

Chronic ER stress down-regulates XIAP by activating the PERK branch of the UPR. PERK attenuates Xiap translation via eIF2α phosphorylation. PERK promotes XIAP degradation via ATF4. CHOP induction and XIAP suppression act in parallel to sensitize cells to ER stress–induced apoptosis.
Abstract
Endoplasmic reticulum (ER) protein misfolding activates the unfolded protein response (UPR) to help cells cope with ER stress. If ER homeostasis is not restored, UPR promotes cell death. The mechanisms of UPR-mediated cell death are poorly understood. The PKR-like endoplasmic reticulum kinase (PERK) arm of the UPR is implicated in ER stress–induced cell death, in part through up-regulation of proapoptotic CCAAT/enhancer binding protein homologous protein (CHOP). Chop−/− cells are partially resistant to ER stress–induced cell death, and CHOP overexpression alone does not induce cell death. These findings suggest that additional mechanisms regulate cell death downstream of PERK. Here we find dramatic suppression of antiapoptosis XIAP proteins in response to chronic ER stress. We find that PERK down-regulates XIAP synthesis through eIF2α and promotes XIAP degradation through ATF4. Of interest, PERK's down-regulation of XIAP occurs independently of CHOP activity. Loss of XIAP leads to increased cell death, whereas XIAP overexpression significantly enhances resistance to ER stress–induced cell death, even in the absence of CHOP. Our findings define a novel signaling circuit between PERK and XIAP that operates in parallel with PERK to CHOP induction to influence cell survival during ER stress. We propose a “two-hit” model of ER stress–induced cell death involving concomitant CHOP up-regulation and XIAP down-regulation both induced by PERK.
INTRODUCTION
The endoplasmic reticulum (ER) is a membrane-bound organelle essential for the folding of virtually all secreted and membrane proteins in eukaryotic cells. Environmental, pathological, or physiological processes that interfere with ER protein folding cause ER stress. Cells activate a set of intracellular signaling pathways, termed the unfolded protein response (UPR), when they are confronted with ER stress. In metazoans, the UPR is controlled by three ER-resident transmembrane proteins that activate distinct transcriptional and translational programs in response to ER stress: inositol-requiring enzyme 1 (IRE1), PKR-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6; Walter and Ron, 2011). The UPR can alleviate ER stress and enhance cell survival by increasing the fidelity of protein folding, promoting ER-associated degradation of misfolded proteins, and attenuating protein translation. If these measures fail to reduce ER stress, UPR signaling switches to promote cell death (Tabas and Ron, 2011). ER stress–induced cell death is implicated in the pathogenesis and progression of diverse diseases, including inflammation, retinal degeneration, diabetes, and infections (Wang and Kaufman, 2012). To begin to dissect the roles of UPR signaling pathways in regulating cell death induced by chronic ER stress, we previously used chemical-genetic tools to selectively and robustly activate IRE1, PERK, or ATF6 in mammalian cells (Lin et al., 2007, 2009; Hollien et al., 2009; Chiang et al., 2012a). By this strategy, we found that chronic activation of the PERK arm, but not the IRE1 or ATF6 arm, of the UPR triggered apoptosis (Lin et al., 2007, 2009). However, the precise mechanisms that promote apoptosis in response to chronic PERK signaling are poorly understood.
The PERK signaling pathway is the key regulator of translation during the UPR (Ron and Harding, 2012). In response to ER stress, PERK activates its cytosolic kinase domain to specifically phosphorylate the α-subunit of eukaryotic translation initiation factor 2 (eIF2α; Harding et al., 1999). Phosphorylated eIF2α inhibits the guanine nucleotide exchange factor, eIF2B, which converts inactive, GDP-bound eIF2 to active, GTP-bound eIF2 (Ron and Harding, 2012). Decreased levels of eIF2-GTP impede formation of the eIF2-GTP-tRNAMet ternary complex required for translation initiation on mRNAs and thereby reduce protein synthesis (Ron and Harding, 2012). Rare mRNAs, such as the Atf4 transcriptional regulator, are translated more effectively when eIF2α is phosphorylated because these mRNAs contain small noncoding upstream open reading frames (uORFs) that inhibit translation of the downstream protein-encoding open reading frame (Harding et al., 2000a). When eIF2 activity is reduced through eIF2α phosphorylation during ER stress, ribosome assembly “skips” these inhibitory uORFs and instead increases translation from the ATF4 protein-encoding ORF (Lu et al., 2004a). By this mechanism, PERK signaling also initiates a distinct transcriptional program during the UPR through production of transcriptional activators such as ATF4 (Harding et al., 2003). PERK's translational and transcriptional outputs profoundly influence cell survival positively and negatively during ER stress. Attenuation of protein synthesis by PERK's phosphorylation of eIF2α reduces ER protein folding demands and ER stress levels. Cells with Perk-null mutations or eIF2α mutated in serine 51 to nonphosphorylatable alanine (eIF2αS/S to eIF2αA/A) both show elevated protein synthesis and increased levels of misfolded proteins and succumb more readily to cell death in response to ER stress (Harding et al., 2000b; Scheuner et al., 2001). Paradoxically, too much PERK signaling also promotes cell death, in part through its induction of the CCAAT/enhancer binding protein homologous protein (CHOP) proapoptotic transcription factor, itself a downstream transcriptional target of ATF4 (Zinszner et al., 1998; Harding et al., 2000a; Rutkowski et al., 2006). Indeed, Chop−/− cells are resistant to pharmacological and in vivo insults that trigger ER stress and cell damage (Oyadomari et al., 2002; Pennuto et al., 2008). However, Chop−/− cells still undergo apoptosis in response to chronic ER stress (Zinszner et al., 1998), and overexpression of CHOP itself does not cause apoptosis (Han et al., 2013). These findings suggest that additional CHOP-independent mechanisms regulate cell death downstream of PERK activation and eIF2α phosphorylation (Zinszner et al., 1998; Cazanave and Gores, 2010; Han et al., 2013)
Inhibitors of apoptosis (IAPs) are a conserved family of cytosolic proteins that regulate cysteine-dependent, aspartyl-specific proteases (caspases)—terminal executioners of cell death in metazoan cells (Salvesen and Duckett, 2002). X-linked IAP (XIAP) is the most potent IAP inhibitor of caspases and is found in all mammalian cell types and tissues (Salvesen and Duckett, 2002; Eckelman et al., 2006). XIAP directly binds and suppresses caspase proteolytic activity through its amino-terminal baculovirus IAP repeat domains (Deveraux et al., 1997). XIAP also contains a carboxyl-terminal RING zinc finger domain with E3 ubiquitin ligase activity that promotes ubiquitination and subsequent proteasomal degradation of substrates that also include caspases (Suzuki et al., 2001; Schile et al., 2008). By these two modes of suppressing caspase activity, XIAP serves as an important guardian against cell death. Because of XIAP's key role in influencing cell survival and apoptosis, multiple transcriptional, translational, and posttranslational mechanisms have evolved to regulate levels of XIAP in the cell (Eckelman et al., 2006; Galban and Duckett, 2010). Here we find a significant down-regulation in XIAP protein levels in response to chronic ER stress. We find that XIAP down-regulation is robustly mediated via translational and posttranslational mechanisms specifically governed by the PERK arm of the UPR via eIF2α phosphorylation and ATF4 activity. Loss of XIAP results in increased sensitivity to ER stress–induced cell death that can be alleviated by restoring XIAP levels. Our findings define an unexpected link between the PERK signaling pathway and a key regulator of intrinsic apoptosis that contributes to cell death induced by chronic ER stress.
RESULTS
Chronic ER stress down-regulates XIAP
We previously found that chronic exposure to ER stress led to down-regulation in the levels of proteins such as spliced XBP-1 and Grp78/BiP both in cell culture and in vivo (Lin et al., 2007). Both spliced XBP-1 and Grp78/BiP are important regulators of ER homeostasis, and their loss sensitizes cells to apoptosis (Lee et al., 2005; Luo et al., 2006; Lin et al., 2007). These findings suggested that reductions in levels of other regulators of cellular homeostasis in response to chronic ER stress might sensitize cells to apoptosis. To examine this possibility, we screened for proteins whose levels dropped significantly as a function of the duration of ER stress. One protein whose levels progressively fell in cells exposed to ER stress was XIAP. In HEK293 cells exposed to either tunicamycin or thapsigargin, XIAP steady-state protein levels significantly and progressively declined over the course of 48 h of drug treatment (Figure 1A). The decline in XIAP correlated with increase in apoptosis, as evident by cell morphology and induction of molecular markers of apoptosis such as cleaved poly(ADP-ribose) polymerase (PARP; Figure 1A). We next examined whether other cell types showed similar declines in XIAP protein levels. Similar to our findings in HEK293 cells, we found sharp and progressive drops in XIAP levels in all mammalian cell types we examined—HeLa, mouse embryonic fibroblast (MEF), Huh7, and HCT116—in response to chronic exposure to tunicamycin or thapsigargin (Figure 1, B–E). By contrast to the sharp decline seen in XIAP protein levels in cells exposed to chronic tunicamycin or thapsigargin, we saw no drop in Xiap mRNA levels (Figure 1F). Finally, we examined whether XIAP protein levels dropped in vivo in an animal model of P23H rhodopsin–induced retinal degeneration. Rhodopsin is a transmembrane light chromophore expressed solely in rod photoreceptors of the eye, and the P23H mutation is the most common cause of autosomal dominant retinitis pigmentosa in North America (Dryja et al., 1990). The P23H mutation leads to rhodopsin protein misfolding in the ER, induction of ER stress, and progressive death of rod photoreceptors with ensuing vision loss (Sung et al., 1991; Illing et al., 2002; Price et al., 2011; Chiang et al., 2012b). We collected retinas from wild-type and transgenic animals expressing P23H rhodopsin at postnatal day 90, an age when photoreceptors were undergoing cell death (Figure 1G, left), and also found significantly reduced XIAP levels in P23H rhodopsin retinas compared with control (Figure 1G, middle). Taken together, our findings demonstrated that down-regulation of XIAP protein is a conserved consequence of chronic ER stress in vitro and in vivo and that this drop correlates with the emergence of cell death.
FIGURE 1:

Chronic ER stress down-regulates XIAP protein but not Xiap mRNA. HEK293 (A), HeLa (B), MEF (C), Huh7 (D), and HCT116 (E) cells were treated with 5 μg/ml tunicamycin (Tm) or 500 nM thapsigargin (Tg) for the indicated hours. Cell lysates were immunoblotted for XIAP or cleaved PARP (cPARP). XIAP levels after Tm or Tg treatment from three different experiments were quantified by densitometry and are shown relative to XIAP levels in untreated cells (A–C). Asterisks indicate statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001). Actin levels served as a protein loading control (A–E). (F) HEK293 cells were treated with Tm or Tg for indicated durations, and Xiap mRNA levels were measured by quantitative PCR (qPCR) and are shown relative to levels in untreated cells. (G) Retinas from wild-type (left) or P23H rhodopsin transgenic (right) rats were collected at postnatal day 90, and retinal protein lysates were probed for XIAP. HSP90 levels served as a protein loading control. A representative immunoblot from five different experimental animals is shown.
The PERK arm of the UPR down-regulates XIAP
The UPR is an attractive mechanism to regulate XIAP levels through its transcriptional, translational, and posttranslational programs activated in response to ER stress. To determine which UPR signaling pathway, if any, regulates XIAP, we used chemical-genetic constructs to specifically and sensitively activate IRE1, PERK, or ATF6 individually (Figure 2A). To selectively activate IRE1, we used an IRE1 construct bearing an isoleucine-to-glycine substitution at residue 642, which enabled selective regulation of IRE1’s RNase function by the small molecule 1NM-PP1 (Lin et al., 2007; Hollien et al., 2009). To selectively activate PERK, we used a PERK fusion construct that joined the kinase domain of PERK to FK506-binding domains that enabled artificial dimerization and activation of Fv2E-PERK in the presence of the small molecule AP20187 (Lu et al., 2004a, b; Lin et al., 2009). To selectively activate ATF6, we expressed the 373–amino acid bZIP transcriptional activator domain of ATF6 (ATF6(373)) under the control of a tetracycline-responsive element (Hiramatsu et al., 2011; Chiang et al., 2012a). We previously demonstrated robust and selective activation of IRE1, PERK, or ATF6 using these chemical-genetic constructs in diverse mammalian cell types (Lin et al., 2007, 2009; Hiramatsu et al., 2011).
FIGURE 2:

The PERK arm of the UPR down-regulates XIAP protein but not Xiap mRNA. (A) Schematic of chemical-genetic constructs to selectively activate individual UPR pathways in stable mammalian cell lines. IRE1 pathway is selectively activated using an IRE1[I642G] with 1NM-PP1 binding site. PERK pathway is selectively activated using Fv2E-PERK with AP20187 dimerizing chemical. ATF6 pathway is selectively activated using a tetracycline-inducible ATF6(373) (TetON-ATF6(373)). (B) Stable HEK293 cell lines bearing IRE1[I642G], Fv2E-PERK, or TetON-ATF6a (373) were treated with 5 μM 1NM-PP1, 1 nM AP20187, or 1 μg/ml doxycycline (Dox) for the indicated hours. Cell lysates were probed for XIAP or cleaved PARP (cPARP). (C) Stable HeLa cell lines bearing Fv2E-PERK or tetracycline-repressible ATF6(373) (TetOFF-ATF6(373)) were treated with 1 nM AP20187 or Dox-free media for the indicated duration, and cell lysates were probed for XIAP or cleaved PARP. (D) Cells stably expressing Fv2E-PERK were treated with AP20187 for the indicated durations, and Xiap mRNA levels were measured by qPCR and are shown relative to levels in untreated cells. (E) Schematic of GADD34 and salubrinal's effects on PERK signaling. (F) HEK293 cells were transfected with GADD34. Then 5 μg/ml Tm or 500 nM Tg was added, and cell lysates were immunoblotted for phosphorylated eIF2α levels. Total eIF2α levels served as a protein loading control. (G) GADD34-transfected cells were treated with Tm or Tg as indicated. Cell lysates were probed for XIAP. (H) Cells were treated with salubrinal as indicated. Cell lysates were probed for XIAP and phospho-eIF2α (eIF2α-P). For B, C, G, and H, actin served as a protein loading control.
Using these tools, we saw a pronounced down-regulation of XIAP in both HEK and HeLa cells after PERK activation that was similar in kinetics and magnitude to the down-regulation of XIAP seen with tunicamycin or thapsigargin treatment (compare Figure 2, B and C, with Figure 1). Furthermore, in both HEK293 and HeLa cells, we saw that down-regulation of XIAP by artificial PERK activation correlated with onset of cell death as evident by morphological changes and emergence of cleaved PARP, similar to what we observed with extended tunicamycin and thapsigargin treatment (Figure 2, B and C, middle, and Figure 1). By contrast, we saw no effect on XIAP levels or induction of cleaved PARP after selective activation of IRE1’s RNase function (Figure 2B). We saw mild down-regulation of XIAP after ATF6 activation in HEK293 cells expressing tetracycline-inducible ATF6(373) (Figure 2B) but not in HeLa cells expressing tetracycline-repressible ATF6(373) (Figure 2C). These divergent effects between the tetracycline (tet)-inducible and tet-repressible ATF6 systems arise from negative effects of extended doxycycline exposure itself (unpublished data), and ATF6 signaling itself might also have a causal role in reduction of XIAP protein level in HEK293 cells. Taken together, these findings indicate that the PERK signaling arm of the UPR is the key UPR pathway responsible for down-regulating XIAP. Of interest, we saw no drop in Xiap mRNA levels after selective PERK activation, indicating that down-regulation of XIAP did not arise from reduced transcription (Figure 2D).
A key proximal step in PERK signal transduction is phosphorylation of eIF2α. To further test the role of PERK signal transduction in regulating XIAP levels, we reduced levels of phosphorylated eIF2α by GADD34 phosphatase expression (Novoa et al., 2001) or increased phosphorylated eIF2α levels by salubrinal addition (Boyce et al., 2005; Figure 2E). When we expressed GADD34, we partially ameliorated the down-regulation of XIAP induced by tunicamycin or thapsigargin (Figure 2G) concomitant with blocking the increase in the levels of phosphorylated eIF2α normally seen after PERK activation by ER stress (Figure 2F). Conversely, when we increased levels of phosphorylated eIF2α with salubrinal, we saw the opposite effect, with a corresponding drop in XIAP levels (Figure 2H). Taken together, these findings show that PERK signaling via phosphorylation of eIF2α is an important step in regulating XIAP levels.
eIF2α phosphorylation attenuates Xiap mRNA translation
Next we dissected how downstream steps in PERK signal transduction regulated XIAP. To investigate how PERK's control of translation via eIF2α phosphorylation affected XIAP, we introduced the Fv2E-PERK system into MEF cells defective for eIF2α phosphorylation to selectively activate PERK signaling without input from other signaling processes (e.g., other eIF2α kinases or UPR pathways; Scheuner et al., 2001). In wild-type eIF2αS/S cells stably expressing Fv2E-PERK (S/SFv2E-PERK), AP20187 addition resulted in rapid activation of the PERK pathway, as evidenced by phosphorylation shift of Fv2E-PERK and phosphorylation of eIF2α (Figure 3A). By contrast, in mutant eIF2αA/A cells stably expressing Fv2E-PERK (A/AFv2E-PERK), AP20187 addition triggered strong phosphorylation shift of Fv2E-PERK without production of phosphorylated eIF2α (Figure 3A). When we examined XIAP levels in S/SFv2E-PERK or A/AFv2E-PERK cells, we found that AP20187 triggered pronounced XIAP down-regulation (>90%) in wild-type eIF2α S/S cells, but XIAP down-regulation was completely abrogated in mutant eIF2α A/A cells (Figure 3A). Phosphorylated eIF2α down-regulated XIAP through attenuation of XIAP protein synthesis, as we saw a substantial drop in newly synthesized XIAP protein in wild-type eIF2α S/S cells but no reduction in XIAP synthesis in mutant eIF2α A/A cells after induction of ER stress or AP20187 addition in 35S metabolic-labeling XIAP synthesis studies (Figure 3, B–D). These findings indicated that eIF2α was required for the down-regulation of XIAP caused by PERK activation and that phosphorylation of eIF2α triggered XIAP translational attenuation, contributing to the drop in steady-state XIAP levels seen with chronic ER stress and chronic PERK activation.
FIGURE 3:
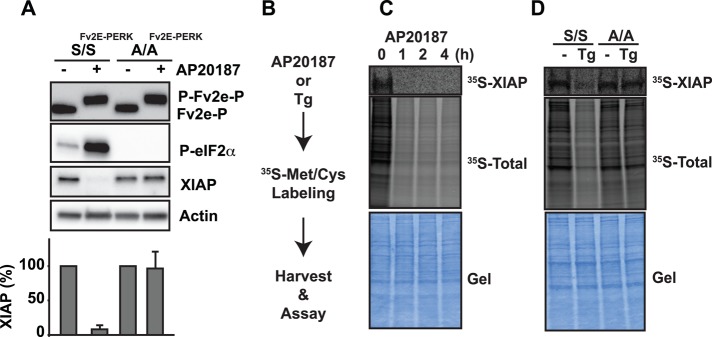
PERK signaling attenuates XIAP translation through eIF2α phosphorylation. (A) Wild-type eIF2αS/S MEFs expressing Fv2E-PERK (S/SFv2E-PERK) and mutant eIF2αA/A MEFs expressing Fv2E-PERK (A/AFv2E-PERK) were treated with 1 nM AP20187, and cell lysates were probed for Fv2E-PERK, phosphorylated eIF2α, or XIAP. Actin served as a loading control. XIAP levels after AP20187 treatment from three different experiments were quantified by densitometry and are shown relative to XIAP levels in untreated cells. (B) Flowchart for metabolic labeling experiments in C and D. (C) Cells expressing Fv2E-PERK were pretreated with AP20187 for the indicated hours and metabolically labeled with 100 μCi of [35S]Met/Cys for 1 h. XIAP was immunoprecipitated, and 35S-labeled XIAP was visualized by phosphorimaging. Middle, total 35S-labeled protein levels after AP20187 treatment. (D) eIF2αS/S and eIF2αA/A MEFs were pretreated with 500 nM Tg for 1 h and labeled with 100 μCi of [35S]Met/Cys. XIAP was immunoprecipitated, and 35S-labeled XIAP was visualized by phosphorimaging. Middle, total 35S-labeled protein levels after Tg treatment. (C, D) Bottom, Coomassie-stained gel as loading control.
ATF4 promotes XIAP protein degradation
PERK signaling also induces a transcriptional program via induction of ATF4 and CHOP transcription factors. To test the effects of ATF4 and CHOP on XIAP, we transduced cells with adenovirus carrying these transcription factors (Figure 4A). Surprisingly, we found pronounced down-regulation of XIAP after ATF4 expression but not that of CHOP (Figure 4A). eIF2α was not phosphorylated in these studies, indicating that ATF4’s effects on XIAP did not occur via translational attenuation. To further test the role of ATF4 in regulating XIAP levels in response to PERK signaling, we introduced Fv2E-PERK into ATF4−/− MEF cells. In wild-type ATF4+/+ cells stably expressing Fv2E-PERK (ATF4+/+Fv2E-PERK), AP20187 addition resulted in phosphorylation shift of Fv2E-PERK and strong phosphorylation of eIF2α (Figure 4B and Supplemental Figure S1). By contrast, ATF4−/− MEF cells expressing Fv2E-PERK (ATF4−/−Fv2E-PERK) also showed strong Fv2E-PERK and eIF2α phosphorylation after AP20187 addition but failed to induce downstream transcriptional targets (Figure 4B and Supplemental Figure S1). When we examined XIAP levels in wild-type ATF4+/+ cells, we found that selective PERK activation triggered pronounced (>90%) down-regulation in XIAP levels (Figure 4B). However, we saw only partial down-regulation (∼50%) in mutant ATF4−/− cells (Figure 4B) after selective PERK activation despite identical Fv2E-PERK activation and phosphorylated eIF2α production in both ATF4+/+ and ATF4−/− MEF cells (Figure 4B). To further confirm the significance of ATF4 in XIAP down-regulation, we used short hairpin RNA (shRNA) to knock down Atf4 in HEK293 cells stably expressing Fv2E-PERK and then examined effects on XIAP after ATF4 knockdown and PERK activation. In HEK293 cells expressing control shRNAs, we saw pronounced reduction of XIAP after addition of AP20187 and activation of the PERK pathway (Figure 4C). By contrast and similar to our findings in the ATF4−/− MEF cells, we found that Atf4 knockdown in HEK cells significantly attenuated XIAP down-regulation after AP20187 addition and PERK pathway activation (Figure 4C). These findings indicated that ATF4 was important for down-regulating XIAP in response to PERK activation by ER stress.
FIGURE 4:

ATF4 but not CHOP promotes XIAP degradation. (A) ATF4 or CHOP were adenovirally transduced into HeLa cells for 24 h, and XIAP, ATF4, or CHOP expression was detected by immunoblotting. (B) Wild-type ATF4 MEFs expressing Fv2E-PERK (ATF4(+/+)Fv2E-PERK) and ATF4-knockout MEFs expressing Fv2E-PERK (ATF4(−/− )Fv2E-PERK) were treated with 1 nM AP20187, and cell lysates were probed for Fv2E-PERK, phosphorylated eIF2α, or XIAP. Changes in XIAP levels were quantified by densitometry from three experimental replicates and are shown relative to XIAP levels in untreated cells. Asterisk indicates statistical significance. (C) HEK293 cells stably expressing Fv2E-PERK were transfected with scrambled nonsilencing control (shCont) or Atf4-specific (shATF4) shRNAs. Cells were treated with 1 nM AP20187, and cell lysates were probed for phosphorylated eIF2α, ATF4, and XIAP. XIAP levels were quantified as before and are shown related to XIAP levels in untreated cells. Asterisk indicates statistical significance. (D) XIAP was immunoprecipitated from cells transduced with ATF4, and ubiquitin (top) or XIAP (bottom) were detected by immunoblotting. (E) HeLa cells were adenovirally transduced ATF4 for 18 h, treated with 50 μg/ml cycloheximide, and harvested at indicated time periods. Cell lysates were probed for XIAP (left), and XIAP levels were quantified and plotted from three experimental replicates (right). (F, G) Cells expressing Fv2E-PERK were treated with 1 nM AP20187, 10 μM MG132 (F), or 1 μM lactacystin (G) as indicated. Cell lysates were probed for XIAP. (H) Cells expressing Fv2E-PERK were treated with 1 nM AP20187 in the presence or absence of 25 μM pancaspase inhibitor zVAD as indicated. Cell lysates were probed for XIAP or cleaved PARP. Actin served as a loading control as indicated.
Next we examined how ATF4 down-regulates XIAP. Microarray and RNA sequencing studies predict that ATF4’s transcriptional targets include a number of ubiquitin ligases (Harding et al., 2003; Lange et al., 2008; Han et al., 2013). Therefore we examined whether ATF4 down-regulated XIAP by promoting its degradation through the ubiquitin-proteasome degradation system. We found pronounced ubiquitination of XIAP after ATF4 overexpression (Figure 4D), suggesting that ATF4 was promoting the destabilization and degradation of XIAP during PERK pathway activation. Indeed, when we examined XIAP protein stability after cycloheximide inhibition of protein synthesis, we found significantly faster decline in XIAP protein levels after overexpression of ATF4 (Figure 4E). To further test whether proteasomal XIAP protein degradation was involved in PERK-induced XIAP down-regulation, we blocked proteasome function using MG132 or lactacystin. We found that proteasomal inhibition prevented the down-regulation of XIAP induced by PERK activation (Figure 4, F and G). Caspases have also been reported to degrade XIAP (Deveraux et al., 1999). To test whether caspases played a role in XIAP down-regulation, we blocked caspase proteolysis using the pancaspase inhibitor zVAD. When we artificially activated PERK, we significantly inhibited production of caspase-cleaved PARP protein, but, in contrast, XIAP levels still dropped and were unaffected by zVAD addition (Figure 4H). Taken together, these studies show that ATF4 promotes XIAP degradation and that the PERK signaling pathway down-regulates XIAP by two modes: 1) attenuation of de novo XIAP synthesis via eIF2α phosphorylation and 2) degradation of existing XIAP via ATF4 induction.
XIAP levels determine cell survival during ER stress
In our studies, down-regulation of XIAP correlated with the emergence of cell death arising from chronic ER stress. To establish causality between XIAP levels and ER stress-induced cell death, we compared the sensitivities of human colon carcinoma cell line HCT116 Xiap+/+ and HCT116 Xiap−/− cells (Krieg et al., 2009; Figure 5A) to apoptosis triggered by extended tunicamycin and thapsigargin treatment. We saw increased PARP cleavage by immunoblotting and increased cell death by flow cytometry in HCT116 Xiap−/− cells compared with HCT116 Xiap+/+ cells in response to tunicamycin and thapsigargin (Figure 5, B and C). To further test the significance of XIAP in influencing ER stress-induced cell death, we reintroduced human XIAP back into HCT116 Xiap−/− cells by stable overexpression (Figure 5D) and compared sensitivities to tunicamycin and thapsigargin. In HCT116 Xiap−/− cells reconstituted with XIAP, we saw significantly decreased PARP cleavage and decreased 7-aminoactinomycin D (7-AAD) labeling compared with the parental HCT116 Xiap−/−-knockout cells in response to ER stress–inducing agents (Figure 5, E and F). These studies show that ambient intracellular XIAP levels prevent ER stress–induced cell death and that XIAP down-regulation triggered by extended ER stress sensitizes cells to apoptosis.
FIGURE 5:

XIAP enhances resistance to ER stress–induced cell death independent of CHOP. (A) HCT116 XIAP+/+ (XIAP WT) or XIAP−/− (XIAP KO) cell lysates were probed for XIAP. (B) XIAP WT or XIAP KO cells were treated with 5 μg/ml Tm or 100 nM Tg. Cell lysates were probed for cleaved PARP. (C) XIAP WT or XIAP KO cells were treated with Tm or Tg. The number of 7-AAD positive cells was quantified by FACS and is shown relative to untreated cells. (D) Cell lysates from XIAP WT, XIAP KO, and XIAP KO cells stably reconstituted with human XIAP (XIAP Re) were probed for XIAP. (E) XIAP KO and XIAP Re cells were treated with Tm or Tg. Cell lysates were probed for cleaved PARP. (F) XIAP KO or XIAP Re cells were treated with Tm or Tg. The number of 7-AAD positive cells was quantified by FACS and is shown relative to untreated cells. (G) CHOP wild-type (WT) or knockout (KO) MEFs were treated with tunicamycin for 24 h at the indicated concentrations, and ATF4, CHOP, and cleaved PARP were detected by immunoblotting. (H) CHOP-KO MEFs and CHOP-KO MEFs expressing human XIAP were treated with tunicamycin as indicated. XIAP and PARP were detected by immunoblotting. Human myc-XIAP (top) has higher mobility than native murine XIAP (bottom). (A–H) Actin served as a protein loading control as indicated.
CHOP is induced by PERK in response to ER stress, and CHOP-knockout (KO) cells are partially resistant to ER stress–induced cell death (Zinszner et al., 1998; Figure 5G). We found no changes in XIAP levels after CHOP overexpression (Figure 4A) or in CHOP-KO cells. Of interest, when we overexpressed XIAP in CHOP-KO cells, we were able to further increase their resistance to ER stress–induced apoptosis (Figure 5H). These findings demonstrate that XIAP's antiapoptotic effects can operate independently of those caused by absence of CHOP.
Chronic PERK activation down-regulates multiple IAP-family proteins
XIAP is a member of the IAP protein family, several of which have also been experimentally demonstrated to regulate cell survival in response to diverse stresses (Salvesen and Duckett, 2002). We examined how extended PERK signaling affected other IAP family members. Similar to the down-regulation we saw with XIAP, we observed down-regulation for cIAP1, cIAP2, livin, survivin, and neuronal apoptosis inhibitory protein (NAIP) after extended PERK signaling (Figure 6). These findings indicated that in addition to down-regulating XIAP, chronic PERK signaling also down-regulated related IAP-family proteins. This broad down-regulation of the IAP protein family likely further increases susceptibility to cell death seen with chronic ER stress.
FIGURE 6:
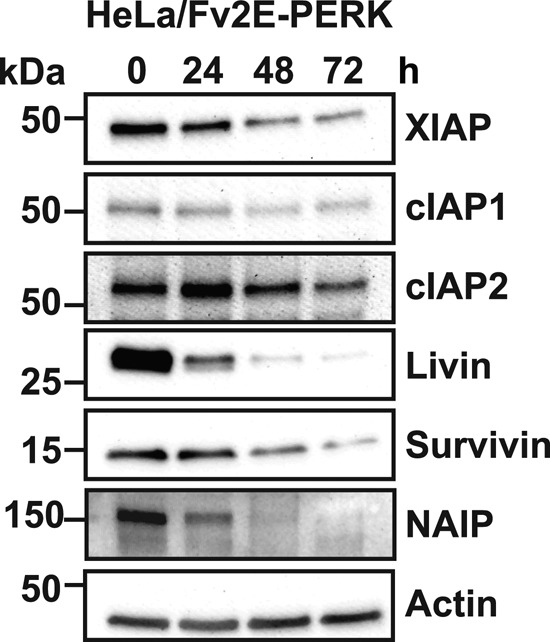
PERK signaling down-regulates multiple IAP-family proteins. HeLa cells stably expressing Fv2E-PERK were exposed to AP20187 for the indicated durations. Cell lysates were probed for XIAP, cIAP1, cIAP2, livin, survivin, and NAIP. Actin levels are shown as a loading control.
DISCUSSION
Metazoan cells respond to ER stress by activating intracellular UPR signaling pathways. IRE1, PERK, and ATF6 signaling initially provides an opportunity for cells to cope with and adapt to ER stress by enhancing the protein-folding capabilities of the ER and reducing the protein-folding load on the ER. However, if these measures fail to improve ER protein-folding fidelity, reduce ER stress, or restore ER homeostasis, the UPR “switches” from the initial adaptive phase to a maladaptive program that culminates in cell death. The precise molecules and mechanisms that link IRE1, PERK, or ATF6 at the ER membrane to activation of apoptotic pathways in the cytosol or other cellular compartments in response to irreparable ER stress are poorly understood. Here we identify a new molecular mechanism operating during the maladaptive phase of the UPR that links chronic ER stress to activation of the cytosolic cell death machinery. Specifically, we find that chronic ER stress removes an important brake against caspase activity through down-regulation of XIAP. Our findings demonstrate that the PERK arm of the UPR is the central mechanism driving XIAP down-regulation through translational attenuation mediated by phosphorylated eIF2α and degradation promoted by ATF4. Of interest, PERK activation leads to the production of proapoptotic CHOP transcriptional activator using the same signal transduction machinery: up-regulation of Chop translation by eIF2α phosphorylation and direct Chop transcriptional induction by ATF4 (Zinszner et al., 1998; Harding et al., 2000a; Rutkowski et al., 2006). We propose that the PERK pathway's up-regulation of CHOP coupled with its concomitant down-regulation of XIAP/IAPs creates a proapoptotic “double hit” that sensitizes cells to ER stress–induced cell death (Figure 7).
FIGURE 7:
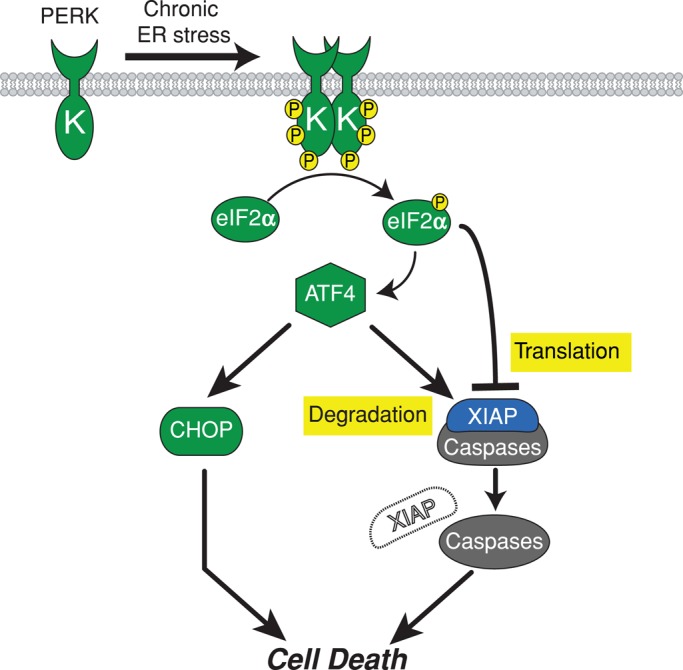
PERK promotes cell death through concomitant up-regulation of proapoptotic CHOP and down-regulation of antiapoptotic IAPs. Chronic ER stress activates PERK, leading to phosphorylation of eIF2α. Phosphorylation of eIF2α leads to production of ATF4 and attenuation of Xiap translation. ATF4 transcriptionally up-regulates proapoptotic CHOP and promotes XIAP degradation. The induction of CHOP coupled with loss of XIAP (double-hit model) sensitizes cells to apoptosis in response to chronic ER stress.
The mechanism of CHOP-induced cell death is under extensive study. In our studies, we found no effect of CHOP expression on XIAP levels, indicating that CHOP does not regulate cell death through direct modulation of XIAP. Prior studies identified CHOP as transcriptionally up-regulating proapoptotic BH3 proteins Bim and Puma (Reimertz et al., 2003; Puthalakath et al., 2007). However, these findings were seen in other experimental cell systems (Cazanave et al., 2010) and suggest that the mechanism of CHOP-induced cell death may vary across cell types. By contrast, we observed XIAP reduction in response to ER stress in all mammalian cell lines that we studied, and XIAP reduction inversely correlated with the emergence of apoptosis in all of these cell types. Therefore this molecular event—the down-regulation of XIAP—appears to be well-conserved step in the induction of ER stress–induced cell death.
IAPs were first identified in baculovirus, where they prevent infected cells from dying when they are generating immense numbers of newly enveloped virions. XIAP is the best-studied IAP in mammals and prevents apoptosis by directly binding and blocking the proteolytic activities of caspases and also promoting ubiquitination and degradation of target proteins, including caspases themselves. Given XIAP's important role in governing cell life/death fates, cells have evolved multiple mechanisms to regulate XIAP. The stress-activated NF-κB transcription factor has been shown to induce Xiap transcription (Stehlik et al., 1998). Human Xiap has a splicing variant that contains an internal ribosome entry site (IRES) in its 5′ untranslated region, and translation from the IRES-containing Xiap mRNA can be positively and negatively regulated by a dynamic ribonucleoprotein (RNP) complex containing heterogeneous nuclear RNP (hnRNP) C1/C2, HuR, La autoantigen, mdm2, hnRNP A1, and polypyrimidine tract-binding protein (Holcik et al., 2003; Riley et al., 2010). XIAP protein is directly bound and inhibited by proteins, including SMAC/DIABLO, Omi/Htra2, and XAF (Salvesen and Duckett, 2002). XIAP function and stability are further regulated by posttranslational modifications such as ubiquitination and S-nitrosylation (Nakamura et al., 2010; Vucic et al., 2011). XIAP protein is a proteolytic target of activated caspases (Deveraux et al., 1999). Our studies reveal that the maladaptive phase of the UPR taps into the PERK pathway to down-regulate XIAP by inhibiting de novo XIAP synthesis, as well as by promoting destabilization and proteasomal degradation of extant XIAP protein. Given the many diverse modes of regulating XIAP function, additional mechanisms that negatively down-regulate XIAP during the maladaptive phase of the UPR are also likely to exist.
Of interest, positive up-regulation of IAPs may also contribute to the adaptive phase of the UPR. Induction and activation of cIAP1, cIAP2, and XIAP were observed in response to acute ER stress and PERK signaling in some cell types (Warnakulasuriyarachchi et al., 2004; Hamanaka et al., 2009; Bevilacqua et al., 2010; Muaddi et al., 2010). These increased cellular levels of IAPs likely contribute to the adaptive phase of the UPR by inhibiting or enhancing degradation of caspase-3 and potentially other ER stress–activated caspases. cIAP1 was reported to promote the ubiquitination and ensuing degradation of the proapoptotic CHOP transcription factor that is induced by PERK signaling (Qi and Xia, 2012). Elimination of CHOP by IAP-mediated ubiquitination could further create a cellular environment conducive for the adaptive phase of the UPR and survival of ER stress. Coupled with our findings that chronic ER stress leads to down-regulation of XIAP, dynamic modulation of IAPs is an important determinant by which UPR switches between adaptive and maladaptive programs.
ER stress contributes to the pathogenesis and progression of many human diseases. Our findings identify several sequential intracellular signaling steps between dysfunction in the ER and down-regulation of an important inhibitor of apoptosis in the cytosol, beginning with chronic PERK signaling to translational attenuation and posttranslational destabilization of XIAP, all occurring concomitantly with PERK signaling's translational and transcriptional induction of CHOP. Targeting chronic PERK activation to prevent loss of XIAP and induction of CHOP could be beneficial in preventing the cell death and tissue injury observed in many ER stress–associated degenerative diseases. Indeed, dramatic enhancement of neuronal survival and function was seen after pharmacological inhibition of PERK in mice with prion disease associated with ER stress (Moreno et al., 2013). Alternatively, accentuating the loss of XIAP could help target malignant cancers by reducing their resistance to ER stress–induced cell death.
MATERIALS AND METHODS
Cell culture and chemicals
HEK293, HeLa, HCT116, Huh7, and MEF cells were maintained in DMEM (Corning Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (FBS; Corning Cellgro), 100 U of penicillin (Sigma-Aldrich, St. Louis, MO), and 100 μg/ml streptomycin (Sigma-Aldrich) at 37°C and 5% CO2. ATF4−/− MEF cells were further supplemented with 55 μM 2-mercaptoethanol (Invitrogen, Carlsbad, CA), and nonessential amino acids (Invitrogen) as previously described (Han et al., 2013). The generation of HEK293 cells stably expressing IRE1[I642G], Fv2E-PERK, or tetracycline-inducible ATF6(373) was previously described (Lin et al., 2007, 2009; Hiramatsu et al., 2011). The generation and characterization of MEFs carrying mutated eIF2α (S51A/S51A: eIF2αA/A), deletion of ATF4 (ATF4-KO), or deletion of CHOP (CHOP-KO) were previously described (Scheuner et al., 2001; Han et al., 2013). HCT116 cells deleted for XIAP (HCT116-XIAP(−/−)) cells were generated as previously described (Cummins et al., 2004).
To generate HeLa cells bearing Fv2E-PERK, HeLa-tet-off-FRT cells were cotransfected with pEF5/FRT/Fv2E-PERK and pOG44 plasmids, and stable HeLa/Fv2E-PERK clones were established. To generate HeLa cells bearing ATF6(373), HeLa-tet-off-FRT cells were transfected with pTRE-3xFLAG-ATF6(373), and stable clones were established. To generate eIF2αA/A, eIF2αS/S, ATF4-WT, and ATF4-KO MEF cells bearing Fv2E-PERK, MEFs were transduced with pBabe-puro-Fv2E-PERK, and stable clones were selected with 2.5 μg/ml puromycin (Corning Cellgro). To reconstitute XIAP in HCT116-XIAP(−/−) cells, pBabe-puro-XIAP was transduced, and stable clones were selected with puromycin. pLVX-puro-myc-XIAP was transduced into CHOP-KO MEF cells and selected with puromycin for establishment of CHOP-KO XIAP cells.
IRE1[I642G] was specifically activated by the ATP analogue 1NM-PP1 (Calbiochem, San Diego, CA) as previously described (Lin et al., 2007). Fv2E-PERK was specifically activated by AP20187 (ARIAD Pharmaceuticals, Cambridge, MA). Doxycycline (Sigma-Aldrich) was used to regulate ATF6(373) expression as previously described (Chiang et al., 2012a). Salubrinal was used as previously described (Boyce et al., 2005). Adenovirus-mediated ATF4 or CHOP transduction into HeLa cells was carried out as described previously (Han et al., 2013).
Western blotting and immunoprecipitation
For Western blotting analysis, total protein lysate was prepared with SDS lysis buffer (62.5 mM Tris-HCl, pH 6.8, 2% [wt/vol] SDS, 10% glycerol), and protein concentration was measured by Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL). From 10 to 20 μg of protein was mixed with LDS sample buffer (Invitrogen) containing 100 mM dithiothreitol and incubated at 70°C for 5 min before loading onto 4–15% gradient SDS–PAGE gel (Bio-Rad, Hercules, CA). After SDS–PAGE, protein was electrically transferred to polyvinylidene fluoride membrane (Bio-Rad) in 1× Tris/glycine containing 20% methanol buffer for 1 h, and membrane was subsequently incubated for 1 h at room temperature in 5% nonfat dry milk in pH 7.6 Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBS-T). Primary antibody was diluted in 5% bovine serum albumin (Cell Signaling Technology, Danvers, MA) in TBS-T, and membrane was incubated overnight at 4°C with gentle rotation. After incubation, the membrane was washed three times with 0.1% TBS-T for 5 min. After incubation with 1:3000 diluted horseradish peroxidase (HRP)–conjugated anti-mouse or anti-rabbit secondary antibody (Cell Signaling Technology) for 1 h at room temperature, membrane was washed three times with 0.1% TBS-T at room temperature, and HRP-catalyzed chemiluminescence was visualized with UVP BioSpectrum Imaging System (UVP, Upland, CA). Primary antibodies and dilutions used for were as follows: mouse anti-XIAP antibody, 1:1000 (BD Transduction Laboratories, San Diego, CA); rabbit anti–phospho eIF2α (Ser-51), 1:1000 (Cell Signaling); rabbit anti–cleaved PARP, 1:1000 (Cell Signaling); rabbit anti–cIAP-1, 1:1000 (Cell Signaling); rabbit anti–cIAP-2, 1:1000 (Cell Signaling); rabbit anti-livin, 1:1000 (Cell Signaling); rabbit anti-survivin, 1:1000 (Cell Signaling); rabbit anti-PARP, 1:1000 (Cell Signaling); mouse anti–FLAG M2, 1:5000 (Sigma-Aldrich); rabbit anti-HSP90, 1:1000 (GeneTex, San Antonio, TX); mouse anti-actin, 1:20,000 (Millipore, Bedford, MA); rabbit anti–FKBP-12 (Thermo Scientific); rabbit anti-NAIP, 1:1000 (Abcam, Cambridge, MA); mouse anti-CHOP, 1:1000 (Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-CREB2, 1:1000 (Santa Cruz Biotechnology); and mouse anti-ubiquitin, 1:1000 (Santa Cruz Biotechnology).
For XIAP immunoprecipitation, cell lysates were prepared in NP-40 lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 0.2% NP-40, 10% glycerol) supplemented with protease inhibitor cocktail (Sigma-Aldrich) and incubated on ice for 15 min. Lysate was centrifuged at 16,000 × g for 20 min at 4°C, and supernatant was immunoprecipitated with anti-XIAP (0.5 μg/sample; BD Transduction Laboratory) conjugated to 30 μl of Protein G Dynabeads (Invitrogen) with overnight incubation with gentle rotation at 4°C. Beads were washed five times with ice-cold phosphate-buffered saline (PBS) containing 0.02% Tween-20, and protein was eluted by boiling for 5 min in 30 μl of 2× LDS sample buffer.
Metabolic labeling and cycloheximide assay
For radiolabeling studies, cells were transferred to prewarmed methionine/cysteine (Met/Cys)-free DMEM (Corning Cellgro) with 10% dialyzed fetal bovine serum (Invitrogen), 100 U of penicillin (Sigma-Aldrich), 100 μg/ml streptomycin (Sigma-Aldrich), and 2 mM GlutaMAX (Invitrogen), and 100 μCi of [35S]Met/Cys was added for 1 h. Cells were harvested, and protein lysates and immunoprecipitation were performed as described. Lysates were resolved by SDS–PAGE, stained with SimplyBlue Safe Staining (Invitrogen), and dried on gel dryer (Bio-Rad) at 70°C for 20 min. Radioisotope incorporation was visualized by phosphorimaging (Typhoon Phosphor-Imager; GE Healthcare, Piscataway, NJ).
For evaluation of XIAP protein stability, a cycloheximide assay was carried out. HeLa cells were pretreated with or without Ad-Atf4 for 18 h and treated with 50 μg/ml cycloheximide to in inhibit protein synthesis. Cells were periodically harvested, and protein lysates were subjected to Western blotting for XIAP.
Molecular biology
For quantitative PCR analysis of mRNA levels, total RNA was extracted with RNeasy Mini Kit (Qiagen, Germantown, MD) according to the manufacture's instruction. A 1-μg amount of RNA was reverse transcribed with iScript cDNA synthesis kit (Bio-Rad), and cDNA samples were subjected to PCR amplification with following gene-specific primer sets: human Xiap, 5′-AGAACACAGGCGACACTTTC-3′ and 5′-CCGTGCTTCATAATCTGCCA-3′; human Rpl19, 5′-ATGTATCACAGCCTGTACCTG-3′ and 5′-TTCTTGGTCTCTTCCTCCTTG-3′; mouse Chop, 5′-ACGGAAACAGAGTGGTCAGTGC-3′ and 5′- CAGGAGGTGATGCCCACTGTTC-3′; and mouse Rpl19, 5′- ATGCCAACTCCCGTCAGCAG-3′ and 5′-TCATCCTTCTCATCCAGGTCACC-3′.
Quantitative PCR was performed with iQ SYBR Green Supermix (Bio-Rad) on CFX96 real-time PCR (Bio-Rad). Analysis of the relative level of mRNA was normalized with the value of Rpl19, whose expression level is not affected by ER stress.
The shRNA fragments for nonsilencing (shCont) and human ATF4 (shATF4) were cloned into LMP vector at XhoI and EcoRI sites, and LMP-shCont and LMP-shATF4 were stably transfected into HEK/Fv2E-PERK cells.
Fluorescence-activated cell sorting cell death quantification
Cells were treated with tunicamycin or thapsigargin, gently trypsinized, collected into fluorescence-activated cell sorting (FACS) tubes, and centrifuged at 500 × g for 5 min at room temperature. Cell pellets were washed with FACS buffer (PBS, pH 7.4, 1% FBS, 0.1% sodium azide) and resuspended in FACS buffer supplemented with 1 μg/ml 7-AAD. The amount of dead cells incorporating 7-AAD was quantified by BD FACS Canto II (BD Biosciences).
Animals and retinal histologic and biochemical analyses
All animals were maintained in accordance with Institutional Animal Care and Use Committee guidelines. P23H rhodopsin transgenic rat lines were created previously (www.ucsfeye.net/mlavailRDratmodels.shtml). P23H (line 2) transgenic rats mated with wild-type Sprague–Dawley animals, and retinas were collected from postnatal-day-90 animals and age-matched Sprague–Dawley littermate controls. For histological analysis, animals were killed and perfusion fixed. Eyes were bisected, postfixed, and embedded in an epoxy resin. One-micrometer-thick sections were cut through the optic nerve head and stained with toluidine blue. For biochemical analysis, retinas were removed from enucleated eyes and lysed in SDS lysis buffer.
Statistical analyses
All results are presented as mean ± SD from at least three to eight individual samples per experimental condition. Student's t tests were performed to determine p values for paired groups. A value of p < 0.05 was considered significant. *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
Acknowledgments
We thank S. Field, J. Reed, G. Salvesen, B. Vogelstein, M. Kamps, J. Bui, K. Iwaisako, C. Koumenis, L. Wiseman, and S. Lowe for helpful reagents and critical comments on the manuscript. These studies were supported by National Institutes of Health Grants EY001919, EY002162, EY006842, EY020846, DK042394, DK088227, and HL052173, the Crohn's and Colitis Foundation of America (CCFA), and the Foundation Fighting Blindness. N.H. was supported by a Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad.
Abbreviations used:
- ATF4
activating transcription factor 4
- CHOP
C/EBP homologous protein
- eIF2α
alpha subunit of eukaryotic translation initiation factor 2
- ER
endoplasmic reticulum
- PERK
PKR-like endoplasmic reticulum kinase
- UPR
unfolded protein response
- XIAP
X-linked inhibitor of apoptosis protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-11-0664) on March 12, 2014.
REFERENCES
- Bevilacqua E, et al. eIF2alpha phosphorylation tips the balance to apoptosis during osmotic stress. J Biol Chem. 2010;285:17098–17111. doi: 10.1074/jbc.M110.109439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol. Gastrointest Liver Physiol. 2010;299:G236–G243. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazanave SC, Gores GJ. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin Lipidol. 2010;5:71–85. doi: 10.2217/clp.09.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Hiramatsu N, Messah C, Kroeger H, Lin JH. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci. 2012a;53:7159–7166. doi: 10.1167/iovs.12-10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Messah C, Lin JH. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol Biol Cell. 2012b;23:758–770. doi: 10.1091/mbc.E11-08-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins JM, Kohli M, Rago C, Kinzler KW, Vogelstein B, Bunz F. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 2004;64:3006–3008. doi: 10.1158/0008-5472.can-04-0046. [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galban S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010;17:54–60. doi: 10.1038/cdd.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Bobrovnikova-Marjon E, Ji X, Liebhaber SA, Diehl JA. PERK-dependent regulation of IAP translation during ER stress. Oncogene. 2009;28:910–920. doi: 10.1038/onc.2008.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000a;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000b;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hiramatsu N, Joseph VT, Lin JH. Monitoring and manipulating mammalian unfolded protein response. Methods Enzymol. 2011;491:183–198. doi: 10.1016/B978-0-12-385928-0.00011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcik M, Gordon BW, Korneluk RG. The internal ribosome entry site-mediated translation of antiapoptotic protein XIAP is modulated by the heterogeneous nuclear ribonucleoproteins C1 and C2. Mol Cell Biol. 2003;23:280–288. doi: 10.1128/MCB.23.1.280-288.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, Knoefel WT, Reed JC. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA. 2009;106:14524–14529. doi: 10.1073/pnas.0907131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Zhang Y, Ron D, Walter P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS One. 2009;4:e4170. doi: 10.1371/journal.pone.0004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004a;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004b;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Translat Med. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- Muaddi H, Majumder M, Peidis P, Papadakis AI, Holcik M, Scheuner D, Kaufman RJ, Hatzoglou M, Koromilas AE. Phosphorylation of eIF2alpha at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol Biol Cell. 2010;21:3220–3231. doi: 10.1091/mbc.E10-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, et al. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BA, Sandoval IM, Chan F, Simons DL, Wu SM, Wensel TG, Wilson JH. Mislocalization and degradation of human P23H-rhodopsin-GFP in a knockin mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52:9728–9736. doi: 10.1167/iovs.11-8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath H, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Qi Y, Xia P. Cellular inhibitor of apoptosis protein-1 (cIAP1) plays a critical role in beta-cell survival under endoplasmic reticulum stress: promoting ubiquitination and degradation of C/EBP homologous protein (CHOP) J Biol Chem. 2012;287:32236–32245. doi: 10.1074/jbc.M112.362160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley A, Jordan LE, Holcik M. Distinct 5’ UTRs regulate XIAP expression under normal growth conditions and during cellular stress. Nucleic Acids Res. 2010;38:4665–4674. doi: 10.1093/nar/gkq241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Harding HP. Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harbor Perspect Biol. 2012;4(p11):A013177. doi: 10.1101/cshperspect.a013177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schile AJ, Garcia-Fernandez M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–2266. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Takahashi R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci USA. 2001;98:8662–8667. doi: 10.1073/pnas.161506698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnakulasuriyarachchi D, Cerquozzi S, Cheung HH, Holcik M. Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J Biol Chem. 2004;279:17148–17157. doi: 10.1074/jbc.M308737200. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.