

Abstract
The type I interferon system is integral to human antiviral immunity. However, inappropriate stimulation or defective negative regulation of this system can lead to inflammatory disease. We sought to determine the molecular basis of genetically uncharacterized cases of the type I interferonopathy Aicardi-Goutières syndrome, and of other patients with undefined neurological and immunological phenotypes also demonstrating an upregulated type I interferon response. We found that heterozygous mutations in the cytosolic double-stranded RNA receptor gene IFIH1 (MDA5) cause a spectrum of neuro-immunological features consistently associated with an enhanced interferon state. Cellular and biochemical assays indicate that these mutations confer a gain-of-function - so that mutant IFIH1 binds RNA more avidly, leading to increased baseline and ligand-induced interferon signaling. Our results demonstrate that aberrant sensing of nucleic acids can cause immune upregulation.
Ion Gresser and colleagues first drew attention to the possibility that inappropriate exposure to type I interferon might be detrimental in the mammalian system1-3. More recently, it has been proposed that Mendelian disorders associated with an upregulation of type I interferon represent a novel set of inborn errors of immunity - resulting from either inappropriate stimulation or defective negative regulation of the type I interferon response pathway4.
Aicardi-Goutières syndrome (AGS: MIM 225750) is an inflammatory disease particularly affecting the brain and skin, occurring due to mutations in any of the genes encoding the DNA exonuclease TREX15, the three non-allelic components of the RNase H2 endonuclease complex6, the deoxynucleoside triphosphate triphosphohydrolase SAMHD17, and the double-stranded RNA editing enzyme ADAR18. Some patients with AGS do not harbor mutations in any of these six genes. AGS patients consistently demonstrate increased expression of gene transcripts induced by type I interferon, a so-called interferon signature9. A similar upregulation of interferon-induced transcripts is seen in the immuno-osseous dysplasia spondyloenchondromatosis (SPENCD)10.
In order to identify further monogenic type I interferonopathies we set out to determine the molecular basis of genetically uncharacterized cases of AGS, and of other patients with undefined neurological and immunological features also demonstrating an upregulated type I interferon response. Here we show that gain-of-function mutations in IFIH1 result in a range of human disease phenotypes, in which an induction of type I interferon signaling is likely central to their pathogenesis.
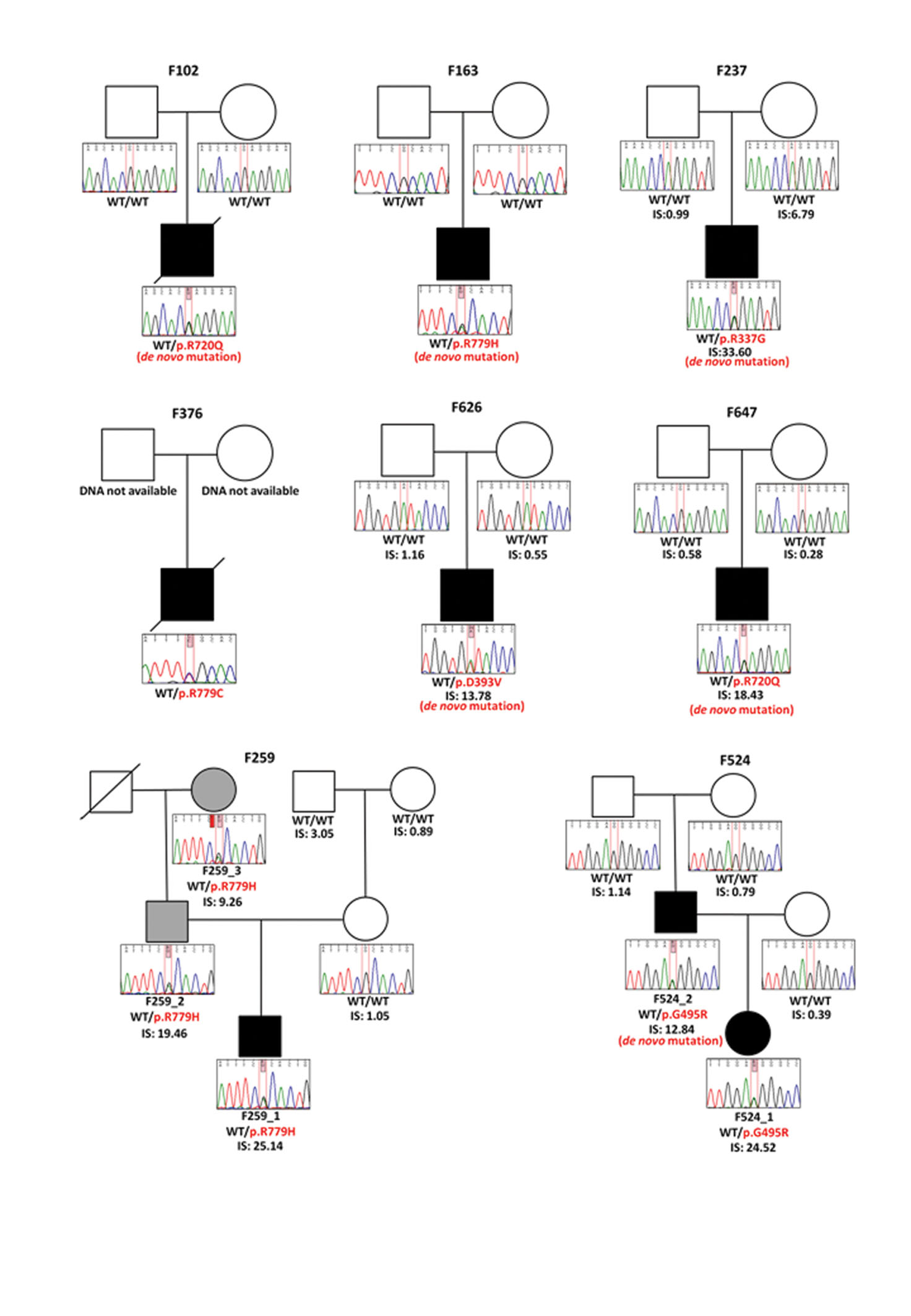
We undertook whole exome sequencing (Supplementary Table 1) in three patients (F102, F163 and F259) with a clinical diagnosis of AGS, based on neuro-radiological criteria and an upregulation of cerebrospinal fluid interferon activity and / or interferon stimulated genes (ISGs) in peripheral blood (Supplementary Table 2), all of whom screened negative for mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR1. Having excluded common polymorphisms listed in publically available databases, we noted that all three patients carried a single rare variant (Arg720Gln in F102, and Arg779His in both F163 and F259)(Table 1) in IFIH1, encoding a cytoplasmic helicase that mediates induction of an interferon response to viral RNA 11. We then screened IFIH1 in other TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR1 mutation-negative patients with a phenotype indicative of AGS, and in patients with a variety of neuro-immunological features in whom we had recorded the presence of an interferon signature in peripheral blood in the absence of apparent infection (Supplementary Tables 2 and 3). In this way we identified a further five probands heterozygous for a rare IFIH1 variant (Arg337Gly in F237, Arg779Cys in F376, Gly495Arg in F524, Asp393Val in F626 and Arg720Gln in F647). In total we observed six rare variants in eight probands, with two pairs of unrelated probands each sharing the same substitution (Arg720Gln and Arg779His)(Fig. 1, Supplementary Fig. 1). All mutation-positive probands were born to non-consanguineous parents.
Table 1.
Ancestry and sequence alterations in IFIH1 mutation-positive families
| Family | Ancestry | Inheritance | Nucleotide alteration |
Exon | Amino acid alteration |
Domain | EVS† allele frequency |
|---|---|---|---|---|---|---|---|
| F102 | European Italian | de novo | c.2159G>A | 11 | p.Arg720Gln | Hel 2 | 0/13006 |
| F163 | European French | de novo | c.2336G>A | 12 | p.Arg779His | Hel 2 | 0/13006 |
| F237 | White American | de novo | c.1009A>G | 5 | p.Arg337Gly | Hel 1 | 0/13006 |
| F259 | European Italian | Inherited* | c.2336G>A | 12 | p.Arg779His | Hel 2 | 0/13006 |
| F376 | White British | n/a | c.2335C>T | 12 | p.Arg779Cys | Hel 2 | 0/13006 |
| F524 | White British | de novo ** | c.1483G>A | 7 | p.Gly495Arg | Hel 1 | 0/13006 |
| F626 | European Italian | de novo | c.1178A>T | 6 | p.Asp393Val | Hel 1 | 0/13006 |
| F647 | Mixed white Irish / Ukranian | de novo | c.2159G>A | 11 | p.Arg720Gln | Hel 2 | 0/13006 |
Mutation in affected child inherited from mutation-positive clinically asymptomatic father. The proband’s paternal grandmother also carries the mutation and is clinically asymptomatic. All three mutation-positive individuals demonstrate a robust interferon signature
Mutation occurred de novo in affected male, who has then transmitted mutation to his affected daughter
n/a not available
Exome Variant Server (http://evs.gs.washington.edu/EVS/)
Fig. 1. Schematic representation of the human IFIH1 gene.

(a) IFIH1 spans 51,624bp of genomic sequence on chromosome 2q24.2 (163,123,589 - 163,175,213). Neighboring genes are also shown. (b) Position of identified variants within the genomic sequence of IFIH1. Exons are numbered within the boxes. Numbers given above the gene indicate the exon boundaries using cDNA numbering. (c) Schematic illustrating the position of protein domains and their amino acid boundaries within the IFIH1 1025 amino acid protein. CARD denotes caspase activation recruitment domain. Hel denotes helicase domains, where Hel1 and Hel2 are the two conserved core helicase domains, and Hel2i is an insertion domain that is conserved in the RIG-I like helicase family. P denotes pincer or bridge region which connects Hel2 to the C-terminal domain (CTD) involved in binding double stranded RNA.
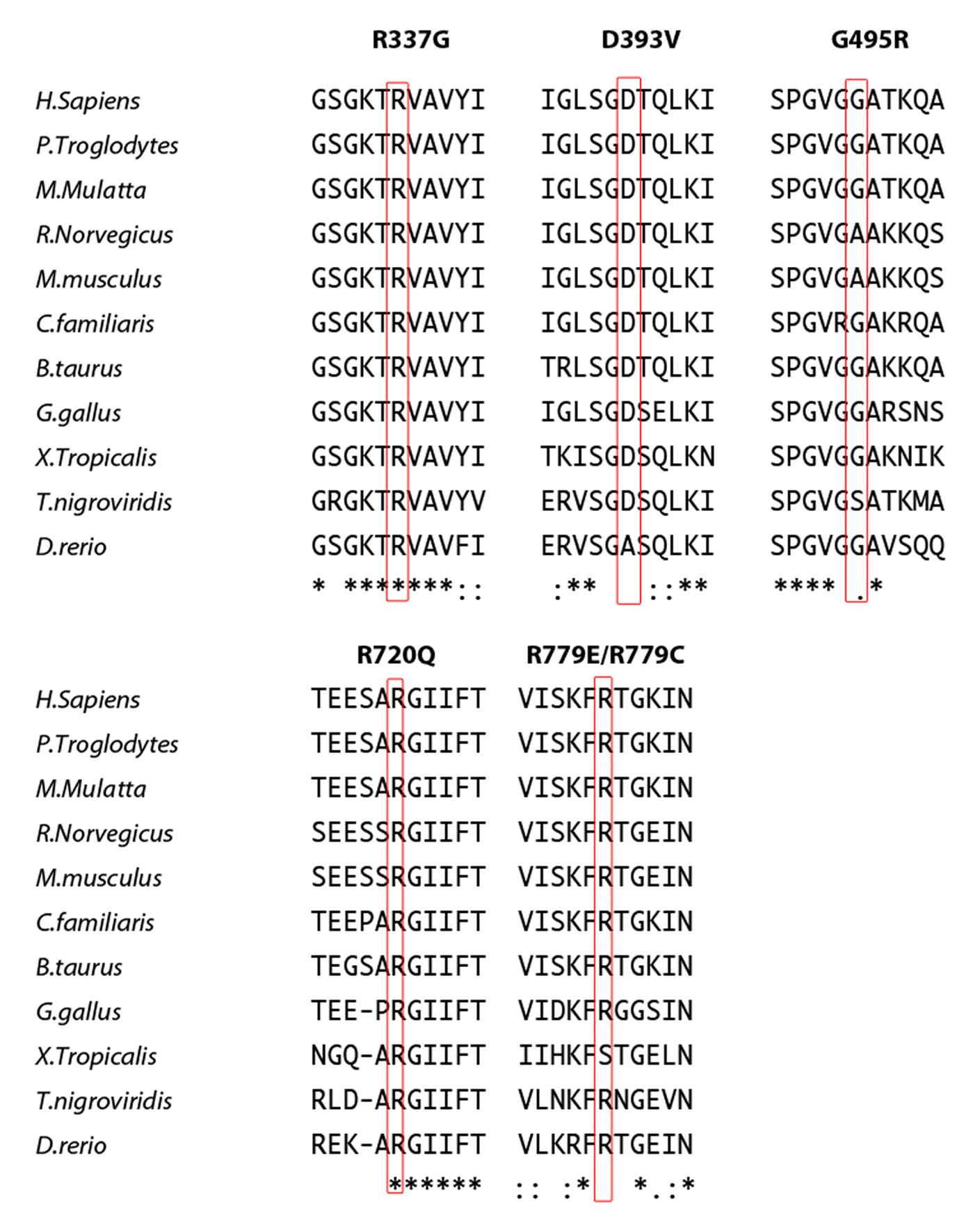
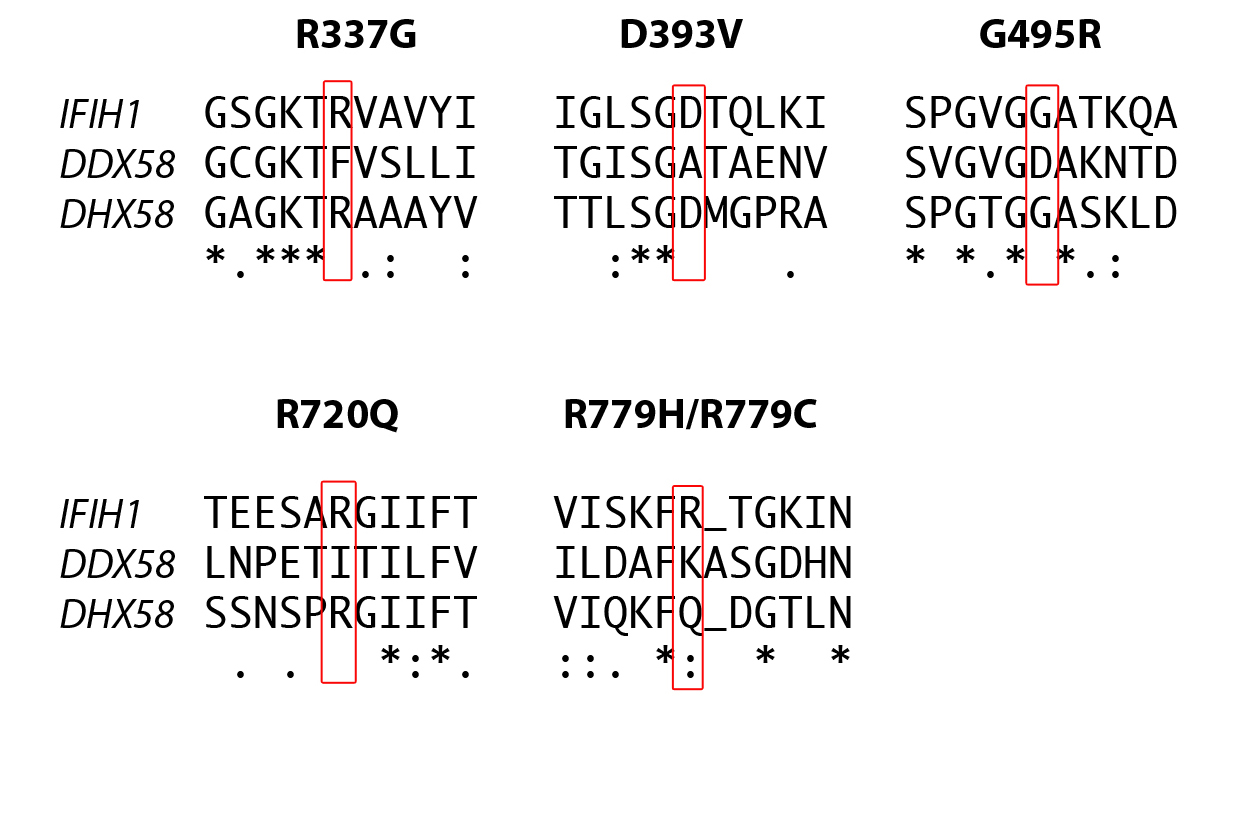
The identified variants of interest were confirmed on Sanger sequencing, and were considered likely pathogenic on the basis of species conservation (Supplementary Figs. 2 and 3), the output of pathogenicity prediction packages (Supplementary Table 4), and absence from the NHLBI ESP database of more than 13,000 control alleles and an in-house collection of >300 exomes. Parental samples were available for seven of the eight probands. In five of these seven the variant was not present in either parent, and genotyping of microsatellite markers was consistent with stated paternity and maternity, thus indicating that the mutations had arisen de novo (Supplementary Table 5). In the remaining two cases (F259_1, F524_1) the variant seen in the proband had been paternally inherited (Supplementary Fig. 1). In family F259 the variant had been transmitted by the proband’s paternal grandmother (F259_3) to her son (F259_2), whilst in family F524 the mutation was shown to have occurred de novo in the father (F524_2).
The majority of IFIH1 mutation-positive probands (F102, F163, F259_1, F376 and F647) demonstrated a clinical picture typical of neonatal AGS (Supplementary Note, Supplementary Table 2). In contrast, two patients (F237 and F626) were developmentally normal until the second year of life, at which point they experienced rapid neuro-regression. Of particular note, both affected individuals (F524_1 and F524_2) in family F524 present a distinct phenotype of (dominantly inherited) spastic paraparesis. The finding of normal neuroimaging in F524_2 at the age of 29 years suggests that further patients with unexplained spasticity might harbor mutations in IFIH1 or other AGS-related genes.
To define the relationship between IFIH1 mutation status and interferon induction in vivo, we tested for an interferon signature in IFIH1 mutation-positive subjects and their mutation-negative relatives. Samples were available from five families (F237, F259, F524, F626, F647). All eight mutation-positive individuals assayed, at a total of 22 data-points, demonstrated a robust upregulation of ISGs (median relative quantification (RQ): 17.43, Interquartile Range (IQR): 12.27 – 25.77) compared with 12 IFIH1 mutation-negative family members, assayed on 17 occasions (median RQ: 0.89, IQR: 0.52 – 1.12), and a previously standardized set of 29 control individuals (median RQ: 0.93, IQR: 0.57 – 1.30)(Fig. 2, Supplementary Fig. 4). Where measured serially, positivity for an interferon signature was sustained over time (e.g. patient F524_2 was assayed on five occasions over an 18 month period).
Fig. 2. Quantitative reverse transcription PCR (qPCR) of a panel of six interferon stimulated genes (ISGs) in whole blood measured in IFIH1 mutation-positive probands and mutation-negative relatives and interferon scores in mutation-positive individuals, mutation-negative relatives and controls.

(a – e) Bar graphs showing relative quantification (RQ) values for a panel of six interferon stimulated genes (ISGs) (IFI27, IFI44L, IFIT1, ISG15, RSAD2, SIGLEC1) measured in whole blood in five AGS families, compared to the combined results of 29 healthy controls. RQ is equal to 2−ΔΔCt, with −ΔΔCt ± standard deviations (i.e. the normalized fold change relative to a calibrator). Each value is derived from three technical replicates. Family / patient number followed by mutation status are given in the first brackets. Numbers in second brackets refer to decimalized age at sampling, followed by interferon score calculated from the median fold change in relative quantification value for the panel of six ISGs. Colors denote individuals, with repeat samples (biological replicates) denoted by different bars of the same color. (f) Interferon score in all patients, relatives and controls calculated from the median fold change in relative quantification (RQ) value for a panel of six interferon-stimulated genes (ISGs). For participants with repeat samples, all measurements are shown. Black horizontal bars show the median interferon score in mutation-positive, mutation-negative and control individuals. Data analyzed by one-way ANOVA using Bonferroni’s multiple comparison test (**** p<0.0001).
IFIH1 (interferon induced with helicase C domain 1), also known as MDA5 (melanoma differentiation-associated protein 5), is a 1025 amino acid cytoplasmic viral RNA receptor. IFIH1 belongs to the RIG-I-like family of cytoplasmic DExD/H box RNA receptors and activates type I interferon signaling through an adaptor molecule, MAVS (mitochondrial antiviral signaling protein). IFIH1 consists of N-terminal tandem caspase activation recruitment domains (2CARD) involved in activating MAVS, a central helicase domain responsible for RNA-binding and RNA-dependent ATP hydrolysis, and a C-terminal domain serving as an additional RNA binding domain (Fig. 1). MDA5 uses long viral double-stranded (ds) RNA as a platform to cooperatively assemble a core filament, in turn promoting stochastic assembly of the 2CARD oligomers for signaling to MAVS12-14. The IFIH1 filament then undergoes end-disassembly upon ATP hydrolysis13, thus regulating the stability of the filament in a dsRNA-length dependent manner, a potential mechanism to suppress aberrant signal activation in response to short (< ~ 0.5 kb) cellular dsRNAs.
To understand the pathogenicity of the IFIH1 mutations observed, we investigated the interferon beta reporter stimulatory activity of wild-type and mutant IFIH1 in human embryonic kidney 293T cells. 293T cells express low levels of endogenous viral RNA receptors, including IFIH1 - evidenced by low interferon production upon stimulation with dsRNA (Fig. 3a), allowing comparison of the signaling activity of ectopically expressed receptors. As expected, signaling of wild-type IFIH1 was induced only upon addition of the long (> 1 kb) dsRNA analog polyinosinic-polycytidylic acid (polyI:C), and not by short, 162 bp, dsRNA (Fig. 3a). Minimal activity was seen in the absence of exogenous RNA (Fig. 3a). As with wild-type IFIH1, all six IFIH1 mutants displayed robust signaling in response to polyI:C (Fig. 3a). Additionally, these mutants exhibited a marked induction of interferon signaling in response to 162 bp dsRNA (Fig. 3a), and demonstrated ~ 4 - 10 fold higher levels of basal signaling activity even in the absence of exogenous ligand (Fig. 3b). As for polyI:C-induced signaling of wild-type IFIH1, the basal, and induced, signaling activity of the six mutants was significantly diminished upon the introduction of additional mutations into the RNA binding site (His927Ala), the filament protein:protein interface (Ile841Arg/Glu842Arg), and the 2CARD (Arg21Ala/Lys23Ala)(Fig. 3b)13, suggesting that the basal signaling activity of these mutants is induced by as-yet undefined endogenous dsRNA.
Fig. 3. IFIH1 mutants activate the interferon signaling pathway more efficiently than wild-type IFIH1.

(a) Interferon beta (IFNβ) reporter activity (mean ± SD, n = 3) of Flag-tagged wild-type and mutant IFIH1 with and without stimulation with poly I:C or 162 bp dsRNA in HEK293T cells. The results are representative of three independent experiments. *P<0.005, **P<0.05 and ***P <0.5 (one tailed, unpaired t test, compared with wild-type values). Below are the anti-Flag (F7425, Sigma-Aldrich) and anti-actin (A5441, Sigma-Aldrich) western blots indicating the expression levels of IFIH1 and the internal control (actin), respectively. (b) IFNβ reporter activity (mean ± SD, n = 3) of mutant IFIH1 with and without additional mutations (H927A, I841R/E842R or R21A/K23A) that disrupt RNA binding, filament formation or 2CARD signal activation by IFIH1. Reporter activity was measured in the absence (top) and presence (bottom) of poly I:C stimulation in HEK293T cells. 10 and 20 ng IFIH1 expression constructs were used with and without poly I:C, respectively. The results are representative of three independent experiments. *P<0.005, **P<0.05 (one tailed, unpaired t test). Below are western blots showing the expression levels of wild-type and R337G IFIH1 with and without H927A, I841R/E842R and R21A/K23A. (c) Mapping of the mutated residues (red spheres) onto the structure of IFIH1Δ2CARD (grey) bound by dsRNA (blue) and ATP analog (green). PDB:4GL2.
Mapping of the mutated residues onto the crystal structure of the 2CARD deletion construct (Δ2CARD)14 showed that these mutations are located on the surface of the RNA- and ATP-binding sites in Hel1 and Hel2 (Fig. 3c). In particular, Arg337 is in direct contact with the adenine base of ATP, stabilizing the IFIH1:ATP interaction. Substitution of Arg337 by Gly could diminish ATP binding and hydrolysis activity of IFIH1. Asp393 and Gly495 are within the contact distance of the bound RNA. Removal of the negatively charged Asp393 (by Asp393Val), and incorporation of the positively charged Arg495 (by Gly495Arg) near the RNA phosphate backbone could increase the intrinsic affinity of IFIH1 for dsRNA. Arg720 and Arg779 are near the ATP binding site, but are also in proximity to the protein:protein interface in the filament (Fig. 3c). The location of the mutated residues in or near the RNA- and ATP-binding sites or filament interface led us to hypothesize that the observed mutations might enhance the stability of the IFIH1 filament by increasing the intrinsic affinity between IFIH1 and dsRNA, or between IFIH1 molecules in the filament, or by decreasing the efficiency of ATP hydrolysis, and thus filament disassembly rate.
To examine these possibilities we purified individual mutants of the Δ2CARD construct, which is both necessary and sufficient for dsRNA binding, filament formation and ATP hydrolysis14. As previously described12,13, electrophoretic mobility shift assay (EMSA) showed that wild-type Δ2CARD cooperatively binds dsRNA in the absence of ATP, but accumulates intermediate size complexes upon addition of ATP (Fig. 4a). This observation is consistent with our previous finding that ATP hydrolysis induces rapid cycles of filament disassembly and reassembly12,13. Interestingly, the population of these intermediate complexes was significantly diminished with all six mutants, in particular with Arg337Gly, showing few or no such complexes. Measurement of ATP hydrolysis activity demonstrated that, with the exception of Arg337Gly, all five mutants hydrolyze ATP as well as wild-type (Fig. 4b). This finding rules out a lack of ATP hydrolysis as a reason for these five mutants to assemble filaments more cooperatively than wild-type. Quantitative analysis of the RNA-bound IFIH1 fraction from EMSA revealed that all six mutants bind RNA more efficiently than wild-type, both in the presence and absence of cellular levels (2 mM) of ATP (Fig. 4c). These results suggest ATP-independent mechanisms, i.e. tighter RNA binding and / or more stable protein-protein interaction, as likely responsible for the observed stability of the IFIH1 filament in vitro (Fig. 4a) and higher signaling activity in cells (Fig. 3a-b).
Fig. 4. IFIH1 mutants form filaments.

(a) Electrophoretic mobility shift assay (EMSA) of purified wild-type and mutant IFIH1 with 112 bp dsRNA. Gel images are representative of three independent experiments. (b) ATP hydrolysis activity (mean ± SD, n = 3) of wild-type and mutant IFIH1. Shown below is the SDS-PAGE analysis (Coomassie stain) of the purified wild-type and mutant IFIH1 used in Fig. 3. (c) Fraction of IFIH1-occupied sites on 112 bp dsRNA, measured from three independent EMSA performed in the presence and absence of ATP. *P<0.0002 (one tailed, unpaired t test), calculated using the values at 160 nM IFIH1. Bound fraction was calculated as in ref 12, and fitted with the Hill equation29. The dissociation constants (Kd) obtained from curve fitting are shown on the right.
Here, we describe six heterozygous IFIH1 mutations in a total of eleven individuals from eight families, where mutation-positive status is consistently associated with an induction of type I interferon activity. The finding of de novo mutations in six families, and the dominant inheritance of a clinical (F524) and / or biochemical phenotype (F524, F259) in two families, strongly suggests that these mutations are pathogenic in the heterozygous state, and that IFIH1 represents a seventh gene, mutations in which are associated with the AGS phenotype. Although AGS is most typically inherited as an autosomal recessive trait, rare examples due to dominant mutations in TREX115 and ADAR18 have been described.
A striking feature in family F259 is the marked clinical discordance between the affected child (F259_1) and his clinically asymptomatic Arg779His mutation-positive father (R259_2) and paternal grandmother (F259_3)(Supplementary Fig. 1), in spite of upregulation of type I interferon signaling in all three (Fig. 2). Thus, mutation-positivity, and positivity for an interferon signature, is not necessarily sufficient to develop an overt clinical phenotype. Notably, the same Arg779His mutation, dominantly transmitted across three generations in family F259, was seen to occur de novo in the proband of family F163. We and others have described both severe neurological disease and non-penetrance / age-dependent penetrance in the context of a recurrent Gly1007Arg mutation in ADAR1, which can also be dominantly inherited or occur as a new mutation16-18. Such clinical variability might be explained by modifying genetic factors or differential exposure to pathogens.
A majority of individuals affected with the autoimmune disease systemic lupus erythematosus (SLE) demonstrate an interferon signature19,20, and polymorphisms in IFIH1 confer an increased risk of developing SLE21. In keeping with this, two patients in our cohort (F376 and F524_1) experienced significant immunological disturbance consistent with lupus. That the majority of IFIH1 mutation-positive patients had no overt lupus phenotype again suggests the importance of modifying genetic or environmental factors, and is concordant with a multi-copy Ifih1 transgenic mouse line demonstrating chronically elevated levels of type I interferon, insufficient by itself to initiate autoimmunity22.
Nejentsev et al.23 described two canonical splice-site variants, a nonsense variant, and a missense substitution in IFIH1 as protective against type I diabetes (T1D)(Supplementary Table 6). They postulated that these, presumed loss-of-function, alleles attenuate the immune response to enterovirus, a factor implicated in the pathogenesis of T1D. Unlike the variants reported in that study, all of the mutations described here are missense substitutions conferring a gain of function, and none have been documented in the NHLBI ESP database. To date, none of our IFIH1 mutation-positive patients have developed T1D.
Studies of the AGS-related proteins TREX1, the RNase H2 complex, SAMHD1 and ADAR1, suggest that an inappropriate accumulation of self-derived nucleic acids can induce type I interferon signaling24,25. The finding of IFIH1 mutations in the similar context implicates the aberrant sensing of nucleic acids as a cause of immune upregulation. The observation of enhanced baseline and ligand-induced type I interferon signaling by all six mutant alleles is consistent with our observation of increased interferon activity / ISG transcripts in every mutation-positive individual tested. It is also consistent with our biochemical analyses, showing that these mutants bind dsRNAs more avidly and tightly than wild-type, albeit to a varying degree, indicating that even small differences in binding can result in a significant biological phenotype. These mutations provide new insights into the function of IFIH1, which might be useful in designing therapies to potentiate host antiviral innate immunity.
The dependence of mutant basal signaling activity on dsRNA binding and filament formation suggests the presence of as-yet undefined endogenous dsRNA capable of stimulating mutant, but not wild-type, receptor. In light of observed clinical non-penetrance, and the rapid-onset of neurological regression in the second year of life in two patients, we cannot dismiss the possibility that exogenous, viral-derived RNA26, also plays a role in the disease process. We note the description of an N-ethyl-N-nitrosourea induced IFIH1 missense mutation in a mouse model demonstrating upregulated interferon signaling and an autoimmune phenotype27. In contrast to the mutations that we describe, signaling by this mutant was ligand independent.
We have previously reported an interferon signature as a reliable biomarker for AGS9 and SPENCD10. The current study further emphasizes the value of searching for an interferon signature as a screening tool to identify other type I interferonopathies4. The recognition of such diseases is not just of academic interest, since defining a disturbance of type I interferon as primary to the pathogenesis of a phenotype suggests the possibility of anti-interferon / anti-inflammatory therapies28.
Methods
Affected individuals and families
All affected individuals included in this study either had a clinical and neuro-radiological diagnosis of Aicardi-Goutières syndrome associated with an upregulation of cerebrospinal fluid interferon and / or interferon stimulated genes in peripheral blood (an interferon signature), or a neuro-immunological phenotype in the presence of an interferon signature in peripheral blood recorded in the absence of any obvious infection.
Clinical information and samples were obtained with informed consent. The study was approved by the Leeds (East) Research Ethics Committee (reference number 10/H1307/132).
Exome sequencing
Genomic DNA was extracted from lymphocytes from affected individuals and parents by standard techniques. For whole exome analysis, targeted enrichment and sequencing were performed on 3 μg of DNA extracted from the peripheral blood of three individuals (F102, F163, F259). Enrichment was undertaken using the SureSelect Human All Exon kits following the manufacturer’s protocol (Agilent, Santa Clara, CA, USA) and samples were pair-end sequenced on either an IlluminaHiSeq 2000 or SOLiD platform. Sequence data were mapped using BWA (Burrows-Wheeler Aligner) against hg18 (NCBI36) human genome as a reference. Variants were called using SOAPsnp and SOAPindel (from the Short Oligonucleotide Analysis Package) with medium stringency (Supplementary Table 1).
Sanger sequencing
Primers were designed to amplify the coding exons of IFIH1 (Supplementary Table 7). Purified PCR amplification products were sequenced using BigDye™ terminator chemistry and an ABI 3130 DNA sequencer. Mutation description is based on the reference cDNA sequence NM_022168.2, with nucleotide numbering beginning from the first A in the initiating ATG codon.
Gene expression analysis
Total RNA was extracted from whole blood using a PAXgene (PreAnalytix) RNA isolation kit. RNA concentration was assessed using a spectrophotometer (FLUOstar Omega, Labtech). Quantitative reverse transcription PCR analysis was performed using the TaqMan Universal PCR Master Mix (Applied Biosystems), and cDNA derived from 40ng total RNA. The relative abundance of target transcripts, measured using TaqMan probes for IFI27 (Hs01086370_m1), IFI44L (Hs00199115_m1), IFIT1 (Hs00356631_g1), ISG15 (Hs00192713_m1), RSAD2 (Hs01057264_m1) and SIGLEC1 (Hs00988063_m1) was normalized to the expression level of HPRT1 (Hs03929096_g1) and 18s (Hs999999001_s1) and assessed with the Applied Biosystems StepOne Software v2.1 and DataAssist Software v.3.01. For each of the six probes, individual (patient and control) data were expressed relative to a single calibrator (control C25). As previously described, the median fold change of the six ISGs, when compared to the median of the combined 29 healthy controls, was used to create an interferon score for each patient9. RQ (relative quantification) is equal to 2−ΔΔCt i.e. the normalized fold change relative to a control. When a patient was assayed on more than one occasion, the data for repeat measurements were combined to calculate a mean value (using Applied Biosystems DataAssist software v.3.01).
Statistics
In the absence of a normal distribution, ISG levels and interferon scores were log transformed and analyzed using parametric testing (one way ANOVA). Bonferroni’s or Dunnett’s multiple comparison tests were applied as detailed in Figure 2 and Supplementary Figure 4. Interferon scores for each group were expressed as the median (interquartile range: IQR). Statistics were calculated using GraphPad Prism version 5.0d for Mac OS X (San Diego California USA www.graphpad.com).
Microsatellite genotyping
To confirm maternity and paternity, informative polymorphic microsatellite markers on chromosomes 3 (D3S3640, D3S3560), 11 (D11S4205, D11S913, D11S987, D11S1889), and 20 (D20S847, D20S896, D20S843) were genotyped using DNA from F102, F524_2, F647, F626, F237, F163 and respective parents, as well as an unrelated control sample. DNA samples were amplified by standard PCR (primer sequences available on request). Each amplicon was mixed with HiDi™Formamide (AppliedBiosystems) and 500ROX™SizeStandard (AppliedBiosystems) and run on the GeneticAnalyzer3010 capillary electrophoresis system. Results were analyzed with the GeneMapper v4.1 (AppliedBiosystems) software.
Protein modeling
The IFIH1 substitutions Arg337Gly, Asp393Val, Gly495Arg, Arg720Gln, Arg779His and Arg779Cys all fall within the helicase domain of the protein. Molecular graphics figures were generated with PyMOL (Schrodinger) using the PDB coordinate (ID:4GL2).
Interferon reporter assay
The pFLAG-CMV4 plasmid encoding IFIH1 was described elsewhere12. Indicated mutations were introduced using KAPA HiFi DNA polymerase. HEK293T cells (ATCC) were maintained in 24-well plates in Dulbecco’s modified Eagle medium (Cellgro) supplemented with 10% heat-inactivated fetal calf serum and 1% penicillin/streptomycin. At ~95% confluence, cells were co-transfected with pFLAG-CMV4 plasmids encoding wild-type or mutant IFIH1 (5 ng, unless mentioned otherwise), Interferon beta (IFNβ) promoter driven firefly luciferase reporter plasmid (200 ng) and a constitutively expressed Renilla luciferase reporter plasmid (pRL-CMV, 20 ng) by using lipofectamine2000 (Life) according to the manufacturer’s protocol. The medium was changed 6 hours post-transfection, and cells were subsequently stimulated with poly I:C (0.5 μg/ml, Invivogen) or in vitro transcribed 162 bp dsRNA (0.5 ug/ml) using lipofectamine2000. Cells were lysed 24 hours post-stimulation and IFNβ promoter activity was measured using the Dual Luciferase Reporter assay (Promega) and a Synergy2 plate reader (BioTek). Firefly luciferase activity was normalized against Renilla luciferase activity. Error bars represent standard deviation of three independent experiments. For western blot, anti-FLAG (F7425, Sigma-Aldrich) and anti-actin (A5441, Sigma-Aldrich) primary antibodies were used together with anti-rabbit IgG-HRP (sc-2004, Santa Cruz Biotechnology) and anti-mouse IgG-HRP (sc-358917, Santa Cruz Biotechnology) secondary antibodies, respectively.
Protein and RNA preparation
Wild-type and variants of IFIH1Δ2CARD were expressed from pET50 (Novagen) as a 6xHis tagged NusA fusion protein in BL21(DE3) as previously described for wild-type IFIH112. Briefly, cells were lysed by Emulsiflex and proteins were purified by a combination of Ni-NTA affinity, heparin affinity and size exclusion chromatographies (SEC) in buffer A (20 mM Hepes, pH 7.5, 150 mM NaCl, and 2 mM DTT). The NusA-tag was removed by HRV 3C cleavage for all proteins. Sequences of the 162 and 112 bp dsRNAs were taken from the first 150 and 100 nt of the IFIH1 gene, flanked by 5′gggaga and tctccc3′. dsRNAs were prepared as previously described12. Briefly, two complementary strands of dsRNA were co-transcribed using T7 RNA polymerase and the duplex RNA was separated from individual strands by 8-10% polyacrylamide gel electrophoresis, followed by electroelution. The 3′ end of purified 112 bp dsRNA was subsequently labeled with fluorescein hydrizide as previously described14.
Electrophoretic Mobility Shift Assay (EMSA) and ATP hydrolysis assay
Assays were performed as previously reported12. Briefly, 3′-fluorescein labeled 112 bp (20 nM)14 was incubated with protein (40 - 160 nM) in buffer B (20 mM Hepes, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2 and 2 mM DTT) in the presence and absence of 2 mM ATP, and the complex was analyzed on Bis-Tris native PAGE (Life). Fluorescent gel images were recorded using the scanner FLA9000 and analyzed with Multigauge (GE Healthcare). Curve fitting was performed using the program KaleidaGraph (Synergy). For ATP hydrolysis assays, IFIH1 (0.3 μM) was incubated with ATP (2 mM) and 112 bp dsRNA (0.6 μM) in buffer B at 37°C. Use of an excess amount of 112 bp dsRNA (0.6 μM corresponds to 4.8 μM IFIH1-binding sites as each 112 bp dsRNA can accommodate 8 IFIH1 molecules) simplifies the comparison between wild-type and mutant IFIH1 by focusing on the intrinsic ATP hydrolysis activities, independent of dsRNA binding12. Reactions were quenched at 0 min and 5 min with 50 mM EDTA, and the level of released phosphate was measured using GreenReagent (Enzo Lifescience) at OD600.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We sincerely thank the participating families for the use of genetic samples and clinical information, and all clinicians who contributed samples and data not included in this manuscript. We thank Diana Chase for proof-reading the manuscript. We thank Gigi Notarangelo for helpful discussion. Y.d.T.D. holds a Novartis Foundation post-doctoral fellowship. S.H. holds a Pew scholarship and Career Development award from Boston Children’s Hospital. Y.J.C. acknowledges the Manchester Biomedical Research Centre, Manchester Academic Health Sciences Centre, the Greater Manchester Comprehensive Local Research Network, the Great Ormond Street Hospital Children’s Charity, the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement 241779, and the European Research Council (GA 309449).
The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Footnotes
The authors declare that they have no competing financial interests.
URLs
UCSC Human Genome Browser: http://genome.ucsc.edu/
Ensembl: http://www.ensembl.org/
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/
Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA, http://snp.gs.washington.edu/EVS/ (accessed 17 October 2013)
PolyPhen2: http://genetics.bwh.harvard.edu/pph2/
SIFT: http://sift.jcvi.org/www/SIFT_enst_submit.html
MutationTaster: http://www.mutationtaster.org/
Clustal Omega: www.ebi.ac.uk
Protein Data Bank: http://www.pdb.org/
Alamut: www.interactive-biosoftware.com/
References
- 1.Gresser I, Tovey MG, Maury C, Chouroulinkov I. Lethality of interferon preparations for newborn mice. Nature. 1975;258:76–8. doi: 10.1038/258076a0. [DOI] [PubMed] [Google Scholar]
- 2.Gresser I, et al. Progressive glomerulonephritis in mice treated with interferon preparations at birth. Nature. 1976;263:420–2. doi: 10.1038/263420a0. [DOI] [PubMed] [Google Scholar]
- 3.Gresser I, et al. Interferon-induced disease in mice and rats. Ann N Y Acad Sci. 1980;350:12–20. doi: 10.1111/j.1749-6632.1980.tb20602.x. [DOI] [PubMed] [Google Scholar]
- 4.Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238:91–8. doi: 10.1111/j.1749-6632.2011.06220.x. [DOI] [PubMed] [Google Scholar]
- 5.Crow YJ, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–20. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 6.Crow YJ, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–6. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 7.Rice GI, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–32. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rice GI, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–8. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice GI, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12:1159–69. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briggs TA, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. 2011;43:127–31. doi: 10.1038/ng.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goubau D, Deddouche S, Reis ESC. Cytosolic sensing of viruses. Immunity. 2013;38:855–69. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peisley A, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A. 2011;108:21010–5. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peisley A, et al. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc Natl Acad Sci U S A. 2012;109:E3340–9. doi: 10.1073/pnas.1208618109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu B, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–89. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 15.Rice G, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–5. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tojo K, et al. Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation. Mov Disord. 2006;21:1510–3. doi: 10.1002/mds.21011. [DOI] [PubMed] [Google Scholar]
- 17.Livingston JH, et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2013;51:76–82. doi: 10.1136/jmedgenet-2013-102038. [DOI] [PubMed] [Google Scholar]
- 18.Kondo T, et al. Dyschromatosis symmetrica hereditaria associated with neurological disorders. J Dermatol. 2008;35:662–6. doi: 10.1111/j.1346-8138.2008.00540.x. [DOI] [PubMed] [Google Scholar]
- 19.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–23. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cen H, et al. Association of IFIH1 rs1990760 polymorphism with susceptibility to autoimmune diseases: a meta-analysis. Autoimmunity. 2013;46:455–62. doi: 10.3109/08916934.2013.796937. [DOI] [PubMed] [Google Scholar]
- 22.Crampton SP, Deane JA, Feigenbaum L, Bolland S. Ifih1 gene dose effect reveals MDA5-mediated chronic type I IFN gene signature, viral resistance, and accelerated autoimmunity. J Immunol. 2012;188:1451–9. doi: 10.4049/jimmunol.1102705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nejentsev S, et al. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:R130–6. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stetson DB. Endogenous retroelements and autoimmune disease. Curr Opin Immunol. 2012;24:692–7. doi: 10.1016/j.coi.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deddouche S, et al. Identification of an LGP2-associated MDA5 agonist in picornavirus-infected cells. Elife. 2014;3:e01535. doi: 10.7554/eLife.01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Funabiki M, et al. Autoimmune Disorders Associated with Gain of Function of the Intracellular Sensor MDA5. Immunity. 2014 doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 28.Crow YJ, et al. Therapies in Aicardi-Goutieres syndrome. Clin Exp Immunol. 2014;175:1–8. doi: 10.1111/cei.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herries DG. Enzyme structure and mechanism. second edition W H Freeman; New York: 1985. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.