker responses in tumour were performed for future trials of LY2157299 in glioblastoma and other cancer populations.
METHODS
The model was updated after completion of each cohort during the first-in-human dose (FHD) study. The flexible design allowed continuous assessment of PK variability by recruiting the required number of patients in each cohort. Based on 30% inhibition of TGF-β RI kinase phosphorylates (pSMAD), biologically effective exposures were anticipated to be reached from 160 mg onwards. The therapeutic window was predicted, based on animal data, to be between 160 and 360 mg.
RESULTS
No medically significant safety issues were observed and no dose limiting toxicities were established in this study. Observed plasma exposures (medians 2.43 to 3.7 mg l−1 h, respectively) with doses of 160 mg to 300 mg were within the predicted therapeutic window. Responses, based on the MacDonald criteria, were observed in these patients.
CONCLUSIONS
A therapeutic window for the clinical investigation of LY2157299 in cancer patients was defined using a targeted PK/PD approach, which integrated translational biomarkers and preclinical toxicity. The study supports using a therapeutic window based on a PK/PD model in early oncology development.
Keywords: PK/PD model, TGF-β inhibitor, therapeutic window
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
TGF-β signalling has been recognized as an important regulator of tumour growth in advanced cancers for the last 20 years. However, due to severe non-monitorable toxicities in animal studies, no small molecule TGF-β inhibitor has thus far progressed into clinical trials in oncology patients.
WHAT THIS STUDY ADDS
Although similar cardiovascular toxicities were observed in animal studies for LY2157299 monohydrate, we applied a pharmacokinetic/pharmacodynamic model to establish prospectively a therapeutic window. Using a therapeutic window approach we identified doses and exposures where we observed responses in glioblastoma patients with this potential first in class treatment. This study supports using a therapeutic window approach based on a pharmacokinetic/pharmacodynamic model in early oncology development.
Introduction
The transforming growth factor β (TGF-β) superfamily of ligands has an important role in the regulation of a variety of physiological processes [1]. TGF-β signalling has been recognized as an important regulator of tumour growth in advanced cancers [2,3]. Inhibiting TGF-β signalling is a novel approach that will simultaneously inhibit tumour spread and neo-angiogenesis and improve the host's antitumour immune response. There are three known human TGF-β ligands [2]: TGF-β1, TGF-β2, and TGF-β3. Of all the isoforms, TGF-β1 is most prevalent. TGF-β signalling is initiated when TGF-β ligands engage TGF-β type-I (RI) and type-II (RII) receptors. This induces phosphorylation of the TGF-β receptor kinases [4]. The TGF-β RI kinase phosphorylates SMAD2 (pSMAD2) and SMAD3 (pSMAD3) result in the formation of SMAD complexes, which are subsequently translocated into the nucleus to stimulate gene transcription of TGF-β responsive genes [5]. Hence, changes in pSMAD2 and pSMAD3 levels can be used to determine activity of the TGF-β signalling or inhibitors to this pathway.
Adenosine triphosphate (ATP)-mimetic drugs, which target the kinase domain of TGF-β RI, have been developed [6]. These ATP-mimetic drugs are small molecule inhibitors designed to block specifically the phosphorylation of SMAD2 and SMAD3 at the TGF-β RI kinase site [1]. As the kinase domains of the nodal type I receptor, activin receptor-like kinase 7 (ALK7) and ALK4 are similar to that of TGF-β R1, the specificity of such small molecule inhibitors is not complete. This may explain some of the observed toxicities, which are considered severe, in animals administered such TGF-β small molecule inhibitors [7]. One of these ALK5-inhibitor programmes was terminated because of the heart valve lesions induced in animals [8]. Their lead ALK5 inhibitor demonstrated an incidence of haemorrhagic, degenerative and inflammatory lesions in heart valves, which occurred during a 10 day dose range finding study. Additional 10 day toxicological studies with six other ALK5 inhibitors at high doses were performed, but again heart valve lesions were observed. The authors reported that no lesions were found either when there was inadequate level of exposure or when they used a much less potent ALK5 compound with no real pharmacological activity. Hence, they concluded that this cardiac toxicity was a class-wide pharmacological effect preventing this class of inhibitors from moving to clinical investigation [8]. Another group also reported similar histopathological findings in all four heart valves for their two ALK5 inhibitors at all doses [9]. As a result of these toxicities observed in animals, drug development programmes aimed at identifying TGF-β-receptor kinase inhibitors did not progress beyond animal studies [8,9].
We investigated several ALK5 inhibitors [6] from which LY2157299 monohydrate (identified as LY2157299 in the text below) was selected for clinical investigation based on its unique properties in animal toxicology studies. Although we also found similar cardiovascular toxicities for our ALK5 inhibitors [10], we were able to apply a pharmacokinetic/pharmacodynamic (PK/PD) model to establish prospectively a therapeutic window for LY2157299. This PK/PD model was updated after completion of each cohort during the first-in-human dose (FHD) study. Due to the relatively narrow window and non-monitorable (specifically, heart valve lesions) preclinical toxicity, we were careful to characterize systematically between patient variation in exposure. The model was used to simulate population plasma exposures and biomarker responses in tumour for future trials of LY2157299 in glioblastoma and other cancer populations.
Methods
Preclinical data and modelling
The primary objective of preclinical PK/PD modelling of LY2157299 in preparation for the phase 1 study was to estimate a pharmacologically effective and tolerable dose range in humans.
As previously reported in mice [11], we combined plasma concentrations, biomarker response data (percentage of pSMAD2 and pSMAD3) in tumour and efficacy data in terms of tumour size. It was concluded that 30% inhibition over 24 h, combined with maximum response (Emax) of 50%, was required to achieve preclinical efficacy. There were also preclinical toxicity constraints, which are described in detail in [10]. Briefly, valvulopathy was a consistent finding in both dog and rat. Short duration (1 month or less) non-clinical toxicology studies in rats and dogs identified the heart valve as a target organ of toxicity for LY2157299. A toxicology study in rats using intermittent dosing (2 weeks on/2 weeks off schedule for 3 months) established a no-observed-effect level of 50 mg kg−1 for the cardiovascular changes. Preclinical PK i.v. data from mouse, rat and dog studies, using allometric scaling, were used to predict the human i.v. PK parameters for clearance, volume and absorption. Human i.v. clearance and volume of distribution were predicted to be 20.9 l h−1 (8.11–53.76) and 94.57 l (44.13–202.69), respectively, given as mean (95% confidence intervals [CI]). Mice and rat models [11] enabled prediction of the targeted inhibition of SMAD phosphorylation under different dosing regimens. We used the predicted PK and PD from preclinical information (Calu6 mouse model) to simulate the average inhibition of pSMAD in tumour over 24 h (areas under the efficacy curves, AUE/24) was estimated after once daily and twice daily administration of LY2157299.
Predicting therapeutic dose range
Simulations were performed to predict expected inhibition of pSMAD in tumour for a variety of doses to define the prospective dose range in patients. Using the previously proposed preclinical PK/PD model, clearance, volume, absorption and bioavailability terms were substituted by predicted values in humans. Bioavailability was assumed to be 50%. This assumption was based on biopharmaceutical data and absolute bioavailability observed in rats of 73%. In addition, the PD parameters, such as kout, Imax and IC50 [11], were assumed to be similar to those in animals. Applying these assumptions, the predicted inhibitions of pSMAD in tumour in humans across a range of doses are presented in Figure 1. Specifically, following 200mg once daily and twice daily, the AUE/24 was 29.71% and 35.64% with Emax at 69.10 and 55.48, respectively. The predicted AUE/24 and Emax following 400 mg once daily were 40.86% and 81.51 and 400 mg twice daily were 50.77% and 71.27, respectively. Similar conclusions for the required dose range in humans were reached using MX1 data in mice and rats. Following a twice daily dosing regimen, the average percentage of inhibition of pSMAD over 24 h was always higher compared with a daily dosing regimen. The comparative percentage inhibition of pSMAD over a 24 h period between once daily and twice daily doses for same total dose day−1 (40 mg and 500 mg) are plotted in Figure 1. The top panels show inhibition of pSMAD following twice daily and the bottom panels following once daily dosing.
Figure 1.

Simulated % of inhibition (median and 5th, 95th percentiles) of pSMAD in tumour in humans from Calu6 models in mice after once daily and twice daily administration of total daily doses of 40 and 500 mg LY2157299
Predicting a safe therapeutic dose range
In addition to the PK/PD model, we included the animal toxicity information to determine whether the expected antitumour efficacy of LY2157299 would fall within or outside of the toxicology risk range. The toxicity of LY2157299 was characterized in repeat and intermittent dose non-clinical safety studies up to 6 months' duration in rats and dogs [10]. The no-observed-effect level (NOEL) in rat for the cardiovascular changes in the 3 month intermittent dosing study was 50 mg kg−1. In dog, the no-observed-effect level in the 30 day study was 20 mg kg−1. Margins of safety for cardiac changes were derived using the no-observed-effect level exposures in the toxicology studies and the exposures predicted from the PK/PD model for the clinical starting dose (40 mg), a dose predicted to be within the biologically effective dose range (240 mg) and the highest proposed dose (360 mg). The systemic exposures [area under the plasma concentration–time curve (AUC(0,24 h)) (μg ml−1 h)] from the toxicology studies in male and female Fischer 344 rats (n = 5 in each group) following 50 mg kg−1 3 months' intermittent dosing (2 weeks on/2 weeks off) on day 83 were 8.01 and 20.77, respectively. The margin of safety to the NOEL for cardiovascular changes, based on exposure, to the midpoint of the predicted biologically efficacious dose range (240 mg) and highest anticipated clinical doses (360 mg) were 1.4 and 0.95, respectively.
A dose escalation scheme (Table 1) was proposed, starting with a total daily dose of 40 mg and increasing up to (potentially) 360 mg. Using the scaled human PK model we projected median (20th and 80th percentile) exposures at each dose level, assuming dose proportionality in patients. We anticipated that we should start reaching biologically effective exposures from cohort 3 (160 mg) onwards. An unacceptable risk was defined as the probability of exceeding 10.96 mg l−1 h being greater than 20%.
Table 1.
Y2157299 proposed dose-escalation scheme for clinical study
| Dose level | Total daily dose (mg day−1) | % increase | Predicted median AUC (20th – 80th percentile range) (mg l−1 h) |
|---|---|---|---|
| 1 | 40 | – | 0.93 (0.76–1.22) |
| 2 | 80 | 100 | 1.86 (1.51–2.44) |
| 3 | 160 | 100 | 3.73 (3.03–4.87) |
| 4 | 240 | 50 | 5.59 (4.54–7.31) |
| 5 | 360 | 50 | 8.38 (6.81–10.96) |
The dose levels in bold are doses that will be administered intermittently (2 weeks on/2 weeks off) based on study protocol amendment. Dose levels 1 and 2 were administered prior to amendment. AUC, area under the plasma concentration vs. time curve.
Combining, anticipated biologically effective exposures and exposure associated with toxicity allowed us to define a therapeutic window (Figure 2), which would justify a safe evaluation in patients. Due to this relatively narrow window and non-monitorable preclinical toxicity, we were careful to characterize systematically between patient variation in exposure. Driven by the necessity to understand fully exposure variation between patients, we expanded cohorts for PK once we were in the desired dose range. It was also evident that this safe therapeutic window could only be achieved in humans if the PK variability was low to moderate.
Figure 2.
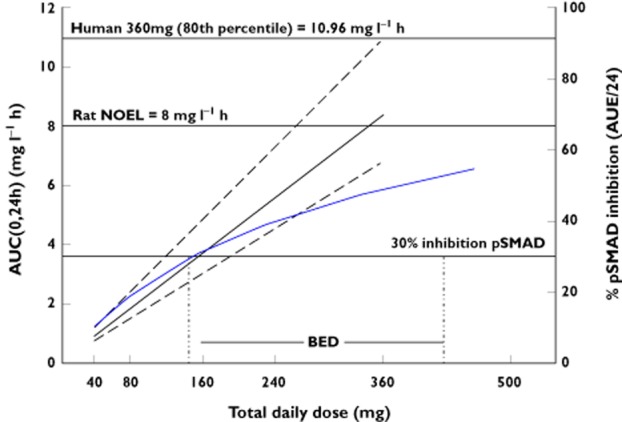
Predicted therapeutic window, based on preclinical information. Dose range (x-axis) to be explored in patients. On the left y-axis the predicted human exposure, based on animal data, is plotted and on the right y-axis the % pSMAD inhibition in tumour is given. The therapeutic window we plan to investigate within is enclosed between the two horizontal black lines, 30% inhibition pSMAD and rat NOEL = 8.0. The solid blue curve illustrates how % pSMAD inhibition changes with dose. At around 160 mg total daily dose, the required inhibition (30%) of pSMAD in tumour is reached; the % inhibition increases slightly until it reaches a plateau of approximately 50% at 360 mg total daily dose. The solid black line is the median exposure (AUC(0,24 h) which increases proportionally with dose and the broken lines are the 20th and 80th percentiles of predicted exposure. —, Median AUC; – – –, 20th & 80th percentile AUC;  , Calu6 mouse tumour
, Calu6 mouse tumour
FHD clinical trial design
The study was conducted in accordance with the principles for human experimentation as defined in the most recent version of the Declaration of Helsinki and was approved by the Investigational Review Board. Informed consent (ICD) was obtained from each patient after they had been told the potential risks and benefits, as well as the investigational nature of the study.
LY2157299 was administered orally as a tablet on an empty stomach. Patients in cohorts 1 and 2 were administered LY2157299 as a continuous treatment and took a single total daily dose of LY2157299 on day 1 and no drug on day 2. This was done in order to obtain a full patient pharmacokinetic profile after a single dose of LY2157299, which was compared with the same patient's profile at the last day of study treatment in a cycle. Before dosing of cohort 3, unexpected toxicities (based on preliminary findings) were observed in non-clinical toxicology 6 month continuous oral administration of LY2157299 studies in rats and dogs. As the result of the 3 month non-clinical study in rats [10], an intermittent dosing regimen with a 28 day cycle (14 days on/14 days off) was used starting in cohort 3 in patients with glioma. The intermittent dosing regimen for a cycle (28 days) was defined as 2 weeks of LY2157299 twice daily administration followed by 2 weeks without LY2157299 administration. The planned duration of treatment, in the absence of disease progression or dose-limiting toxicity, was two treatment cycles of LY2157299. After the second cycle, if the patients did not have progressive disease based on RECIST [12] or MacDonald criteria [13], they could continue treatment as long as they signed a second ICD. During monotherapy with LY2157299, there was a cohort-by-cohort safety and PK analysis to allow an informed decision prior to each dose escalation and no intra-patient dose escalation was allowed. A minimum of three patients were enrolled per dose level. The highest dose of LY2157299 administered was not to exceed 360 mg day−1, as discussed in the previous section, unless the observed exposure (AUC) in patients was less than predicted based on the non-clinical PK model or there were compelling reasons to escalate beyond this dose. Due to systemic exposure being directly related with non-monitorable toxicity in animals, between patient variability on exposure had to be low to moderate, i.e. below 50%. Should the exposure variability be any higher, an unacceptably higher proportion of patients would be at risk of exceeding the predefined exposure limit.
Venous blood samples of approximately 4 ml were drawn into sodium heparinized tubes for measurement of LY2157299. Plasma samples collected were assayed for LY2157299 using a validated liquid chromatography/mass spectrometry (LC/MS) method. The lower limit of quantification was 0.05 ng ml−1 and the upper limit of quantification was 1000.00 ng ml−1. Samples above the limit of quantification were diluted and reanalyzed to yield results within the calibrated range. The inter-assay accuracy (% relative error) during validation ranged from −3.4% to 6.0%. The inter-assay precision (% relative SD) during validation. ranged from 3.7% to 8.5%. LY2300559 was stable for up to 42 days when stored at approximately −20°C and was stable for up to 481 days when stored at approximately −70°C.
PK analysis methods
Preliminary non-compartment results from the monotherapy part of the FHD study of LY2157299 have been presented previously [14,15]. Here we present a population PK evaluation of LY2157299 given as an intermittent dosing in 30 patients with glioma at a dose range of 80, 120 and 150 mg twice daily (morning and evening) [cohorts 3 (15 patients), 4 (six patients) and 5 (nine patients)]. In addition, the population PK analysis included three patients from cohort 1 (40 mg day−1) and four patients from cohort 2 (80 mg day−1). In cohort 3, 15 patients were evaluable for PK in cycle 1 and nine patients were evaluable for PK in cycles 1 and 2. In cohort 4, four out of six patients were evaluable for PK in both cycles 1 and 2 and in cohort 5, seven out of nine patients were evaluable for PK in both cycles 1 and 2.
Non-compartmental PK analyses were performed using WinNonLin Professional Version 5.3 (Pharsight Corporation, Mountain View, CA, USA). All population PK analyses were performed using non-linear mixed effects modelling in nonmem, version 7.2 [16,17], using first order conditional estimation (FOCE) method. The assumptions on the population PK model were as described in the Appendix. Demographic characteristics were tested as potential covariates on PK parameters and posterior predictive checks were carried out.
Results
From five dose escalation cohorts, a total of 717 observations from 37 patients were obtained. Observed concentrations against time for LY2157299 at each dose level are shown in Figure 3A. Figure 3B shows dose-normalized observed plasma concentration data across cohorts, cycles, and on day 1 and at steady-state (day 14). Figure 3C shows histograms of patient characteristics.
Figure 3.

LY2175299 plasma concentrations in patients for all five dose levels. (A) Observed plasma concentrations at steady-state (day 14) for all five cohorts (dose levels). Full symbols represent cycle 2 observations. Each symbol is a single concentration at given time for a patient. (B) Individual dose-normalized observed plasma concentrations vs. time. (C) Histograms of patient characteristics included in pharmacokinetic analyses. A and B △, 300 mg; 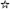 , 240 mg; ○, 160 mg; □, 80 mg;
, 240 mg; ○, 160 mg; □, 80 mg;  , 40 mg
, 40 mg
Non-compartmental analyses
LY2157299 was rapidly absorbed and plasma concentrations were measurable for at least 48 h. At steady-state, on day 14, the median time to maximum concentration (tmax,ss) ranged from 0.5 to 2 h post-dose, independent of dose. Both the maximum observed concentration at steady-state (Cmax,ss) and exposure increased with dose. Formal assessment of time-linear kinetics, that is, whether the observed exposure (AUC(0,∞)), day 1 and AUC(0,∞), day 14 were similar, was not possible. However, no accumulation of LY2157299 in the five cohorts was observed over the 14 day twice daily dosing regimen. From the statistical analysis of the PK parameters, the estimated ratios of geometric means for the AUC(0, ∞) and Cmax,ss between 40 mg and 300 mg daily were 5.61 (90% CI 3.80, 8.30) and 3.99 (90% CI 2.43, 6.54), respectively. For a doubling of dose, the fold increases for AUC(0,∞) and Cmax,ss were 1.81-fold with corresponding 90% CIs (1.58, 2.07) and 1.61-fold with 90% CIs (1.36, 1.91), respectively. This suggests dose proportional PK for at least a doubling dose within the studied dose range of 40 mg to 300 mg, particularly for AUC(0,∞). Within and between patient coefficients of variation were estimated as 29% and 42% for AUC (0,∞) at steady-state, respectively, and 31% and 55% for Cmax,ss, pooled across the five cohorts from the dose proportionality analysis.
LY2157299 population PK model in patients with glioblastoma
A first order absorption linear two compartment model with elimination from the central compartment provided the best fit to the plasma data with parameter estimates listed in Table 2. The oral mean population apparent clearance of LY2157299 was 38 l h−1 with acceptable between patient variability at 46%. Between-occasion (in the two different treatment cycles) variability on apparent clearance was small, estimated at 18%. The goodness of fit of the model to the data for LY2157299 was assessed and deemed acceptable. The model fit was assessed in terms of population-weighted residuals. The population weighted and individual weighted residuals appeared to be distributed evenly around the group mean predicted concentrations and the majority of the weighed residuals lay between ± 2 SDs from the mean.
Table 2.
Pharmacokinetic parameters in final population model
| Parameter description | Population estimate (%SEE) | Inter-patient variability (%SEE) |
|---|---|---|
| Rate of absorption | ||
| Parameter for Ka (h−1) | 2.24 (20) | 76 (36) |
| Clearance | ||
| Parameter for CL/F (l h−1) | 38.4 (8.4) | 46 (36) |
| Volume of distribution for first compartment | ||
| Parameter for V1/F (l) | 100 (10) | 42 (34) |
| Volume of distribution for second compartment | ||
| Parameter for V2/F (l) | 92.8 (12) | 48 (33) |
| Inter-compartment clearance | ||
| Parameter for Q (l h−1) | 9.2 (13) | 40 (57) |
| Between-occasion variability CL_IOV | 18 (65) | |
| Residual error (additive) | 0.15 (20) | |
| Residual error (proportional) | 51 (10) |
CL/F, apparent clearance; Ka, absorption rate constant; SEE, standard error of the estimate; V/F, apparent volume of distribution.
LY2157299 was rapidly absorbed into the systemic circulation with an absorption rate constant of 2.2 h−1, with some variation of 76% between patients, which could either reflect a real observation or (more probably) be due to sparse sampling during the absorption phase. The model predicted individual patient concentration–time profiles and exposures well (Figure 4). Visual inspection, followed by piecewise covariate search, did not reveal any significant demographic covariates. The model was also used to define optimal sparse PK sampling times, using the software PopDes [18], for subsequent trials requiring sparse blood draws.
Figure 4.
Steady-state (day 14) observed LY2157299 plasma concentrations and model-simulated median, 5th, 20th, 80th and 95th percentiles following administration of 150 mg twice dailyand 80 mg twice daily LY2157299 doses.  , time vs. median;
, time vs. median;  , 5–95 percentiles;
, 5–95 percentiles;  , 20–80 percentiles;
, 20–80 percentiles;  , observed concentrations
, observed concentrations
Biomarkers and efficacy
The trial was designed to collect pSMAD in plasma as a surrogate for tumour tissue, as a biomarker for pharmacological activity. Unfortunately, results were not reliable given problems with the assay. Based on the MacDonald criteria, response was observed in four patients in cohorts 3 and 5. The responses all proceeded to disease stabilization and were detected, except for one patient, after at least four cycles of treatment. During the course of the study no medically significant safety issues were observed and no dose-limiting toxicity was established at the doses that were explored.
Discussion
Population PK/PD models are routinely utilized nowadays in later phases of drug development, where sparse PK samples and therapeutic drug monitoring are the norm. Such models are required by regulators [19,20] and recommendations based on them regularly feature on drug labels [21]. In later phases of oncology development, quantitative assessment is often supported through use of PK/PD models, for instance in non-small cell lung carcinoma [22]. A recent publication by a NIH working group from 2011 recommends the quantitative integration of our growing understanding of cellular and tissue level networks, where therapeutic and toxic effects of drugs can best be understood at a systems level. There are very limited examples in oncology, where such models with early translational biomarkers are used in preclinical phases of drug development [23,24].
We used a targeted PK/PD approach during the early development of LY2157299. This approach integrated biomarkers and preclinical toxicity and allowed us to define prospectively a therapeutic window for the clinical investigation of LY2157299 in cancer patients. This involved several steps, specifically in vivo preclinical evaluation of: (i) target inhibition, (ii) pharmacological efficacy and (iii) toxicity profile. In each of these three evaluations it was important to have a detailed understanding of the in vivo exposure necessary to modulate the target, which allows the designing of the optimal dosing regimen required for evaluation of traditional antitumour efficacy and can be used to reduce off-target effects. Based on preclinical data, we defined a therapeutic window for exposure ranges to be investigated in patients. Scaling the animal PK/PD model to humans, we calculated required doses and exposures with optimal escalation schema to effectively and safely reach concentrations within the therapeutic range. During the clinical evaluation phase of LY2157299, we prospectively, cohort by cohort, evaluated PK. There was no excessive accumulation of LY2157299 as it approached steady-state with a twice daily regimen. Additionally, with the favourable study design of collecting PK during the first two cycles of treatment, we estimated low between occasion variability of 18%, i.e. there were minor changes in the PK profile in the same patient during different cycles of LY2157299 administrations. Observed plasma exposures for cohorts 3, 4 and 5 were within the predicted therapeutic window, where we anticipated efficacy and were below exposures associated with toxicity in animals. Based on the MacDonald criteria, response was observed in four patients in cohorts 3 and 5. The responses all proceeded to disease stabilization and were detected, except for one patient, after at least four cycles of treatment.
The preclinical PK/PD model, which simulates pSMAD tumour levels in rats [11], is now updated with the population PK model in patients. Using the same doses as those administered to patients, we simulated probable tumour pSMAD–time profiles. These median simulated pSMAD profiles in tumours following 80 mg twice daily and 150 mg twice daily administration of LY2157299 are plotted in Figure 5. Currently, there are no observations of pSMAD levels in tumours in patients. There are qualitative assessments of ID1 levels of one patient with glioblastoma from this trial [25], where a salvage surgical resection was indicated for one patient after 2 months of treatment. The optimal measurement of the exposure that TGF-β RI encounters is to measure concentrations of LY2157299 in the cytoplasm of the tumour. For patients with glioblastoma the molecule has to pass though the blood–brain barrier, but the only measures of concentrations of LY2157299 we currently have are plasma exposure. However, as the patients were at steady-state on day 14, where the dose administered is equal to the amount of LY2157299 eliminated from the body and distribution equilibrium was reached, we may assume that the observed total plasma concentrations (Figure 5) were similar to those in tumour. The maximum observed concentrations of LY2157299 following administration of 150 mg twice daily were at 800 and 990 ng ml−1, which were just above 2 μm (774 ng ml−1). Using LY2157299 concentrations of 10, 2, 0.2 and 0.02 μm in reference [25], specifically shown in Figure S3, showed that there was a clear response of pSMAD by immunoblotting at 10 and 2 μm. In our simulation (Figure 5), following 150 mg twice daily administration of LY2157299, concentrations were at the 2 μm level with corresponding maximum pSMAD inhibition of 60%, maintained above 50% over close to 8 h. During treatment in the study, LY2157299 was administered twice daily, hence at 12 h another dose was administered and continuous maintenance of required pSMAD inhibition was achieved. Future studies of target-related modulation of pSMAD or other TGF-β-associated biomarkers may be conducted in other studies, where pre-and post-treatment biopsies are more easily obtained than in patients with glioblastoma. Also concentrations of LY2157299 in cerebrospinal fluid will help to understand the rate this compound crosses the blood–brain barrier. Both of these measures, although useful, are invasive and difficult to obtain on a routine basis.
Figure 5.

Observed plasma concentration time profiles following 150 and 80 mg twice daily administration of LY2157299 and simulated pSMAD inhibition in tumour in patients. △, 150 mg twice daily;  , 80 mg twice daily; —, 80 mg twice daily; – – –, 150 mg twice daily
, 80 mg twice daily; —, 80 mg twice daily; – – –, 150 mg twice daily
A quantitative, PK/PD driven approach was undertaken very early in drug development, during candidate identification of a small molecule inhibiting the TGF-β RI kinase pathway administered orally [24]. This ensured that although we encountered similar cardiovascular preclinical pathology [10] to other investigators [8,9], an appropriate therapeutic window was identified between the exposure (and target-linked biomarker) required for efficacy and the exposure associated with toxicity in animals. Using a therapeutic window is not a novel idea and it has been used to establish utility functions in late phases of drug development and post-marketing in a number of therapeutic areas [26], including oncology [27]. Although utilizing such an approach early in development is unusual, it is now recommended as beneficial [28].
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare IG, ALC, AS, NSP, CPM, JMY and MML had support from Eli Lilly for the submitted work, IG, ALC, AS, NSP, CPM, JMY and MML were employees with Eli Lilly in the previous 3 years and there are no other relationships or activities that could appear to have influenced the submitted work.
We are grateful to the patients who took part in this trial.
References
- 1.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-β signaling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;12:1011–1022. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- 2.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;2:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 3.Wakefield LM, Roberts AB. TGF-β signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–29. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 5.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-β responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 6.Sawyer JS, Anderson BD, Beight D, Cambell RM, Jones ML. Synthesis and activity of new aryl-and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46:3953–3956. doi: 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
- 7.Gorelik L, Flavell RA. Abrogation of TGF beta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 8.Robinson S, Pool R, Giffin R Emerging Safety Science Workshop. Institute of Medicine of the National Academies. Washington, DC: The National Academies Press; 2007. [Google Scholar]
- 9.Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, Steel MJ, Roberts RR, Roberts A, Heier A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. 2011;39:916–924. doi: 10.1177/0192623311416259. [DOI] [PubMed] [Google Scholar]
- 10.Stauber AJ, Zimmermann JL, Berridge BR. 2006. Pathobiology of a Valvulopathy in Fischer 344 Rats Given a Transforming Growth Factor-b RI Kinase Inhibitor. Society of Toxicology, 45th Annual Meeting and Tox Expo, (abstr 290)
- 11.Bueno L, deAlwis DP, Pitou C, Glatt, Aarons L. Semi-mechanistic modeling of the tumor growth inhibitory effects of LY2157299, a new type I receptor TGF-β kinase antagonist, in mice. Eur J Cancer. 2008;44:142–150. doi: 10.1016/j.ejca.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Nat Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 13.Macdonald DR, Cascino TL, Schold SC, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 14.Calvo-Aller E, Baselga J, Glatt S, Cleverley A, Lahn M, Arteaga CL, Rothenberg ML, Carducci MA. First human dose escalation study in patients with metastatic malignancies to determine safety and pharmacokinetics of LY2157299, a small molecule inhibitor of the transforming growth factor-beta receptor I kinase. J Clin Oncol. 2008;26(Suppl) abstr 14554. [Google Scholar]
- 15.Rodon J, Baselga J, Calvo E, Seoane J, Brana I, Sicart E, Gueorguieva I, Cleverley A, Lahn MMF, Pillay S, Holdhoff M, Blakeley JO, Carducci MA. First Human Dose (FHD) Study of the oral transforming growth factor-beta (TGF-β) receptor I kinase inhibitor LY2157299 in patients with treatment-refractory malignant glioma. J Clin Oncol. 2011;29(Suppl) abstr 3011. [Google Scholar]
- 16.Beal S, Sheiner LB. The NONMEM system. Am Stat. 1980;34:118–119. [Google Scholar]
- 17.Beal S, Sheiner LB, editors. NONMEM Project Group: NONMEM Users Guides. San Francisco, CA: University of California at San Francisco; 1998. [Google Scholar]
- 18.Gueorguieva I, Ogungbenro K, Graham G, Glatt S, Aarons L. A program for individual and population optimal design for univariate and multivariate response pharmacokinetic-pharmacodynamic models. Comput Methods Programs Biomed. 2007;86:51–61. doi: 10.1016/j.cmpb.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 19.U.S. Department of Health and Human Services, Food and Drug Administration. 1999. Guidance for Industry: Population Pharmacokinetics. Available at http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133184.pdf (last accessed 29 August 2013)
- 20.U.S. Department of Health and Human Services, Food and Drug Administration. 2003. Guidance for Industry: Exposure-Response Relationships – Study Design, Data Analysis, and Regulatory Applications. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072109.pdf (last accessed 29 August 2013)
- 21.Bhattaram VA, Booth BP, Ramchandani RP, Beasley BN, Wang Y, Tandon V, Duan JZ, Baweja RK, Marroum PJ, Upooor RS, Rahman NA, Sahajwalla CG, Powell JR, Mehta MU, Gobburu JV. 2005. Impact of pharmacometrics on drug approval and labeling decisions: survey of 42 new drug applications. AAPSJ. 7, E503-E512. [DOI] [PMC free article] [PubMed]
- 22.Wang Y, Sung C, Dartois C, Ramchadani R, Booth BP, Rock E, Gobburu J. Tumor size-survival relationship in non-small cell lung cancer patients to aid early clinical development decision making. Clin Pharmacol Ther. 2009;86:167–174. doi: 10.1038/clpt.2009.64. [DOI] [PubMed] [Google Scholar]
- 23.Simeoni M, Magni P, Cammia C, De Nicolao G, Croci V, Pesenti E, Germani M, Poggesi I, Rocchetti M. Predictive pharmacokinetic-pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 2004;64:1094–1101. doi: 10.1158/0008-5472.can-03-2524. [DOI] [PubMed] [Google Scholar]
- 24.Yingling JM. Targeting the TGF-β RI kinase with LY2157299: a PK/PD-driven drug discovery and clinical development program. 2005. Proc Amer Assoc Cancer Res 46, (abstr SY13-2)
- 25.Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodon L, Folch G, Carmona MA, Pieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, Cuartas I, Raventos C, Martinez-Ricarte F, Poca MA, Garcia-Dorado D, Lahn MM, Yingling JM, Rodon J, Sahuquillo J, Baselga J, Seoane J. TGF-β receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 26.Bonate PL, Howard DR. Pharmacokinetics in Drug Development: Clinical Study Design and Analysis. Arlington, VA: Americal Association of Pharmaceutical Scientists Press; 2004. p. 572. [Google Scholar]
- 27.Rosner G, Muller P, Tang F, Madden T, Borje S. Dose individualization for high-dose anti-cancer chemotherapy. In: D'Argenio D, editor. Advanced Methods of Pharmacokinetic and Pharmacodynamic Systems Analysis. Vol. 3. New York: Kluwer Academic Publishers; 2004. pp. 239–254. [Google Scholar]
- 28.Sorger PK, Allerheiligen SRB, Abernethy DR, Altman RB, Brouwer KLR, Califano A, D'Argenio DZ, Iyenger R, Jusko WJ, Lalonde R, Lauffenburger DA, Shoichet B, Stevens JL, Subramaniam S, van der Graaf P, Vicini P. 2011. Quantitative and Systems Pharmacology in the Post-genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms (NIH White Paper by the QSP Workshop Group)