

Abstract
Aim
The aim of this study was to investigate relationships between flavin-containing mono-oxygenase 3 (FMO3) genotype and phenotype (conversion of odorous trimethylamine into non-odorous trimethylamine N-oxide) in a large Japanese cohort suffering from trimethylaminuria.
Methods
Urinary excretion of trimethylamine and trimethylamine N-oxide was determined for 102 volunteers with self-reporting symptoms of trimethylaminuria. For each we determined the sequence of the entire coding region, plus 1.3 kb of flanking intronic and 2.5 kb of the upstream region of the FMO3 gene. The affect of upstream variants on transcription was determined with a reporter gene assay.
Results
Seventy-eight subjects were diagnosed as suffering from trimethylaminuria, based on urinary excretion of <90% of total TMA as TMA N-oxide. Of these, 13 were classified as severe, 56 as moderate and nine as mild cases, excreting <43%, 48–70% and 73–83% of trimethylamine as trimethylamine N-oxide, respectively. Twenty-seven mutations were identified in FMO3, 15 in the coding region, of which eight abolish or severely impair FMO3 activity (Pro70Leu, Cys197fsX, Thr201Lys, Arg205Cys, Met260Val, Trp388Ter, Gln470Ter and Arg500Ter), and 12 in the upstream region. The mutations segregate into 19 haplotypes, including four different combinations of upstream mutations, each of which reduces transcriptional activity in comparison with the ancestral upstream sequence of FMO3.
Conclusions
Comparisons of genotype and phenotype reveal that severe trimethylaminuria is caused by loss of function mutations in FMO3. For moderate and mild cases the situation is more complex, with most resulting from factors other than FMO3 genotype. Our results have implications for the diagnosis and management of the disorder.
Keywords: FMO3, genetic polymorphism, trimethylamine, trimethylamine N-oxide, urine
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Trimethylaminuria results from reduced capacity to convert trimethylamine to trimethylamine N-oxide, a reaction catalyzed by FMO3.
Mutations of FMO3 are known to cause trimethylaminuria, but an understanding of the phenotypic consequences of different FMO3 genotypes (haplotypes) is lacking.
WHAT THIS STUDY ADDS
Severe trimethylaminuria is caused by mutations that severely impair FMO3 activity.
Most affected individuals present with moderate or mild forms of the disorder, due to factors other than FMO3 genotype.
Although for the majority of sufferers sequencing of FMO3 would not be informative, reduction of trimethylamine burden should prove beneficial.
Introduction
Humans possess five functional forms of flavin-containing mono-oxygenases (FMOs; EC 1.14.13.8), designated FMO1 to FMO5 [1–3]. FMOs catalyze the NADPH-dependent oxidation of a wide range of foreign chemicals that include therapeutic drugs, environmental chemicals and dietary components [4]. An important dietary component is choline, present in many foods, which commensal gut bacteria break down to produce the volatile and odorous chemical trimethylamine (TMA). FMO3 (MIM 136132), present in the liver, catalyzes the N-oxygenation of TMA to produce non-odorous TMA N-oxide [5, 6]. Individuals with defective FMO3 suffer from the disorder trimethylaminuria [TMAuria (MIM 60279)] and excrete excessive amounts of TMA in bodily secretions, including sweat, urine and reproductive fluids [5, 7, 8]. Consequently, they exhibit an unpleasant body and breath odour and suffer the social and psychological problems associated with body malodours [5].
TMAuria is diagnosed by an abnormally high ratio of TMA : TMA N-oxide in the urine [7, 9, 10]. Primary TMAuria is an inherited disorder caused by mutations in the FMO3 gene [11–14]. More than 30 mutations have been identified that abolish or severely reduce FMO3 activity [15–17]. Secondary TMAuria can be caused by environmental factors, often in combination with genetic variants. Some secondary forms are acquired, during adult life, as a consequence of viral hepatitis [5, 18] or of disease states such as liver cirrhosis [19] or ureamia [20]. Others are transient, being associated with menstruation [21, 22], early childhood [23–26] when FMO3 expression is immature [27] or precursor overload [5, 20] and symptoms are more pronounced in those possessing certain combinations of polymorphic variants of FMO3.
In a previous study of 22 Japanese self-reporting for TMAuria [28] we identified 16 mutations of FMO3, which segregated into seven distinct haplotypes, obtained estimates for the ages of the mutations and provided evidence for balancing selection at the FMO3 locus. In the present study, we analyzed a much larger group of Japanese self-reporting with symptoms of TMAuria. Nineteen distinct FMO3 haplotypes were identified and the effects of these on the activity and expression of FMO3 were determined. Our results provide insight into the relationships between FMO3 genotype and TMAuria phenotype and have implications for the diagnosis and management of the disorder.
Methods
Human subjects
One hundred and two unrelated Japanese (50 male and 52 female), ranging in age from 3 to 67 years, were recruited in response to an Internet article, because of self-perception of a fishy body odour. Informed consent was obtained from all subjects or from their parents and the study was approved by the ethics committee of Showa Pharmaceutical University (H22-22-7).
Phenotype analysis
The potential TMAuria sufferers were diagnostically assessed by determining the percentage of total urinary TMA (i.e., TMA + TMA N-oxide) excreted as TMA N-oxide. Concentrations of TMA and TMA N-oxide, in the first sample of urine obtained after an overnight fast, were determined by gas chromatography using a flame ionization detector [29]. Urine samples of women were not collected when they were menstruating. Briefly, TMA concentration was directly analyzed by headspace gas chromatography after samples were made alkaline (pH >12), by addition of 10 m NaOH, and preheated at 95°C for 20 min. TMA N-oxide concentrations were calculated by subtracting the concentration of free TMA from that of total TMA (free TMA + TMA N-oxide) after chemical reduction of TMA N-oxide to TMA through the use of TiCl3. Intra-and inter-assay variations for free and total TMA were within 5%, using previously described gas chromatography conditions [29]. The detection limit for TMA concentration in urine was 0.01 μg ml−1.
DNA analysis
Samples of buccal cells were obtained from each individual by scraping the inside of the mouth with a cotton swab. Genomic DNA was prepared from buccal cells through the use of QIAamp DNA Mini Kits (QIAGEN, Hilden, Germany). Each of the eight coding exons of FMO3 [30], together with some flanking intronic and 3′-untranslated regions (a total of 2.9 kb), plus four sections (a total of 2.5 kb) from within a 3.4-kb region upstream of the FMO3 gene were amplified by PCR in 25 μl reaction mixtures containing 50 ng of genomic DNA, Takara LA-PCR buffer (Takara Bio, Otsu, Japan), 2.0 mm MgCl2, 0.2 mm dNTPs, 5.0 pmol of each primer and 1.0 U Takara LA Taq DNA polymerase (Takara Bio, Otsu, Japan). The PCR conditions consisted of an initial denaturation at 94°C for 1 min, followed by 35 cycles of 94°C for 30 s, 55 or 57°C for 30 s and 72°C for 45 s. Primers for amplification of these regions and for DNA sequencing are described in supplementary material. Both strands of PCR products were directly sequenced using a BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). Excess dye was removed by ethanol precipitation and eluates were analyzed on a PRISM 3730xl DNA analyzer (Applied Biosystems). The sequence of the complete human FMO3 gene described in the GenBank (accession number AL021026) was used as a reference. Allele frequencies for each DNA variant were determined by gene counting. Haplotypes were inferred through the use of SNP Alyze (DYNACOM, Chiba, Japan) and HAP [31] programs.
Construction of FMO3-luciferase reporter gene plasmids
The 5′-flanking region of the FMO3 gene, extending from −5167 to −1764 (the transcriptional initiation site) (numbered relative to the A of the ATG translational initiation codon), was amplified from human genomic DNA of individuals with the appropriate 5′-upstream haplotype class by PCR, using the primers hFMO3 550 + Mlu (5′-CAGTACGCGTAGACATGAGTATCAGGCTAAC-3′) and hFMO3 3953 + Bgl primer (5′-ACTGAGATCTAATGTTCTAGTGAGCCTACATAT-3′). Amplification products were digested with MluI and BglII, then inserted into a MluI/BglII-digested pGL3-Enhancer promoterless luciferase reporter vector (Promega, Madison, WI, USA). Recombinant reporter plasmids were cloned and the identity of FMO3 haplotypes was confirmed by DNA sequencing.
Transient transfection and reporter gene assays
Human HepG2 hepatoma cells were cultured at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum (Biochrom, Berlin, Germany). Cells were plated 24 h before transfection in 24-well plates (0.5 × 105 cells/well). HepG2 cells were co-transfected, through the use of Lipofectamine2000 Reagent (Gibco-BRL, Rockville, MD, USA), pGL3-FMO3 is the test plasmid (400 ng/well), and the thymidine kinase (TK) promoter-driven control renilla luciferase plasmid pRL-TK (40 ng/well) (Promega). Cells were harvested and analyzed for luciferase activities.
Luciferase reporter gene activity was evaluated with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Forty-eight hours after transfection, the cells were washed once with phosphate-buffered saline (pH 7.4) and lysed by passive lysis buffer (100 μl/well). After incubation at room temperature for 15 min, lysates were collected and centrifuged at 15 000 rev min–1 at 4°C for 30 s in an Eppendorf microfuge. Supernatants (10 μl) were mixed with the luciferase reagent (20 μl) and luciferase reporter gene activity was measured in a MultimodeReder TriStar LB 941 (Berthold, Bad Wildbad, Germany). Results were normalized by dividing firefly luciferase activities, derived from the pFMO3 plasmids, by renilla luciferase activity, derived from the control plasmid pRL-TK.
Statistical analysis
To test whether the distribution of haplotypes identified as containing mutations that severely affect FMO3 activity was random with respect to the rank order of values for the percent of total TMA excreted as TMA N-oxide, we carried out a runs test using the function runs.pvalue in the package randomizeBE [32] in R [33]. In this test, individuals were classified as either 1 (homozygous or compound heterozygous for haplotypes containing a mutation that reduces FMO3 activity by at least 50%) or 0 (not homozygous or compound heterozygous for such haplotypes).
Results and discussion
Metabolic capacity of FMO3
One hundred and two Japanese individuals self-reporting with symptoms of TMAuria were analyzed for the metabolic capacity of FMO3, as measured by the percentage of total TMA, i.e., TMA plus TMA N-oxide, excreted in the urine as the N-oxide (Figure 1). Of the 102 individuals, 78 (76%) displayed symptoms of TMAuria, as judged by urinary excretion of <90% of total TMA as TMA N-oxide, with the remaining 24 (24%) being unaffected, with a urinary excretion of >90% of total TMA as the N-oxide [7–9]. TMAuria has been classified as severe or mild, based, respectively, on excretion of <60% or 60–90% of total TMA as TMA N-oxide [7–9]. However, this classification is based on studies of small numbers of individuals and has been refined, as outlined below, to reflect gaps in the spectrum of urinary excretion of total TMA as TMA N-oxide in the large cohort of individuals in the present study (see also Table 2, below). Of the affected individuals, 13 (17%) excreted <43% of total TMA as TMA N-oxide and were classified as severe TMAurics, 56 (72%) excreted 48–70% of total TMA as TMA N-oxide and were classified as moderate TMAurics, and nine (11%) excreted 73–83% of total TMA as TMA N-oxide and were classified as mild TMAurics.
Figure 1.
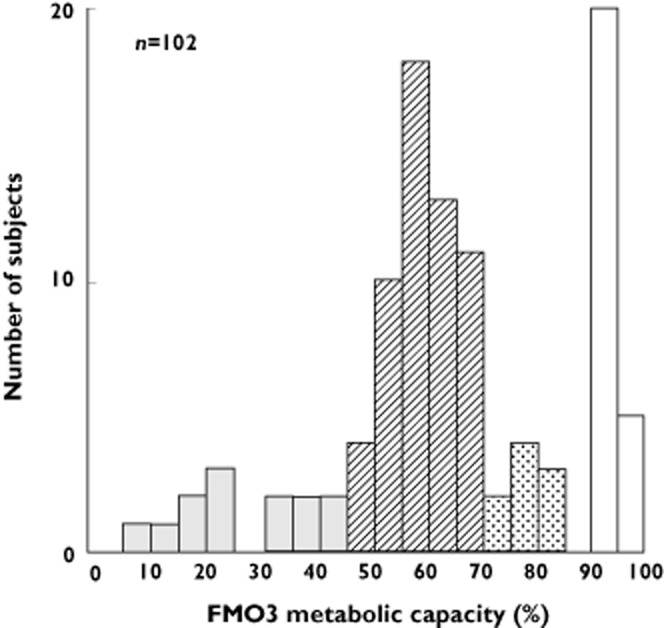
Distribution of metabolic capacity of FMO3 among a Japanese cohort self-reporting for symptoms of TMAuria. Metabolic capacity of FMO3 is expressed as percent of total urinary TMA excreted as TMA N-oxide. Based on the percent of total TMA excreted as TMA N-oxide, individuals were classified as suffering from severe (<43%) (solid bars), moderate (48–70%) (hatched bars) or mild (73–83%) (stippled bars) TMAuria or unaffected (>90%) (open bars)
Table 2.
Genotype and urinary excretion of TMA and TMA N-oxide of individuals of the Japanese cohort
| ID | Gender | Age (years) | Genotype (haplotype pair) | TMA + TMA N-oxide (mmol mol−1 creatinine) | TMA (mmol mol−1 creatinine) | TMA N-oxide (as % of TMA + TMA N-oxide) |
|---|---|---|---|---|---|---|
| Severe TMAuria | ||||||
| 1 | M | 18 | 12/14 | 401 | 363.0 | 9.5 |
| 2 | F | 19 | 19/19 | 84 | 75.7 | 10.2 |
| 3 | M | 22 | 5/13 | 91 | 76.0 | 16.5 |
| 4 | F | 36 | 9/16 | 27 | 22.5 | 17.8 |
| 5 | M | 58 | 17/17 | 51 | 40.3 | 20.6 |
| 6 | M | 6 | 13/19 | 23 | 17.8 | 21.2 |
| 7 | F | 7 | 17/19 | 136 | 106.4 | 21.8 |
| 8 | F | 28 | 1/3 | 59 | 39.5 | 33.1 |
| 9 | M | 7 | 13/19 | 67 | 44.9 | 33.3 |
| 10 | M | 17 | 12/14 | 82 | 50.9 | 38.1 |
| 11 | M | 27 | 13/18 | 110 | 66.2 | 40.0 |
| 12 | F | 22 | 13/13 | 25 | 14.6 | 40.4 |
| 13 | F | 31 | 9/10 | 41 | 23.6 | 42.7 |
| Moderate TMAuria | ||||||
| 14 | M | 21 | 1/9 | 42 | 21.5 | 48.3 |
| 15 | F | 60 | 1/3 | 127 | 64.5 | 49.4 |
| 16 | M | 23 | 1/3 | 167 | 84.0 | 49.7 |
| 17 | M | 35 | 1/1 | 63 | 31.7 | 50.0 |
| 18 | M | 38 | 1/15 | 41 | 20.3 | 50.5 |
| 19 | M | 31 | 1/9 | 175 | 85.9 | 50.9 |
| 20 | M | 42 | 3/13 | 62 | 29.8 | 51.5 |
| 21 | F | 42 | 1/9 | 68 | 32.7 | 51.7 |
| 22 | M | 30 | 7/8 | 84 | 40.2 | 52.1 |
| 23 | F | 30 | 9/16 | 54 | 26.0 | 52.2 |
| 24 | F | 5 | 3/4 | 28 | 13.2 | 53.0 |
| 25 | F | 57 | 1/9 | 30 | 13.9 | 53.1 |
| 26 | F | 36 | 1/4 | 92 | 42.6 | 53.8 |
| 27 | M | 24 | 3/3 | 72 | 32.6 | 54.6 |
| 28 | F | 32 | 1/3 | 56 | 25.2 | 55.2 |
| 29 | F | 29 | 1/15 | 27 | 12.1 | 55.2 |
| 30 | M | 37 | 1/9 | 99 | 43.8 | 55.6 |
| 31 | M | 60 | 9/15 | 49 | 21.2 | 56.6 |
| 32 | M | 33 | 9/15 | 69 | 30.0 | 56.6 |
| 33 | F | 22 | 1/9 | 178 | 76.0 | 57.3 |
| 34 | F | 28 | 3/9 | 55 | 23.0 | 57.8 |
| 35 | M | 24 | 1/17 | 192 | 80.6 | 58.0 |
| 36 | F | 15 | 1/1 | 35 | 14.4 | 58.2 |
| 37 | F | 33 | 1/9 | 29 | 12.2 | 58.2 |
| 38 | M | 40 | 1/1 | 39 | 16.3 | 58.5 |
| 39 | M | 27 | 9/15 | 198 | 82.0 | 58.5 |
| 40 | M | 36 | 1/3 | 109 | 45.4 | 58.5 |
| 41 | F | 41 | 1/1 | 126 | 51.5 | 59.2 |
| 42 | M | 28 | 15/15 | 108 | 44.0 | 59.3 |
| 43 | M | 38 | 1/15 | 87 | 35.3 | 59.4 |
| 44 | M | 30 | 1/15 | 204 | 81.6 | 59.9 |
| 45 | F | 37 | 1/1 | 43 | 17.2 | 59.9 |
| 46 | F | 28 | 14/15 | 382 | 152.1 | 60.2 |
| 47 | M | 28 | 1/1 | 78 | 30.8 | 60.3 |
| 48 | M | 25 | 2/15 | 130 | 51.1 | 60.7 |
| 49 | M | 38 | 1/3 | 139 | 53.2 | 61.7 |
| 50 | M | 30 | 1/1 | 62 | 23.7 | 61.8 |
| 51 | M | 31 | 6/15 | 60 | 22.9 | 62.0 |
| 52 | M | 37 | 1/3 | 27 | 10.2 | 62.4 |
| 53 | M | 26 | 1/1 | 110 | 40.4 | 63.3 |
| 54 | M | 25 | 9/15 | 123 | 44.8 | 63.6 |
| 55 | M | 40 | 1/18 | 68 | 24.7 | 63.9 |
| 56 | F | 32 | 3/9 | 106 | 38.1 | 64.0 |
| 57 | F | 33 | 1/9 | 74 | 26.5 | 64.2 |
| 58 | F | 28 | 1/3 | 129 | 46.2 | 64.2 |
| 59 | F | 36 | 1/15 | 96 | 33.3 | 65.5 |
| 60 | F | 30 | 3/15 | 53 | 18.3 | 65.5 |
| 61 | F | 45 | 1/3 | 54 | 17.9 | 66.9 |
| 62 | M | 32 | 3/15 | 309 | 99.0 | 68.0 |
| 63 | F | 28 | 1/9 | 178 | 56.6 | 68.2 |
| 64 | M | 20 | 9/9 | 93 | 29.3 | 68.5 |
| 65 | F | 38 | 15/15 | 85 | 26.7 | 68.6 |
| 66 | F | 28 | 1/9 | 72 | 22.2 | 69.1 |
| 67 | M | 31 | 1/1 | 174 | 53.4 | 69.3 |
| 68 | M | 38 | 3/9 | 47 | 14.4 | 69.4 |
| 69 | F | 4 | 9/11 | 422 | 127.0 | 69.9 |
| Mild TMAuria | ||||||
| 70 | M | 3 | 1/3 | 385 | 101.3 | 73.7 |
| 71 | F | 31 | 1/1 | 70 | 18.3 | 73.9 |
| 72 | F | 56 | 15/15 | 90 | 22.5 | 75.1 |
| 73 | F | 18 | 3/15 | 60 | 13.5 | 77.3 |
| 74 | M | 6 | 1/3 | 111 | 22.8 | 79.5 |
| 75 | F | 31 | 1/15 | 39 | 7.7 | 80.0 |
| 76 | F | 30 | 1/9 | 121 | 22.1 | 81.7 |
| 77 | F | 67 | 9/9 | 102 | 18.6 | 81.8 |
| 78 | M | 21 | 1/1 | 240 | 42.2 | 82.4 |
| Unaffected | ||||||
| 79 | M | 31 | 1/9 | 287 | 28.7 | 90.0 |
| 80 | F | 41 | 2/4 | 293 | 28.4 | 90.3 |
| 81 | F | 26 | 1/1 | 29 | 2.8 | 90.5 |
| 82 | F | 30 | 1/9 | 320 | 29.3 | 90.8 |
| 83 | F | 32 | 3/15 | 154 | 13.6 | 91.1 |
| 84 | F | 29 | 1/15 | 34 | 3.0 | 91.2 |
| 85 | F | 38 | 3/9 | 96 | 8.4 | 91.2 |
| 86 | F | 10 | 1/3 | 70 | 6.1 | 91.2 |
| 87 | F | 38 | 1/1 | 48 | 4.2 | 91.3 |
| 88 | M | 23 | 3/9 | 360 | 31.1 | 91.4 |
| 89 | F | 44 | 3/9 | 189 | 16.1 | 91.5 |
| 90 | M | 33 | 9/16 | 76 | 5.9 | 92.2 |
| 91 | F | 26 | 3/9 | 96 | 7.4 | 92.3 |
| 92 | F | 31 | 1/4 | 136 | 10.5 | 92.3 |
| 93 | F | 30 | 9/15 | 67 | 5.1 | 92.4 |
| 94 | M | 21 | 15/16 | 31 | 2.2 | 93.0 |
| 95 | F | 34 | 1/15 | 168 | 11.6 | 93.1 |
| 96 | M | 45 | 1/9 | 66 | 4.4 | 93.3 |
| 97 | M | 29 | 1/1 | 96 | 5.4 | 94.4 |
| 98 | F | 31 | 3/9 | 182 | 7.3 | 96.0 |
| 99 | M | 30 | 1/16 | 76 | 2.3 | 97.0 |
| 100 | M | 40 | 2/15 | 225 | 5.5 | 97.6 |
| 101 | M | 52 | 1/14 | 136 | 3.1 | 97.7 |
| 102 | F | 35 | 13/15 | 50 | 0.9 | 98.3 |
ID numbers for individuals of the Japanese cohort self-reporting for symptoms of TMAuria were assigned according to increasing percentage of total urinary TMA excreted as TMA N-oxide. IDs 1–13, 14–69 and 70–78, were diagnosed as suffering, respectively, from severe, moderate or mild trimethylaminuria (TMAuria). IDs 79–102 were diagnosed as unaffected by TMAuria. F, female; M, male; TMA, trimethylamine.
In addition to resulting in TMAuria, compromised FMO3 activity has implications for the ability of an individual to metabolize therapeutic drugs that are substrates of FMO3, as has been found for the non-steroidal anti-inflammatories benzydamine [34] and sulindac [35, 36].
FMO3 nucleotide sequence variation
To gain insight into the genotypes underlying the various phenotypes a total of 5.4 kb of the FMO3 gene were sequenced from each of the 102 individuals studied. This comprised the entire coding region (533 codons, including the stop codon), 1.3 kb of flanking intronic and 3′-untranslated regions and a total of 2.5 kb of 5′-flanking sequence from four separate regions extending from −5259 to −2076, numbered relative to the A of the ATG translational initiation codon. Twenty-six diallelic single-nucleotide polymorphisms (SNPs) and one 2-bp deletion were identified (Table 1). Twenty of the SNPs were transitions and six were transversions. Twelve of the SNPs were in the 5′-flanking region and 14 in the coding region. Twelve of the coding-region SNPs were non-synonymous substitutions. Of these, nine were mis-sense mutations at positions 11203C>T (Pro70Leu) in exon 3, 15036A>G (Asn114Ser) and 15167G>A (Glu158Lys) in exon 4, 15530T>A (Asp198Glu), 15538C>A (Thr201Lys) and 15549C>T (Arg205Cys) in exon 5, 18281G>A (Val257Met) and 18290A>G (Met260Val) in exon 6, and 21443A>G (Glu308Gly) in exon 7 and three were nonsense mutations at positions 21684G>A (Trp388Ter) in exon 7, and 24592C>T (Gln470Ter) and 24682C>T (Arg500Ter) in exon 9. Two synonymous SNPs were present at positions 15136C>T (Ser147Ser) in exon 4 and 21375T>C (Asn285Asn) in exon 7. The 2-bp deletion occurred in exon 5 at 15527_15528delTG (Cys197fsX).
Table 1.
Variant alleles, their frequency in the Japanese cohort and their effect on FMO3 activity
| dbSNP accession number | Variant | Effect on amino acid | †Effect on protein activity | ‡Frequency of derived allele |
|---|---|---|---|---|
| rs6608461 | g.-5109G>C | 0.613 | ||
| rs72637246 | g.-4600T>C | 0.206 | ||
| rs66937021 | g.-3788T>C | 0.206 | ||
| rs1736554 | g.-3606G>A | 0.186 | ||
| rs3754487 | g.-3549C>T | 0.206 | ||
| rs1736555 | g.-3548A>G | 0.600 | ||
| rs3754489 | g.-3544C>T | 0.206 | ||
| rs28363515 | g.-2962T>C | 0.206 | ||
| rs12404183 | g.-2854T>C | 0.206 | ||
| rs1736560 | g.-2650C>G | 0.600 | ||
| rs12404218 | g.-2543T>A | 0.206 | ||
| rs3754491 | g.-2177G>C | 0.206 | ||
| g.11203C>T | P70L [39] | ∼50% reduction [39] | 0.005 | |
| rs186763441 | g.15036A>G | N114S [37, 48] | None [37] | 0.005 |
| rs1800822 | g.15136C>T | S147S | None | 0.186 |
| rs2266782 | g.15167G>A | E158K [11, 38] | Minor [38] | 0.206 |
| rs3832024 | g.15527_15528delTG | C197fsX [13, 37] | Severe [13, 37] | 0.039 |
| g.15530T>A | [D198E]* | 0.039 | ||
| g.15538C>A | T201K [37, 38, 49] | Severe [37, 38] | 0.010 | |
| rs28363549 | g.15549C>T | R205C [13, 28, 37, 38] | ∼50% reduction [13, 37, 38] | 0.020 |
| rs1736557 | g.18281G>A | V257M [38, 40] | None [38, 40] | 0.196 |
| g.18290A>G | M260V [37, 38, 49] | Severe [37, 38] | 0.020 | |
| rs909530 | g.21375T>C | N285N [49] | None | 0.623 |
| rs2266780 | g.21443A>G | E308G [12, 38] | Minor [12, 38] | 0.196 |
| g.21684G>A | W388X [37, 48] | Severe [37] | 0.010 | |
| g.24592C>T | Q470X [37, 48] | Severe [37] | 0.005 | |
| g.24682C>T | R500X [13, 37, 49] | Severe [13, 37] | 0.025 |
Although the FMO3 variant g.15530T>A predicts a D to E change in amino acid residues at position 198, it occurs in cis with FMO3 g.15527_15528delTG, which introduces a stop codon at position 197 of FMO3, and thus would not be represented in the resultant truncated polypeptide.
Effects as reported in the literature: Severe: >90% decrease of enzyme activity; minor: <10% decrease in enzyme activity; none: no effect on enzyme activity.
Refers to frequency of the derived allele in the cohort of individuals self-reporting for symptoms of TMAuria.
Comparison with the FMO3 sequence of the chimpanzee (Pan troglodytes) (NM_001009092.1) revealed that for each diallelic SNP observed in humans, one of the two alleles was present at the corresponding position in the chimpanzee (Table 1). The ancestral state of each SNP was inferred from the chimpanzee sequence. For 22 of the 26 SNPs, the more common human variant corresponded to the inferred ancestral allele. The exceptions were the SNPs at −5109, −3548, −2650 and +21375.
Functional consequences of coding region mutations
All three nonsense mutations and the frameshift mutation are known to result in loss of function of FMO3 catalytic activity [13, 37] (Table 1). Of the mis-sense mutations, T201K [37, 38] and M260V [37, 38] result in loss of function or severe impairment of FMO3 activity, P70L [39] and R205C [13, 37, 38] reduce enzyme activity by ∼50%, E158K [11] and E308G [12] have little effect individually, but have a moderate effect when present in cis [16, 38], whereas N114S [37] and V257M [38, 40] have virtually no effect on catalytic activity (Table 1). The effect of D198E is unknown but, because it occurs in cis with the frameshift mutation C197fsX, it would not be represented in the resultant truncated polypeptide. Both synonymous mutations are common in the general population [17] and, thus, are unlikely to affect either RNA processing or rate of translation.
With the exception of E158K, V257M and E308G, which are present in African, European and Asian populations as polymorphic variants [17], all of the other mis-sense mutations, the nonsense mutations and the frameshift mutation have been reported only in Japanese individuals, suggesting that these rare mutations may have occurred relatively recently in the Japanese population. The age of the R205C mutation has been estimated as 62 kyears (SD = 52 kyears) [28], which supports an origin that post dates the migration of modern humans out of Africa.
Haplotypes and their predicted impact
The 27 sequence variants identified in the sample of 204 chromosomes segregate into 19 distinct haplotypes. These, together with a haplotype composed of the ancestral state of each allele, inferred from the chimpanzee, are shown in Figure 2. The most common haplotype in the sample (haplotype 1), with a frequency of 0.333, is identical to the inferred ancestral haplotype at all but one position (the synonymous SNP at 21375). Haplotypes 1, 2, 3 and 4, with a combined frequency of 0.505, contain no non-synonymous mutations and, thus, each encodes a native FMO3 protein. Haplotypes 6, 7, 15 and 16, with a combined frequency of 0.177, each contain a single SNP (N114S, E158K or V257M) that has little or no effect on enzyme activity and, thus, would encode a protein with essentially normal activity. Haplotypes 8, 9 and 10, with a combined frequency of 0.186, contain two SNPs (E158K and E308G), which, when present together, result in a moderate reduction in enzyme activity. Haplotypes 5 and 14, with a combined frequency of 0.025, each contain a single SNP (P70L or R205C) that reduces enzyme activity by about 50%. Six haplotypes, 11, 12, 13, 17, 18 and 19, with a combined frequency of 0.109, each contain a mutation that would result in the loss or severe impairment of FMO3 activity.
Figure 2.

Haplotypes, their estimated frequencies in the Japanese cohort and their effect on activity and expression on FMO3. The nucleotide positions of the variants are given relative to the A of the ATG translational initiation codon. Nucleotide variants are shown as derived changes in comparison with the chimpanzee sequence. Bases identical to the chimpanzee sequence are indicated by a dash. Haplotypes are numbered arbitrarily. *Although the T > A variant at +15530 predicts a D to E change in amino acid residue at position 198, it occurs in cis with g.15527_15528delTG (C197fsX) and, thus, would not be represented in the resultant truncated polypeptide. 1Refers to frequency of the haplotype in the cohort of individuals self-reporting for symptoms of TMAuria. 2Severe effect: > 90% decrease of enzyme activity; moderate effect: 10–20% decrease in enzyme activity; −: no effect on enzyme activity. 3Moderate effect: 30–45% decrease in transcription in vitro; −: no effect on transcription
The 12 SNPs present in the 5′-flanking region of the FMO3 gene segregate into five distinct haplotype groups (Figure 3). The effect on transcription of each of the variant upstream haplotypes was investigated by measuring its ability to drive expression in vitro of a luciferase reporter gene. In comparison with the ancestral 5′-flanking region present in haplotypes 1, 5, 6, 14 and 19, each of the four variant upstream haplotype groups displayed a decrease in transcriptional activity, ranging from 30–45% (Figure 3). Although the impact of these haplotypes in vivo has not been confirmed, it is possible that they contribute to TMAuria by affecting expression of the FMO3 gene. Thus, although eight haplotypes (1, 2, 3, 4, 6, 7, 15 and 16) encode a native protein or one that has essentially full activity, only haplotypes 1 and 6 might also have normal levels of expression. Therefore, of the 19 haplotypes identified in the sample, 17, with a combined frequency of 0.662, encode proteins that are inactive or have reduced activity, or have potentially compromised expression.
Figure 3.
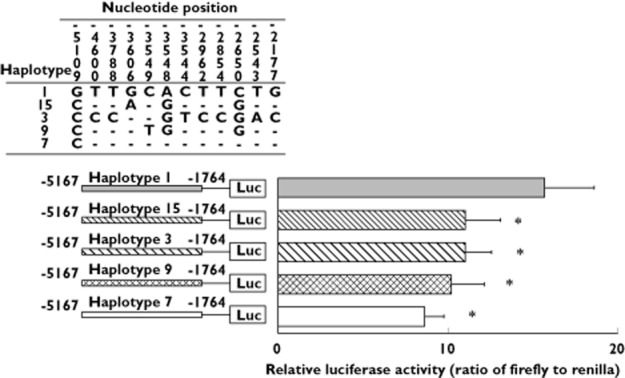
Effect of upstream mutations of FMO3 on transcription. Regions of FMO3 haplotypes extending from −5167 to −1764 (numbered relative to the A of the ATG translational initiation codon) were cloned into a pGL3-Enhancer promoterless firefly luciferase reporter vector [47]. The constructs were used to co-transfect HepG2 human hepatoma cells, along with pRL-TK, which contains a renilla luciferase gene under the control of a thymidine kinase promoter. Upper section: sequence variants and their positions relative to the A of the ATG of the initiation codon. Dashes indicate bases identical to that present in haplotype 1 (the ancestral sequence). The upstream sequence of haplotype 1 is identical to those in haplotypes 5, 6, 14 and 19; that of haplotype 15 is identical to those in haplotypes 2, 17 and 18; that of haplotype 3 is identical to those in haplotypes 10, 13 and 16; that of haplotype 9 is identical to those in haplotypes 4 and 12; and that of haplotype 7 is identical to those in haplotypes 8 and 11. Lower section: transcriptional activity of promoter haplotypes. Horizontal bars indicate the expression of firefly luciferase relative to renilla luciferase. Luc, firefly luciferase gene. Data represent means ± SD of three independent experiments. *P < 0.01, significantly different from transcriptional activity of the ancestral haplotype (haplotype 1) by one-way anova followed by Dunnet's multiple comparison tests
One of the variant upstream haplotype groups, consisting of haplotypes 7, 8 and 11, contains only a single 5′-flanking-region SNP, −5109G>C, which is sufficient to decrease transcription in vitro by 45%. The SNP occurs in all three of the other variant upstream haplotype groups, but the presence in these of additional SNPs has no further effect on transcription, indicating that the reduction of transcriptional activity of each of the variant upstream haplotypes is a consequence of the −5109G>C SNP.
Two other variant upstream haplotypes that reduce transcription in vitro have been reported [41]. One contains the SNPs −2589C>T and −2106G>A, neither of which are present in the individuals of the present study, the other contains −2106G>A and −1961T>C, the position of the latter SNP being outside the region sequenced in the present study.
Another variant haplotype, which contains the SNPs −2650C>G, −2543T>A and −2177G>C, has been reported to increase transcription in vitro by eight-fold [41]. The effect was found to be due largely to the −2177G>C SNP, but was modified by the presence of other SNPs. All three of these SNPs are present in haplotypes 3, 10, 13 and 16 of the Japanese cohort, which comprise a haplotype group that exhibits decreased transcriptional activity. In addition to −2650C>G, −2543T>A and −2177G>C, haplotypes 3, 10, 13 and 16 contain seven other 5′-flanking-region SNPs, including −5109G>C, the presence of which might account for the difference in effect on transcriptional activity of these haplotypes compared with that reported by Koukouritaki et al. [41].
Relationship between TMAuria phenotype and FMO3 genotype
The large number of affected individuals in the cohort and the wide range in the severity of their symptoms presented an opportunity to gain a better understanding of the relationship between TMAuria phenotype and FMO3 genotype (Table 2).
To what extent can an individual's phenotype with respect to TMAuria be accounted for by their FMO3 genotype? Of the 24 unaffected individuals, i.e., those excreting >90% of total TMA as TMA N-oxide, only one, ID 102, has a haplotype that contains a known loss of function mutation. The individual is heterozygous for haplotype 13, which contains C197fsX, and, therefore, is genotypically a carrier. The other haplotype of this individual is 15, which is one that contains upstream variants that might moderately reduce expression. The individual has a very high urinary ratio of TMA N-oxide excretion (98.3%), although this could be a consequence of a relatively low total urinary TMA of 50 mmol mol−1 creatinine. Another individual, ID 101, is heterozygous for haplotype 14, containing R205C, which results in ∼50% reduction of FMO3 activity. However, the other haplotype of this individual is haplotype 1, which would express normal amounts of a fully functional protein. The genotype of ID 101 would, thus, predict a FMO3 metabolic capacity above that associated with a carrier, which is compatible with a high ratio of urinary TMA N-oxide excretion, of 97.7%. Some of the remaining unaffected individuals are hetero or homozygous for haplotypes that contain upstream variants that might reduce expression. In some of these individuals, those with a 3/9 or 9/16 genotype, one of the haplotypes (haplotype 9) also contains E158K/E308G in cis and, thus, would produce a protein with moderately reduced activity. Nevertheless, the genotype of each of these individuals predicts a FMO3 metabolic capacity at least equivalent to that of an asymptomatic carrier, which is reflected by urinary ratios of TMA N-oxide excretion >90%, in one case in the context of high total urinary TMA (360 mmol mol−1 creatinine). Therefore, for all of the individuals diagnosed as unaffected by TMAuria, phenotype is accounted for by FMO3 genotype.
Of the 13 individuals classified as severe TMAurics, i.e., those excreting <43% of total TMA as TMA N-oxide, seven (IDs 2, 5, 6, 7, 9, 11 and 12) are homozygous or compound heterozygous for mutations known to result in loss of function or severe impairment of enzyme activity and, thus, would have no or very little FMO3 activity. Three others, IDs 1, 3 and 10, are heterozygous for haplotypes 12 or 13, which contain mutations that abolish (C197fsX) or severely impair (T201K) enzyme activity. In each of these individuals the other haplotype present is 5 or 14, both of which contain mutations (P70L or R205C) that decrease enzyme activity by about 50%, a genotype that would predict a FMO3 metabolic capacity of about 25% of normal. The relatively large difference between IDs 1 and 10 in the percent of total TMA excreted as the N-oxide (9.5 compared with 38), despite their identical FMO3 genotypes, is a consequence of a marked difference in the total urinary TMA (401 vs. 82 mmol mol−1 creatinine) and the amount of TMA N-oxide excreted is similar, at 38 and 31 mmol mol−1 creatinine, respectively. Furthermore, when individuals were ranked according to the percent of total TMA excreted as TMA N-oxide (Table 2), those homozygous or compound heterozygous for haplotypes containing mutations that severely impair FMO3 activity (IDs 2, 5, 6, 7, 9, 11 and 12) or compound heterozygous for such a haplotype and one containing a mutation that decreases enzyme activity by 50% (IDs 1, 3 and 10) were more clustered than expected by chance, tending to show lower values of urinary ratios of TMA N-oxide excretion (runs test: P = 1.58 × 10−8).
Of the other three severe TMAurics, one (ID 13) was heterozygous for two haplotypes, 9 and 10, predicted to result in similar reductions in both the amount and activity of FMO3. However, two other individuals homozygous for haplotype 9 (ID 64 and ID 77) have considerably higher urinary ratios of TMA N-oxide excretion, 68.5 and 81.8%, respectively. The remaining two individuals classified as severe TMAurics, IDs 4 and 8, have genotypes of 9/16 and 1/3, respectively. In other individuals who have the 9/16 genotype urinary ratios of TMA N-oxide excretion were 52.2% (ID 23) and 92.2% (ID 90) and in those who have the 1/3 genotype the urinary ratios ranged from 49.4–91.2%, although in all but one it is <80%. Therefore, the phenotypes of 10 of the 13 individuals classified as severe TMAurics can be explained by their FMO3 genotypes. Given the severity of their phenotypes, the other three, having no coding region causative mutations, were likely to have unidentified mutations elsewhere in the FMO3 gene that severely affect expression, by perturbing transcription or RNA processing.
Of the 65 individuals classified as either moderate (48–70% urinary excretion of total TMA as TMA N-oxide) or mild (73–83% urinary excretion) TMAurics, none was homozygous or compound heterozygous for mutations known to abolish or severely impair FMO3 activity and only four (IDs 20, 35, 55 and 69) were heterozygous for such mutations. The genotype predicted to have the most severe consequence was found in ID 69, who was heterozygous for haplotype 11, which contains the loss of function mutation Q470X, and for haplotype 9, containing E158K and E308G, together with upstream SNPs, which could result in moderate reductions in both the expression and activity of FMO3. In addition, ID 69 is only 4 years old and, thus, might not have attained fully mature expression of FMO3 [27]. However, this individual, despite having the highest urinary excretion of total TMA (422 mmol mol−1 creatinine) of any in the cohort, excreted a relatively high percentage (69.9%) as the N-oxide.
Eleven of the moderate or mild TMAurics were homozygous for haplotype 1, predicted to express normal amounts of a fully functional protein. The urinary ratio of TMA N-oxide excretion of these individuals varied from 50–82%. Other homozygotes for haplotype 1 (IDs 81, 87 and 97) were classified as unaffected, with urinary ratios >90%. Of the remaining 50 individuals, 35 had genotypes that were present in unaffected individuals.
Therefore, in contrast to unaffected individuals or those with severe TMAuria, for individuals with moderate or mild TMAuria the phenotype is not readily explained by the FMO3 genotype. The fact that none of the 65 mild or moderate TMAurics was homozygous or compound heterozygous for known causative mutations suggests that it is unlikely that these phenotypes are a consequence of unidentified mutations that severely affect activity or expression of FMO3. However, some of these individuals might have unidentified mutations in their FMO3 genes that have moderate effects on transcription or RNA processing. Alternatively, the activity of their FMO3 might be inhibited, for instance, by post-translational modification via nitric oxide-mediated S-nitrosylation [42].
Can FMO3 genotype predict phenotype with respect to TMAuria? Our results indicate that individuals who are homozygous or compound heterozygous for mutations that abolish or severely impair FMO3 activity, or are compound heterozygous for such mutations and for a mutation that decreases enzyme activity by about 50%, will display symptoms of severe TMAuria (Table 2). For other genotypes, the situation is not clear. Although they will not result in severe TMAuria, the same genotype can be associated with moderate or mild forms of the disorder or with an unaffected phenotype. Even individuals homozygous for genes encoding the ancestral, fully functional FMO3 can display symptoms characteristic of moderate or mild TMAuria or be unaffected by the disorder. It is clear that for moderate or mild TMAuria, factors other than FMO3 genotype contribute to the severity of symptoms.
Secondary TMAuria can occur as a result of hepatitis [5, 18] or of liver or kidney disease [19, 20]. However, none of the individuals studied appears to have been affected by these conditions. Other potential contributory factors are gender, age and TMA load. For individuals in the cohort displaying symptoms of moderate or mild TMAuria, no difference was found between males and females in the percentage of total urinary TMA excreted as TMA N-oxide (52.7 ± 14.9, mean ± SD, n = 38 and 53.8 ± 15.1, mean ± SD, n = 31, respectively). (To avoid potential distortion of results due to the transient reduction in TMA metabolism that females can experience during menstruation [21, 22], subjects were not tested during this period). Similarly, for these individuals there was no correlation between the percent of total TMA excreted as the N-oxide and either age or total urinary TMA. Seven of the subjects were children aged between 3 and 7 years old. Of these, three (IDs 6, 7 and 9) were compound heterozygous for known causative mutations of TMAuria. The case of one (ID 69) is discussed above. It is possible that the other three (IDs 24, 70 and 74) were affected by transient childhood TMAuria [23–26], as a consequence of immature expression of FMO3 [27], in the case of ID 70, exacerbated by a high TMA load.
However, for affected individuals a strong correlation was observed between total urinary TMA and the amount excreted as the free amine (r2 = 0.76, P < 0.0001, n = 78). This implies that individuals with moderate or mild TMAuria would benefit from a reduction of total TMA load. Indeed, because of a low total TMA load, 15 (23%) of those diagnosed as moderate or mild TMAurics, on the basis of the percent of total urinary TMA excreted as TMA N-oxide, excreted <20 mmol TMA mol−1 creatinine, which appears to be the threshold for the presence of the fishy body odour associated with the disorder [5]. Consequently, these individuals (IDs 24, 25, 29, 36, 37, 38, 45, 52, 60, 61, 68, 71, 73, 75 and 77) did not exhibit the symptoms of the disorder. Remarkably, of the 13 individuals diagnosed as severe TMAurics, two (IDs 6 and 12), both of whom were either homozygous or compound heterozygous for known causative mutations of TMAuria, excreted <20 mmol TMA mol−1 creatinine (Table 2) (as a consequence of extremely low total TMA loads) and, thus, would be expected to be asymptomatic. At the other end of the TMAuria spectrum, of the 24 individuals diagnosed as unaffected, on the basis of excreting >90% of total TMA as TMA N-oxide, four (17%) (IDs 79, 80, 82 and 88), as a consequence of high total TMA loads, excreted >20 mmol TMA mol−1 creatinine and, thus, would exhibit the fishy odour characteristic of the disorder and hence suffer from secondary TMAuria, as a result of precursor overload [7].
TMA load can be influenced by dietary intake of precursors of TMA, such as choline, which is present in a variety of foods including eggs, offal, legumes, Brassicas and soya products, and TMA N-oxide, present in seafood. Restriction of dietary precursors of TMA would be expected to result in a reduction of TMA load and, consequently, help reduce the severity of symptoms. It is possible that some individuals in the cohort (e.g., IDs 81, 84, 87 and 94) were managing their condition by dietary restriction of TMA precursors, which resulted in their classification as unaffected. However, this is clearly not the case for IDs 79, 80, 82 and 88 (see above and Table 2). Alternatively, some of those diagnosed as unaffected might be suffering from another malodour disorder such as gingivitis, blood-borne halitosis [43] or dimethylglycinuria [44]. Administration of a choline challenge [45] might resolve the phenotypic status of those classified as unaffected.
Other factors that could contribute to an increased burden of TMA include an increase in the intestinal production or absorbance of TMA, as a consequence, respectively, of normal bacterial overgrowth or of gastrointestinal problems that result in increased gastrointestinal permeability, or inflammation, which can down-regulate expression of FMO3 [46]. In addition, there is a growing realization of the importance of gut flora composition to host metabolism and it is likely that differences in the relative abundance, within the intestine, of bacterial species that produce or metabolize TMA would influence the TMA load entering the bloodstream of the human host and counteract the beneficial effects of dietary restriction of precursors of TMA.
In conclusion, our results have implications for the diagnosis and management of TMAuria. Genetic testing can identify individuals affected by the severe primary genetic form of the disorder (caused by loss of function mutations of FMO3) and distinguish these from those affected by milder genetic forms (caused by mutations or polymorphisms that have less severe effects) and those whose symptoms are not due to genetic factors. Based on the Japanese cohort we have investigated, most of those affected with TMAuria, as judged by the percent of total urinary TMA excreted as TMA N-oxide, suffer from cases of moderate or mild forms of the disorder, the majority of which cannot be diagnosed genetically and, consequently, are due to factors other than FMO3 genotype. It is likely, therefore, that FMO3 exon sequencing, which is increasingly being offered to patients after a positive diagnosis based on urinary analysis, would identify causative mutations only for those suffering from the severe form of the disorder and would not be informative for the majority of sufferers. Nevertheless, it would appear that for most cases of TMAuria a reduction of total TMA load, through either dietary restriction of precursors of TMA or manipulation of gut flora, would result in reduced TMA excretion and, thus, prove beneficial to affected individuals.
Competing Interests
All authors have completed the Unified Competing Interest and declare HY had support from Ministry of Education, Science, Sports and Culture of Japan and Takeda Science Foundation, outside the submitted work and no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years.
We thank Norie Murayama and Marie Kozono for their sample handling and Steve LeComber for advice on the runs test. This work was supported in part by the Ministry of Education, Science, Sports and Culture of Japan and the Takeda Science Foundation.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Sequence of oligonucleotides used for amplification and sequencing of upstream regions, coding exons and flanking intronic regions of the human FMO3 gene
References
- 1.Lawton MP, Cashman JR, Cresteil T, Dolphin CT, Elfarra AA, Hines RN, Hodgson E, Kimura T, Ozols J, Phillips IR, Philpot RM, Poulson LL, Rettie AE, Shephard EA, Williams DE, Ziegler DM. A nomenclature for the mammalian flavin-containing monooxygenase gene family based on amino acid sequence identities. Arch Biochem Biophys. 1994;308:254–257. doi: 10.1006/abbi.1994.1035. [DOI] [PubMed] [Google Scholar]
- 2.Phillips IR, Dolphin CT, Clair P, Hadley MR, Hutt AJ, McCombie RR, Smith RL, Shephard EA. The molecular biology of the flavin-containing monooxygenases of man. Chem Biol Interact. 1995;96:17–32. doi: 10.1016/0009-2797(94)03580-2. [DOI] [PubMed] [Google Scholar]
- 3.Hernandez D, Janmohamed A, Chandan P, Phillips IR, Shephard EA. Organization and evolution of the flavin-containing monooxygenase genes of human and mouse: identification of novel gene and pseudogene clusters. Pharmacogenetics. 2004;14:117–130. doi: 10.1097/00008571-200402000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106:357–387. doi: 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell SC, Smith RL. Trimethylaminuria: the fish malodor syndrome. Drug Metab Dispos. 2001;29:517–521. [PubMed] [Google Scholar]
- 6.Lang DH, Yeung CK, Peter RM, Ibarra C, Gasser R, Itagaki K, Philpot RM, Rettie AE. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: selective catalysis by FMO3. Biochem Pharmacol. 1998;56:1005–1012. doi: 10.1016/s0006-2952(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 7.Phillips IR, Shephard EA. Seattle, USA: University of Washington; 1997. Trimethylaminuria. in: GeneReviews at GeneTests:Medical Genetics Information Resource (database online) Copyright 2011 Available at http://www.ncbi.nlm.nih.gov/books/NBK1103/ updated (last accessed 19 February 2013) [Google Scholar]
- 8.Shephard EA, Treacy EP, Phillips IR. Clinical utilty gene card for:Trimethylaminuria. Eur J Hum Genet. 2012;20 doi: 10.1038/ejhg.2011.214. doi: 10.1038/ejhg.2011.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cashman JR, Camp K, Fakharzadeh SS, Fennessey PV, Hines RN, Mamer OA, Mitchell SC, Nguyen GP, Schlenk D, Smith RL, Tjoa SS, Williams DE, Yannicelli S. Biochemical and clinical aspects of the human flavin-containing monooxygenase form 3 (FMO3) related to trimethylaminuria. Curr Drug Metab. 2003;4:151–170. doi: 10.2174/1389200033489505. [DOI] [PubMed] [Google Scholar]
- 10.Mackay RJ, McEntyre CJ, Henderson C, Lever M, George PM. Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin Biochem Rev. 2011;32:33–43. [PMC free article] [PubMed] [Google Scholar]
- 11.Dolphin CT, Janmohamed A, Smith RL, Shephard EA, Phillips IR. Missense mutation in flavin-containing mono-oxygenase 3 gene, FMO3, underlies fish-odour syndrome. Nat Genet. 1997;17:491–494. doi: 10.1038/ng1297-491. [DOI] [PubMed] [Google Scholar]
- 12.Treacy EP, Akerman BR, Chow LML, Youil R, Bibeau C, Lin J, Bruce AG, Knight M, Danks DM, Cashman JR, Forrest SM. Mutations of the flavin-containing monooxygenase gene (FMO3) cause trimethylaminuria, a defect in detoxication. Hum Mol Genet. 1998;7:839–845. doi: 10.1093/hmg/7.5.839. [DOI] [PubMed] [Google Scholar]
- 13.Yamazaki H, Fujita H, Gunji T, Zhang J, Kamataki T, Cashman JR, Shimizu M. Stop codon mutations in the flavin-containing monooxygenase 3 (FMO3) gene responsible for trimethylaminuria in a Japanese population. Mol Genet Metab. 2007;90:58–63. doi: 10.1016/j.ymgme.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Phillips IR, Shephard EA. Flavin-containing monooxygenases: mutations, disease and drug response. Trends Pharmacol Sci. 2008;29:294–301. doi: 10.1016/j.tips.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Hernandez D, Addou S, Lee D, Orengo C, Shephard EA, Phillips IR. Trimethylaminuria and a human FMO3 mutation database. Hum Mutat. 2003;22:201–213. doi: 10.1002/humu.10252. [DOI] [PubMed] [Google Scholar]
- 16.Phillips IR, Francois AA, Shephard EA. The flavin-containing monoooxygenases (FMOs): genetic variation and its consequences for the metabolism of therapeutic drugs. Curr Pharmacogenomics. 2007;5:292–313. [Google Scholar]
- 17.Yamazaki H, Shimizu M. Survey of variants of human flavin-containing monoooxygenase 3 (FMO3) and their drug oxidation activities. Biochem Pharmacol. 2013;58:1588–1593. doi: 10.1016/j.bcp.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Ruocco V, Florio M, Filioli FG, Guerrera V, Prota G. An unusual case of trimethylaminuria. Br J Dermatol. 1989;120:459–461. doi: 10.1111/j.1365-2133.1989.tb04175.x. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell M, Ayesh R, Barrett T, Smith R. Trimethylamine and foetor hepaticus. Scand J Gastroenterol. 1999;34:524–528. doi: 10.1080/003655299750026281. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell S. Trimethylaminuria (fish-odour syndrome) and oral malodour. Oral Dis. 2005;11:10–13. doi: 10.1111/j.1601-0825.2005.01081.x. (Suppl. 1) [DOI] [PubMed] [Google Scholar]
- 21.Zhang AQ, Mitchell SC, Smith RL. Exacerbation of symptoms of fish-odour syndrome during menstruation. Lancet. 1996;348:1740–1741. doi: 10.1016/s0140-6736(05)65872-2. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu M, Cashman JR, Yamazaki H. Transient trimethylaminuria related to menstruation. BMC Med Genet. 2007;8:2. doi: 10.1186/1471-2350-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayatepek E, Kohlmüller D. Transient trimethylaminuria in childhood. Acta Paediatr. 1998;87:1205–1207. doi: 10.1080/080352598750031257. [DOI] [PubMed] [Google Scholar]
- 24.Zschocke J, Kohlmueller D, Quak E, Meissner T, Hoffmann GF, Mayatepek E. Mild trimethylaminuria caused by common variants in FMO3 gene. Lancet. 1999;354:834–835. doi: 10.1016/s0140-6736(99)80019-1. [DOI] [PubMed] [Google Scholar]
- 25.Zschocke J, Mayatepek E. Biochemical and molecular studies in mild flavin monooxygenase 3 deficiency. J Inherit Metab Dis. 2000;23:378–382. doi: 10.1023/a:1005647701321. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu M, Denton T, Kozono M, Cashman JR, Leeder JS, Yamazaki H. Developmental variations in metabolic capacity of flavin-containing monooxygenase 3 in childhood. Br J Clin Pharmacol. 2011;71:585–591. doi: 10.1111/j.1365-2125.2010.03876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koukouritaki SB, Simpson P, Yeung CK, Rettie AE, Hines RN. Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr Res. 2002;51:236–243. doi: 10.1203/00006450-200202000-00018. [DOI] [PubMed] [Google Scholar]
- 28.Allerston CK, Shimizu M, Fujieda M, Shephard EA, Yamazaki H, Phillips IR. Molecular evolution and balancing selection in the flavin-containing monooxygenase 3 gene (FMO3) Pharmacogenet Genomics. 2007;17:827–839. doi: 10.1097/FPC.0b013e328256b198. [DOI] [PubMed] [Google Scholar]
- 29.Yamazaki H, Fujieda M, Togashi M, Saito T, Preti G, Cashman JR, Kamataki T. Effects of the dietary supplements, activated charcoal and copper chlorophyllin, on urinary excretion of trimethylamine in Japanese trimethylaminuria patients. Life Sci. 2004;74:2739–2747. doi: 10.1016/j.lfs.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 30.Dolphin CT, Riley JH, Smith RL, Shephard EA, Phillips IR. Structural organization of the human flavin-containing monooxygenase 3 gene (FMO3), the favored candidate for fish-odor syndrome, determined directly from genomic DNA. Genomics. 1997;46:260–267. doi: 10.1006/geno.1997.5031. [DOI] [PubMed] [Google Scholar]
- 31.Halperin E, Eskin E. Haplotype reconstruction from genotype data using ImPerfect Phylogeny. Bioinformatics. 2004;20:1842–1849. doi: 10.1093/bioinformatics/bth149. [DOI] [PubMed] [Google Scholar]
- 32.Labes D. 2012. randomizeBE: Function to create a random list for crossover studies. Rpackage version 0.3-1. Available at http://CRAN.R-project.org/package=randomizeBE (last accessed 19 February 2013)
- 33.R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2012. ISBN 3-900051-07-0, Available at http://www.R-project.org/ (last accessed 19 February 2013) [Google Scholar]
- 34.Mayatepek E, Flock B, Zschocke J. Benzydamine metabolism in vivo is impaired in patients with deficiency of flavin-containing monooxygenase 3. Pharmacogenetics. 2004;14:775–777. doi: 10.1097/00008571-200411000-00009. [DOI] [PubMed] [Google Scholar]
- 35.Hisamuddin IM, Wehbi MA, Chao A, Wyre HW, Hylind LM, Giardiello FM, Yang VW. Genetic polymorphisms of human flavin monooxygenase 3 in sulindac-mediated primary chemoprevention of familial adenomatous polyposis. Clin Cancer Res. 2004;10:8357–8362. doi: 10.1158/1078-0432.CCR-04-1073. [DOI] [PubMed] [Google Scholar]
- 36.Hisamuddin IM, Wehbi MA, Schmotzer B, Easley KA, Hylind LM, Giardiello FM, Yang VW. Genetic polymorphisms of flavin monooxygenase 3 in sulindac-induced regression of colorectal adenomas in familial adenomatous polyposis. Cancer Epidemiol Biomarkers Prev. 2005;14:2366–2369. doi: 10.1158/1055-9965.EPI-05-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamazaki H, Shimizu M. Genetic polymorphism of the flavin-containing monooxygenase 3 (FMO3) associated with trimethylaminuria (fish odor Syndrome): observations from Japanese patients. Curr Drug Metab. 2007;8:487–491. doi: 10.2174/138920007780866825. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu M, Yano H, Nagashima S, Murayama N, Zhang J, Cashman JR, Yamazaki H. Effect of genetic variants of the human flavin-containing monooxygenase 3 on N-and S-oxygenation activities. Drug Metab Dispos. 2007;35:328–330. doi: 10.1124/dmd.106.013094. [DOI] [PubMed] [Google Scholar]
- 39.Shimizu M, Kobayashi Y, Hayashi S, Aoki Y, Yamazaki H. Variants in the flavin-containing monooxygenase 3 (FMO3) gene responsible for trimethylaminuria in a Japanese population. Mol Genet Metab. 2012;107:330–334. doi: 10.1016/j.ymgme.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 40.Dolphin CT, Janmohamed A, Smith RL, Shephard EA, Phillips IR. Compound heterozygosity for missense mutations in the flavin-containing monooxygenase 3 (FM03) gene in patients with fish-odour syndrome. Pharmacogenetics. 2000;10:799–807. doi: 10.1097/00008571-200012000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Koukouritaki SB, Poch MT, Cabacungan ET, McCarver DG, Hines RN. Discovery of novel flavin-containing monooxygenase 3 (FMO3) single nucleotide polymorphisms and functional analysis of upstream haplotype variants. Mol Pharmacol. 2005;68:383–392. doi: 10.1124/mol.105.012062. [DOI] [PubMed] [Google Scholar]
- 42.Ryu SD, Yi HG, Cha YN, Kang JH, Kang JS, Jeon YC, Park HK, Yu TM, Lee JN, Park CS. Flavin-containing monooxygenase activity can be inhibited by nitric oxide-mediated S-nitrosylation. Life Sci. 2004;75:2559–2572. doi: 10.1016/j.lfs.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 43.Tangerman A. Halitosis in medicine: a review. Int Dent J. 2002;52:201–206. doi: 10.1002/j.1875-595x.2002.tb00925.x. (Suppl. 3) [DOI] [PubMed] [Google Scholar]
- 44.Binzak BA, Wevers RA, Moolenaar SH, Lee YM, Hwu WL, Poggi-Bach J, Engelke UF, Hoard HM, Vockley JG, Vockley J. Cloning of dimethylglycine dehydrogenase and a new human inborn error of metabolism, dimethylglycine dehydrogenase deficiency. Am J Hum Genet. 2001;68:839–847. doi: 10.1086/319520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chalmers RA, Bain MD, Michelakakis H, Zschocke J, Iles RA. Diagnosis and management of trimethylaminuria (FMO3 deficiency) in children. J Inherit Metab Dis. 2006;29:162–172. doi: 10.1007/s10545-006-0158-6. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Chaluvadi MR, Reddy R, Motika MS, Richardson TA, Cashman JR, Morgan ET. Hepatic flavin-containing monooxygenase gene regulation in different mouse inflammation models. Drug Metab Dispos. 2009;37:462–468. doi: 10.1124/dmd.108.025338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimizu M, Murayama N, Nagashima S, Fujieda M, Yamazaki H. Complex mechanism underlying transcriptional control of the haplotyped flavin-containing monooxygenase 3FMO3) gene in Japanese: different regulation between mutations in 5′-upstream distal region and common element in proximal region. Drug Metab Pharmacokinet. 2008;23:54–58. doi: 10.2133/dmpk.23.54. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu M, Tomioka S, Murayama N, Yamazaki H. Missense and nonsense mutations of the flavin-containing monooxygenase 3 gene in a Japanese cohort. Drug Metab Pharmacokinet. 2007;22:61–64. doi: 10.2133/dmpk.22.61. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu M, Fujita H, Aoyama T, Yamazaki H. Three novel single nucleotide polymorphisms of the FMO3 gene in a Japanese population. Drug Metab Pharmacokinet. 2006;21:245–247. doi: 10.2133/dmpk.21.245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence of oligonucleotides used for amplification and sequencing of upstream regions, coding exons and flanking intronic regions of the human FMO3 gene