

TIM4 is a receptor for phosphatidylserine that mediates engulfment of apoptotic cells. Remarkably, it does not require its cytosolic or transmembrane domains to mediate phagocytosis. TIM4 associates with integrins that serve as signal-transducing coreceptors.
Abstract
T-cell immunoglobulin mucin protein 4 (TIM4), a phosphatidylserine (PtdSer)-binding receptor, mediates the phagocytosis of apoptotic cells. How TIM4 exerts its function is unclear, and conflicting data have emerged. To define the mode of action of TIM4, we used two distinct but complementary approaches: 1) we compared bone marrow–derived macrophages from wild-type and TIM4−/− mice, and 2) we heterologously expressed TIM4 in epithelioid AD293 cells, which rendered them competent for engulfment of PtdSer-bearing targets. Using these systems, we demonstrate that rather than serving merely as a tether, as proposed earlier by others, TIM4 is an active participant in the phagocytic process. Furthermore, we find that TIM4 operates independently of lactadherin, which had been proposed to act as a bridging molecule. Of interest, TIM4-driven phagocytosis depends on the activation of integrins and involves stimulation of Src-family kinases and focal adhesion kinase, as well as the localized accumulation of phosphatidylinositol 3,4,5-trisphosphate. These mediators promote recruitment of the nucleotide-exchange factor Vav3, which in turn activates small Rho-family GTPases. Gene silencing or ablation experiments demonstrated that RhoA, Rac1, and Rac2 act synergistically to drive the remodeling of actin that underlies phagocytosis. Single-particle detection experiments demonstrated that TIM4 and β1 integrins associate upon receptor clustering. These findings support a model in which TIM4 engages integrins as coreceptors to evoke the signal transduction needed to internalize PtdSer-bearing targets such as apoptotic cells.
INTRODUCTION
Various forms of apoptosis occur during development, tissue remodeling, and infection, culminating in the death of vast numbers of cells. To maintain homeostasis, these cellular remains are efficiently cleared by professional phagocytes; the importance of this process is emphasized by the occurrence of autoimmune and inflammatory diseases when the phagocytic process is perturbed (Gaipl et al., 2007; Thorp et al., 2008; Korns and Bratton, 2011). Several receptors can initiate the phagocytosis of effete cells, recognizing so-called “eat me” molecular determinants that uniquely identify apoptotic bodies. The glycerophospholipid phosphatidylserine (PtdSer) is perhaps the best-characterized “eat me” signal. In healthy cells, PtdSer is abundant in the inner leaflet of the plasmalemma, but during apoptosis it is quickly redistributed and exposed on the exofacial leaflet, where it is recognized by specific phagocytic receptors such as T-cell immunoglobulin mucin receptor 4 (TIM4; Miyanishi et al., 2007). TIM4 is a heavily glycosylated, PtdSer-binding receptor expressed exclusively by professional phagocytes. The N-terminal immunoglobulin-fold domain of TIM4 possesses a metal ion–dependent pocket that selectively binds PtdSer to mediate the phagocytosis of apoptotic cells. Despite the elucidation of its structure (Santiago et al., 2007), the mode of action of TIM4 has remained enigmatic because its cytosolic domain is devoid of recognizable intracellular signaling motifs. Furthermore, the cytosolic and transmembrane domains of TIM4 are seemingly dispensable for its phagocytic function (Park et al., 2009). This led to the speculation that TIM4 might simply function as a molecular tether that secures PtdSer-bearing targets to the phagocyte surface, where another, bona fide phagocytic signaling receptor then elicits actin rearrangement and the elaboration of pseudopodia required to engulf apoptotic bodies (Toda et al. 2012). Neither the identity of this putative ancillary receptor nor its mode of association with TIM4—if any—is known.
TIM4 is only one of several apoptotic cell receptors on the surface of professional phagocytes. The coexistence of multiple receptors that can bind PtdSer either directly or through bridging proteins, such as lactadherin, complicates the analysis of signaling by TIM4 in its native environment. To circumvent the confounding effects of multiple PtdSer receptors, we used two separate but complementary models to study TIM4 in isolation: 1) a heterologous expression system in which TIM4 was expressed in cells that are not otherwise phagocytic, and 2) primary murine macrophages, in which TIM4 was found to be the primary PtdSer receptor. Our findings indicate that TIM4 is not merely a tether, but instead functions actively in the initiation of phagocytosis by associating with integrins. By bridging PtdSer to integrins, TIM4 elicits stimulation of Src kinases, focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase, which in turn recruit the guanine nucleotide-exchange factor Vav3 that activates the small GTPases RhoA, Rac1, and Rac2, leading to the ingestion of the apoptotic targets.
RESULTS
TIM4 is the main receptor mediating the phagocytosis of PtdSer-bearing targets in murine macrophages
Bone marrow-derived macrophages (BMDMs) from TIM4−/− mice were reported to have a reduced ability to engulf apoptotic bodies (Martinez et al., 2011). This observation suggests that TIM4 may be the primary PtdSer receptor in these cells, which could in principle provide a useful system to study the mode of action of these receptors. To quantitatively assess the contribution of TIM4, we compared the ability of BMDMs isolated from wild-type and TIM4−/− mice to internalize PtdSer-bearing targets. To rule out the participation of phagocytic ligands other than PtdSer potentially present on the surface of apoptotic cells, we used as targets glass beads coated with a mixture of phospholipids: PtdSer (23 mol%) was present where indicated to promote recognition by TIM4. Biotinylated phosphatidylethanloamine (PtdEth; 0.8%) was added to facilitate differentiation between fully internalized beads and those that were incompletely engulfed or merely adherent extracellularly and therefore accessible to fluorescent avidin (see Materials and Methods). The balance (76.2 or 99.2%) was provided by phosphatidylcholine (PtdCho). As shown in Figure 1A, wild-type BMDMs efficiently bound and ingested PtdSer-coated beads, whereas beads containing only PtdCho bound poorly and were rarely internalized. TIM4−/− BMDMs engulfed a much lower number of PtdSer-bearing targets, comparable to the residual uptake of PtdCho-coated beads by wild-type BMDMs (Figure 1B). These observations demonstrate that BMDMs preferentially internalize PtdSer-bearing targets and that TIM4 is the primary receptor for PtdSer in these cells. Thus uptake of PtdSer-coated glass beads by BMDMs provides a suitable model to study the mode of action of TIM4.
FIGURE 1:
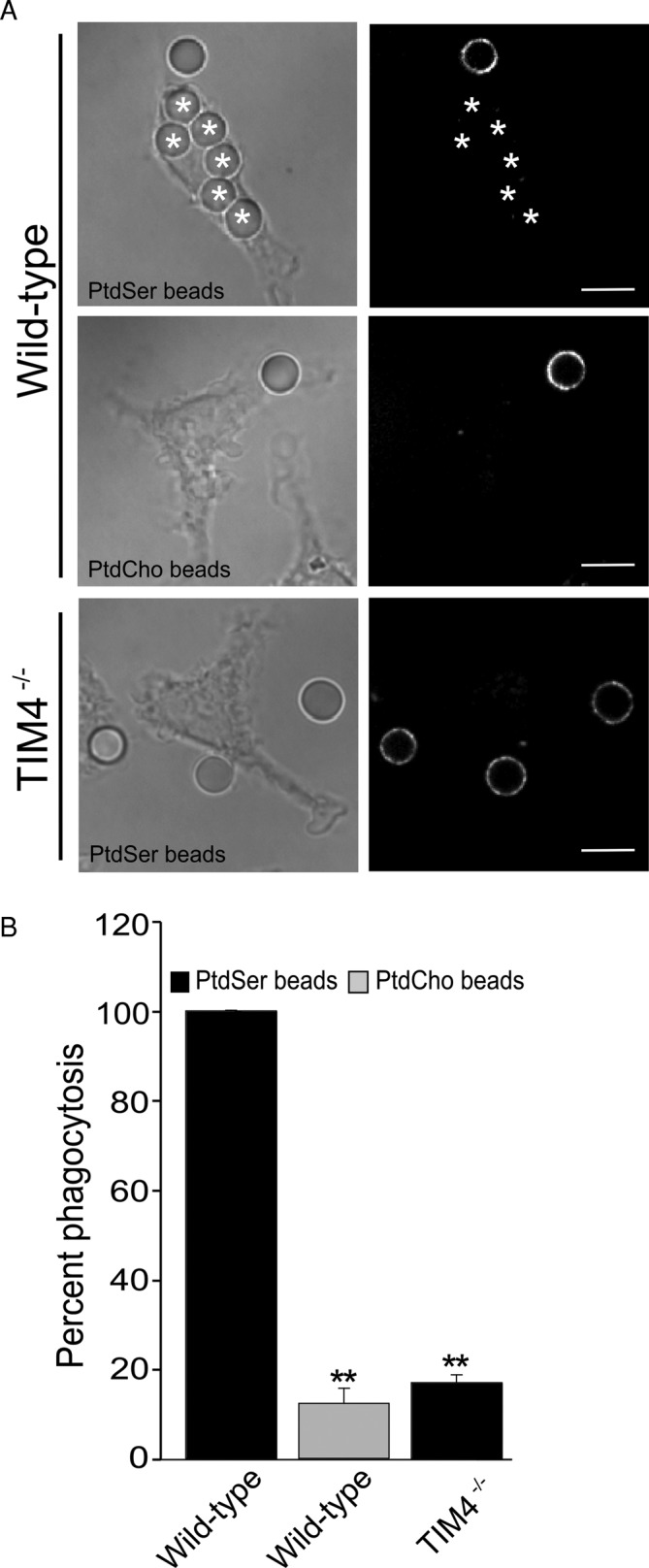
Lack of TIM4 impairs phagocytosis of PtdSer-bearing phagocytic targets in BMDMs. (A) Wild-type and TIM4−/− BMDMs were exposed to phagocytic targets bearing PtdSer, biotinylated PtdEth, and PtdCho or biotinylated PtdEth and PtdCho only, to assess their phagocytic capacity. After 30 min, fluorescent avidin was added to identify external or partially internalized beads (right, shown in white). Internalized beads are highlighted with an asterisk. The representative micrographs, acquired by spinning disk confocal microscopy, show a single XY focal plane. Scale bars, 9.7 μm. (B) Fraction of wild-type or TIM4−/− BMDMs that have fully internalized one or more phagocytic targets coated with the indicated lipids. Data represent mean ± SEM of at least three independent experiments each counting ≥25 cells/condition. **p ≤ 0.01.
Heterologously expressed TIM4 supports the phagocytosis of PtdSer-bearing targets in nonmyeloid (epithelioid) cells
As a complementary system for studying the mode of action of TIM4 without the confounding effects of other PtdSer receptors, we expressed TIM4 in AD293 cells. When untransfected, the kidney-derived AD293 cells (a variant of HEK293 selected for its enhanced adherence to the substratum) lack functional PtdSer receptors and are not phagocytic. Full-length TIM4 was tagged with mCherry to enable identification of the transfected cells. As illustrated in Figure 2B, cells expressing TIM4-mCherry effectively bound and internalized beads bearing PtdSer, whereas beads with only PtdCho bound poorly and were not ingested (Figure 2C). Upward of 90% of the TIM4-expressing cells internalized one or more PtdSer-decorated targets after 30 min, whereas <1% of the transfectants engulfed PtdCho-coated beads devoid of PtdSer. That TIM4 and not another PtdSer receptor intrinsic to AD293 cells was responsible for the engulfment was demonstrated using cells transfected with PM-RFP, a construct consisting of the N-terminus of Lyn, which is targeted to the plasma membrane by virtue of its dual acylation (Teruel et al., 1999). Cells expressing this labeled, membrane-targeted construct failed to internalize PtdSer-coated beads (Figure 2A). Untransfected cells were similarly incapable of engulfment. Quantification of particle binding and engulfment in multiple similar experiments is summarized in Figures 2, D and E, respectively. Together these data demonstrate that expression of TIM4 endows AD293 cells with the ability to efficiently bind and internalize targets in a PtdSer-dependent manner.
FIGURE 2:

Expression of the PtdSer-binding receptor TIM4 confers phagocytic properties to AD293 cells. The nonphagocytic embryonic kidney cell line AD293 was transfected with plasmids encoding a membrane-targeted RFP (PM-RFP; A) or TIM4 fused to mCherry (TIM4-mCherry; B, C). Transfectants were exposed to phagocytic targets bearing PtdSer, biotinylated PtdEth, and PtdCho or biotinylated PtdEth and PtdCho only, to assess their phagocytic capacity. After 30 min, fluorescent avidin was added to identify external or partially internalized beads, shown in blue. White arrows point to representative phagocytic targets that have been completely engulfed and are therefore avidin negative. The representative micrographs, acquired by spinning disk confocal microscopy, show a single XY focal plane. Scale bars, 9.7 μm. (D) Normalized binding capacity of AD293 transfectants expressing PM-RFP or TIM4-mCherry for beads with and without PtdSer. (E) Fraction of PM-RFP or TIM4-mCherry transfectants that have fully internalized one or more phagocytic targets coated with the indicated lipids. Data are means ± SEM of at least three independent experiments each counting ≥ 25 cells/condition. **p ≤ 0.01.
TIM4 is uniquely required for internalization of PtdSer-bearing phagocytic targets
It was shown previously that the cytosolic and transmembrane domains of TIM4 are dispensable for phagocytosis (Park et al., 2009), an observation that we were able to recapitulate using a GPI-anchored form of TIM4 (kindly provided by K. Ravichandran (University of Virginia School of Medicine) and described in Meyers et al., 2005; unpublished data). These observations were interpreted to mean that TIM4 serves only to tether PtdSer-bearing targets to the cell surface, where other PtdSer receptors subsequently and independently induce engulfment (Park et al., 2009; Toda et al., 2012). If this interpretation were correct, the need for TIM4 should be circumvented by tethering the targets to the membrane by other means. To test this notion, we expressed in AD293 cells a mutant FcγRIIA (FcγRIIAmut) receptor that is competent for binding immunoglobulin G (IgG) but is unable to elicit the signaling events necessary for phagocytosis. We then exposed the transfectants to PtdSer-coated glass beads that were also opsonized with IgG. Opsonization was accomplished by adding 0.8 mol% of biotinylated PtdEth to the lipid mixture used to coat the beads, followed by exposure to anti-biotin IgG. Such targets bearing both IgG and PtdSer were readily internalized by cells transfected with either wild-type FcγRIIA or TIM4, indicating that the presence of both ligands did not compromise access of the beads to either receptor (Figure 3, A and D). By contrast, despite promoting the tight binding of particles to the cell surface, FcγRIIAmut failed to stimulate phagocytosis (Figure 3, B and D). The inability to internalize particles was not due to an inhibitory effect of FcγRIIAmut, since cells cotransfected with this mutant receptor and with TIM4 engulfed particles at least as efficiently as those bearing TIM4 alone (Figure 3, C and D).
FIGURE 3:

TIM4 is not merely a tether supporting phagocytosis by other receptors. AD293 transfectants expressing FcγRIIA-GFP (A), FcγPIIAmut-GFP (B), FcγRIIAmut-GFP with TIM4-mCherry (C), and TIM4-mCherry alone (not shown) were incubated for 30 min with phagocytic targets bearing both PtdSer and IgG. (A–C) Representative optical slices obtained by spinning disk confocal microscopy after 30 min of incubation with phagocytic targets. White arrows indicate internalized PtdSer beads. Incompletely internalized targets are marked with fluorescent avidin (blue). Scale bar, 6.8 μm. (D, E) Mean fraction of transfected cells expressing FcγRIIA-GFP, FcγRIIAmut-GFP, TIM4-mCherry, or a combination of these, having internalized one or more PtdSer and IgG-coated targets in the absence (D) or presence (E) of 10% (vol/vol) fetal bovine serum. (F) Individual images obtained from time-lapse video microscopy showing the displacement of the plasma membrane from a TIM4-GFP–expressing cell (top) or a PM-GFP– expressing cell (bottom) around phagocytic targets. PtdSer-coated beads (white asterisk) were forced against the cell surface using an optical tweezer. The position of the PtdSer target is marked with an asterisk. Scale bar, 14.9 μm.
We used a second approach to test whether tight apposition of particles to the membrane, the putative role of TIM4, is sufficient to elicit phagocytosis via a separate endogenous receptor. To this end, we used optical tweezers to press captured PtdSer-bearing beads onto the plasma membrane of AD293 cells. As shown in Figure 3F (see also Supplemental Movie S1), tight apposition of PtdSer-coated beads onto the cell surface was insufficient to promote phagocytosis, despite application of the maximal force applicable by the optical trap (∼100 pN); formation of close contacts between the bead and cell surface was verified by visualizing the plasmalemma using the fluorescent marker PM-green fluorescent protein (GFP). Under comparable conditions, beads brought into contact with TIM4-expressing cells were readily engulfed after zippering of the membrane around the target (Figure 3, E and F, and Supplemental Movie S2).
Together the preceding data indicate that TIM4 contributes directly to signaling phagocytosis and is not merely a passive tether. This conclusion is in apparent conflict with the concept that other receptors, possibly of lower affinity or abundance, bind to PtdSer on targets initially secured by TIM4. Specifically, it has been proposed that by binding lactadherin, integrins are in fact the phagocytic receptors that mediate the uptake of PtdSer-bearing targets (Hanayama et al., 2002; Akakura et al., 2004). It was therefore conceivable that, because our experiments were performed in the absence of serum, the latter mechanism was inoperative, accounting for the inability of tightly apposed beads to undergo phagocytosis. To exclude this possibility, we retested the phagocytic ability of FcγRIIAmut transfectants in the presence of 10% serum. As shown in Figure 3E, the presence of serum did not improve the phagocytic ability of cells expressing the inactive FcγRIIAmut. Also of note is the fact that phagocytosis by TIM4-transfected cells was observed routinely in nominally serum-free medium. Thus the involvement of integrin-lactadherin complexes cannot explain the differences between TIM4 and FcγRIIAmut, nor can the failure of beads pressed onto the cell by the laser tweezers to become internalized. Instead our data imply that TIM4 plays a more direct role in the initiation of phagocytosis.
TIM4-mediated engulfment is an integrin-dependent process
Previous studies of TIM4 function revealed that its extracellular domain suffices to support phagocytosis, an observation that led to the speculation that TIM4 served solely as a molecular tether. On the other hand, our aforementioned data reveal that TIM4 is required for the engulfment of PtdSer-coated particles by a process not replicated by other tethers or by forcing the physical apposition of the beads against the cell surface using laser tweezers. We interpret these results to mean that, rather than functioning exclusively as a tether, TIM4 contributes directly to the initiation of signaling. These considerations prompted us to search for coreceptor molecules that may interact with TIM4 to effect phagocytosis. Although our earlier experiments (Figure 2) ruled out a mechanism involving integrins in conjunction with lactadherin, other modes of engagement of integrins remain possible. We therefore analyzed the dependence on integrins of TIM4-mediated phagocytosis. Extracellular divalent cations, such as Ca2+ and Mg2+, regulate integrin function by stabilizing its active conformation. Treatment with EDTA, a divalent cation chelator, reduced uptake of PtdSer-bearing particles by wild-type BMDMs by >90% relative to control cells (Figure 4A), suggesting that integrins are essential for this process. In addition, the possible functional role of integrins in TIM4-mediated phagocytosis was investigated using arginine, glycine, and aspartic acid (RGD)-containing peptides that competitively inhibit ligand binding to integrins. In the presence of 500 μg/ml of the RGD peptide (equivalent to ∼1 μM), TIM4-mediated phagocytosis of PtdSer-coated beads was reduced by >80% in wild-type BMDMs (Figure 4A) and by ∼59% in TIM4-transfected AD293 cells (Figure 4B) relative to control cells. These data strongly suggested that, although independent of lactadherin, integrins actively participate in TIM4-driven engulfment.
FIGURE 4:

Integrin β1 is required for TIM4-dependent phagocytosis. (A) Wild-type BMDMs were treated with 10 mM EDTA or 500 μg/ml RGD peptide, and uptake of PtdSer-bearing beads was assessed. Data were normalized to untreated control and are means ± SEM of three independent experiments. (B) Effect of RGD peptide on phagocytosis of PtdSer-bearing beads by AD293 TIM4 transfectants. Data were normalized to untreated control and are means ± SEM of three independent experiments. (C) Distribution of integrin β1, detected by immunostaining, in the phagocytic cup of AD293 TIM4 transfectants. The representative micrographs show YZ reconstruction of images obtained by spinning disk confocal microscopy. The white arrow indicates the localization integrin β1 to the TIM4-GFP–positive phagocytic cup. Incompletely engulfed portions of the target were labeled with fluorescent avidin and are shown in blue. Bar, 6.8 μm. (D) Representative fluorescence micrographs showing immunofluorescence staining for endogenous integrin β1 after treatment of AD293 TIM4 transfectants with either scrambled siRNA (left) or integrin β1-specific siRNA (right). Bar, 17.1 μm. (E) Quantitation of the effect of integrin β1-specific and scrambled siRNA on β1 protein expression, from experiments like that in D, quantifying the mean fluorescence intensity of endogenous integrin β1 after immunostaining. Data were normalized to scrambled siRNA–treated cells and are the mean ± SEM of three independent experiments. (F) Effect of integrin β1-specific and scrambled siRNA on TIM4-mediated phagocytosis of PtdSer beads in AD293 TIM4 transfectants. Data were normalized to scrambled siRNA–treated cells and are the mean ± SEM of three independent experiments. (G) Fluorescence micrographs showing confocal images of wild-type BMDMs expressing talin-GFP engulfing PtdSer beads. Asterisks indicate accumulation of talin-GFP at nascent phagosomes. Scale bar, 10 μm. (H) Fluorescence micrographs showing confocal images of AD293 cells expressing TIM4-mCherry and talin-GFP engulfing PtdSer beads. Asterisks, two representative phagocytic cups that can be seen in the YZ reconstruction on top, showing TIM4-mCherry and talin-GFP accumulation. Scale bar, 6.8 μm. **p ≤ 0.01.
We next analyzed the distribution of integrins during the course of TIM4-dependent phagocytosis. Integrin β1, revealed by immunofluorescence using a monoclonal antibody (clone P5D2), was enriched at sites of phagosome formation, where it colocalized with TIM4-GFP (Figure 4C). To evaluate more directly the role of β1 integrins, we used small interfering RNA (siRNA) to down-regulate their expression. Effective knockdown of the integrins was verified by immunofluorescence, which enabled us to evaluate phagocytosis only in those cells in which knockdown could be confirmed. On average, 58.3% reduction in β1 integrin expression relative to scrambled siRNA-treated cells was achieved (Figure 4, D and E). Of importance, TIM4-transfected cells treated with integrin β1 siRNA showed a commensurate (56%) reduction in phagocytosis compared with cells treated with scrambled siRNA (Figure 4F). The contribution of other integrins was studied similarly. Because the available antibodies were not suitable for quantitative immunostaining, real-time PCR was used in these instances to validate the effectiveness of the silencing procedure. siRNA directed to the human integrin β3 and integrin β5 decreased the corresponding mRNA levels by 44.5 and 87.2%, respectively (Supplemental Figure S1A). Knockdown of integrin β3 reduced phagocytosis of PtdSer-bearing targets by 59.3%, whereas knockdown of β5 depressed phagocytosis by 66.4%, relative to scrambled siRNA–treated cells (Supplemental Figure S1B).
The requirement for integrins suggested that effectors of these proteins may be involved in signaling during TIM4-dependent phagocytosis. To test this notion, we assessed talin recruitment to the forming phagocytic cup in wild-type BMDMs transfected with talin-GFP and in AD293 cells transfected with both TIM4 and talin-GFP. As illustrated in Figure 4, G and H, talin-GFP was consistently recruited to sites of phagocytosis in wild-type BMDMs and AD293 transfectants, respectively, consistent with local activation of integrins. Taken together, these data indicate that integrins act as essential intermediates in TIM4-initiated phagocytosis.
TIM4 functions as an integrin ligand
The functional relationship between TIM4 and the integrins prompted us to study the mode by which these proteins are coupled. Because our earlier data ruled out lactadherin as an indirect mediator, we considered the possibility that integrins associate directly with TIM4. This possibility was tested by immunoprecipitating TIM4 and probing the precipitated material for the presence of integrin β1 by Western blotting. The converse experiment—that is, precipitation of integrin β1 followed by immunoblotting for TIM4—was performed in parallel. No evidence for stable association between TIM4 and integrin β1was found using this approach (unpublished data). It is conceivable that the interaction between these molecules is weak or destabilized by the detergent used for immunoprecipitation. As an alternative approach, we studied the relationship between TIM4 and integrin β1 in their native, membrane environment by colocalization analysis at the single-particle level. To this end, we used a construct of TIM4 tagged with the hemagglutinin (HA) epitope (TIM4-HA); by placing the tag at the N-terminus of TIM4, which is exposed extracellularly, we were able to detect the receptors on the surface of intact, living cells without interference from the vast excess of intracellular TIM4. TIM4-HA was expressed in AD293 cells and detected using a subsaturating concentration of anti-HA antibody, followed by a fluorophore-conjugated secondary Fab fragment. In the same cells we immunolabeled the endogenous integrin β1 using a specific monoclonal antibody that recognizes an exofacial epitope (clone AIIB2), followed by a fluorophore-conjugated secondary Fab fragment (Figure 5A). To better visualize the receptors and increase the likelihood of detecting complexes, we induced the formation of small clusters of TIM4 using a Fab2 tertiary antibody that cross-linked the labeled secondary antibody used for this receptor. In separate experiments, a similar strategy was used to cluster the integrin. Using this approach, we could readily detect colocalization of TIM4-HA with integrin β1 at the cell surface (Figure 5, A and C). The statistical significance of this finding was validated by comparing the number of particles found to colocalize in the cells to the average frequency of colocalization in 2000 random distributions of particles at comparable density generated through a Monte Carlo simulation (see Materials and Methods). As shown in Figure 5C, colocalization of TIM4-HA with β1 detected experimentally at the cell surface was statistically significant when clustering was accomplished by cross-linking the receptor or the integrin. Of note, the colocalization did not attain statistical significance in the absence of cross-linking Fab2, likely because at the low concentration of primary antibody used only a small fraction of TIM4-HA and β1 is labeled, minimizing the likelihood of simultaneous labeling of both molecules in a single complex. Use of higher concentrations of primary antibodies is precluded by the resulting inability to distinguish the point spread functions associated with individual molecules.
FIGURE 5:
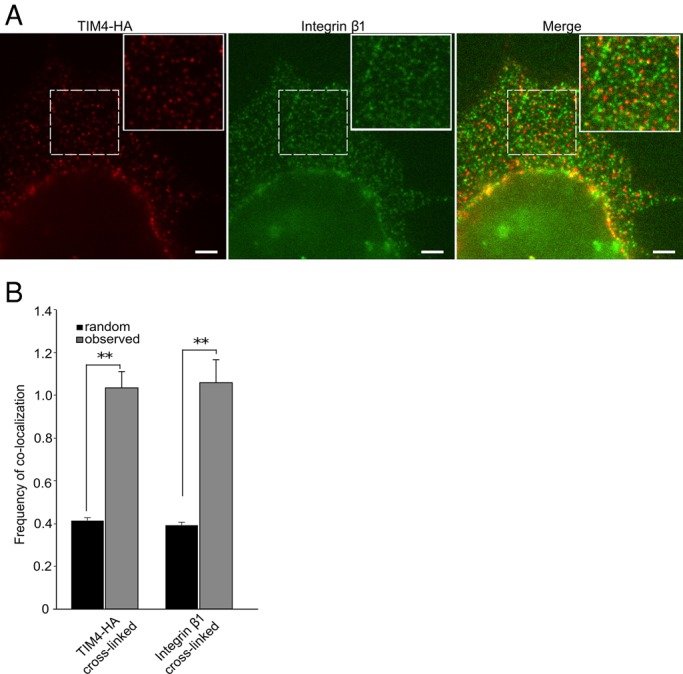
TIM4 engages integrins directly. (A, B) Analysis of the spatial apposition between TIM4 and integrin β1. Cells were labeled with low concentrations of primary antibodies to reveal only a fraction of the epitopes using fluorophore-conjugated secondary Fab fragments (red for TIM4-HA and green for integrin β1), to enable definition of individual features. This was followed by addition of Fab2 fragments to cross-link specifically the secondary Fab fragments recognizing either anti-HA or anti-β1 integrin. The boxes demarcate the region magnified in the inset. Scale bars, 3.3 μm. (B) Comparison of the frequency of colocalization of features in 2000 randomly generated images with comparable particle density (black bars) with the actual frequency of colocalization observed upon image analysis of immunolabeled cells (gray bars) like those in A. Data show results obtained when either TIM4 or integrin β1 was cross-linked. Experimental data shown were obtained from a total of ≥30 images from individual cells from three independent experiments. **p ≤ 0.01.
TIM4-dependent phagocytosis requires focal adhesion kinase, Src-family kinases, and phosphatidylinositol 3-kinase activity
Given the requirement for integrins in TIM4-dependent phagocytosis, we next explored the events downstream of integrin engagement. One of the earliest biochemical events during integrin outside-in signaling is the activation of Src-family tyrosine kinases (Abram and Lowell, 2009). We assessed the contribution of these kinases by treating both wild-type BMDMs and TIM4-transfected AD293 cells with PP1, an inhibitor of Src-family kinases. PP1 reduced uptake of PtdSer-bearing particles by ∼70% in wild-type BMDMs (Figure 6A) and 77% in the AD293 TIM4 transfectants (Figure 6B). In addition to the Src-family kinases, FAK has been reported to be an essential transducer of integrin-initiated signals and is generally believed to act downstream of Src-family kinases (Evangelista et al., 2007; Abram and Lowell, 2009). Indeed, treatment of wild-type BMDMs and TIM4-transfected AD293 cells with PF573228, a pharmacological inhibitor of FAK, impaired phagocytosis by ∼80% in both cell types relative to control vehicle–treated cells (Figure 6, A and B).
FIGURE 6:

TIM4-dependent phagocytosis requires focal adhesion kinase, Src-family kinases, and phosphatidylinositol 3-kinase activity. (A, B) Effect of the Src-family kinase inhibitor PP1 and the FAK inhibitor PF 573228 on TIM4-driven phagocytosis in wild-type BMDMs (A) and AD293 TIM4 transfectants (B). Data are percentage phagocytosis normalized to control (vehicle treated) cells and are means ± SEM of three independent experiments. (C) Representative fluorescence micrographs showing the distribution of PH-Akt-RFP in a TIM4-GFP–expressing AD293 cell engulfing PtdSer beads. Incompletely engulfed targets were labeled with fluorescent avidin and are shown in blue. Bar, 6.8 μm. (D, E) Effect of the PI3K inhibitors wortmannin and LY294002 on TIM4-driven phagocytosis in wild-type BMDMs (D) and AD293 TIM4 transfectants (E). Data were normalized to untreated control and are means ± SEM of three independent experiments. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
Integrins also promote the generation of phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3) by class I phosphoinositide 3-kinases (PI3Ks). To detect the local generation of PtdIns(3,4,5)P3 at sites of TIM4 engagement, we expressed a genetically encoded lipid biosensor consisting of the pleckstrin-homology (PH) domain of Akt fused to red fluorescent protein (RFP). Cotransfection of cells with TIM4-GFP and PH-Akt-RFP revealed robust production of PtdIns(3,4,5)P3 (and/or PtdIns(3,4)P2) at nascent phagocytic cups (Figure 6C). Moreover, treatment of wild-type BMDMs with the potent PI3K inhibitors wortmannin and LY294002 reduced particle uptake by ≥90% (Figure 6D), and in AD293 TIM4 transfectants LY294002 virtually eliminated particle uptake (>99% inhibition; Figure 6E). These data demonstrate that TIM4-dependent phagocytosis requires Src-family kinase and FAK activity in addition to the generation of PtdIns(3,4,5)P3 at sites of phagosome formation, consistent with the involvement of integrins in the signaling process.
TIM4-dependent phagocytosis is an actin-dependent process that requires Rac1, Rac2, and RhoA
Although phagocytosis is generally accepted to be an actin-dependent process, the signaling events involved in regulation of actin dynamics differ, depending on the type of receptor(s) engaged. Indeed, different GTPases and nucleating agents are believed to mediate actin polymerization in integrin-mediated (e.g., complement receptor) versus FcγR-mediated phagocytosis. These observations compelled us to explore the mechanisms that operate during TIM4-dependent phagocytosis.
As illustrated in Figure 7, A and B, pretreatment of both wild-type BMDMs and TIM4-transfected AD293 cells with latrunculin B or cytochalasin D virtually eliminated internalization, confirming the actin dependence of the process. We proceeded to study the pathway underlying actin remodeling during TIM4-mediated phagocytosis. We initially used Clostridium difficile toxin B to assess the involvement of Rho-family GTPases; toxin B glucosylates and thereby inactivates most members of the Rho family (Jank et al., 2007). As illustrated in Figure 7, A and B, phagocytosis was markedly inhibited by the toxin in both wild-type BMDMs and AD293 TIM4 transfectants. Rac1, Rac2, and Cdc42 drive actin polymerization when phagocytosis is triggered by Fcγ and other types of receptors (Massol et al., 1998; Hoppe and Swanson, 2004; Koh et al., 2005; Hall et al., 2006; Utomo et al., 2006; Park et al., 2007). We therefore assessed whether they are activated also in the course of TIM4 stimulation, using a probe consisting of the p21-binding domain of p21-activated kinase (PBD-PAK). In the BMDMs, PBD-PAK markedly accumulated in TIM4-induced phagocytic cups, where it colocalized with filamentous actin (Figure 7C).
FIGURE 7:
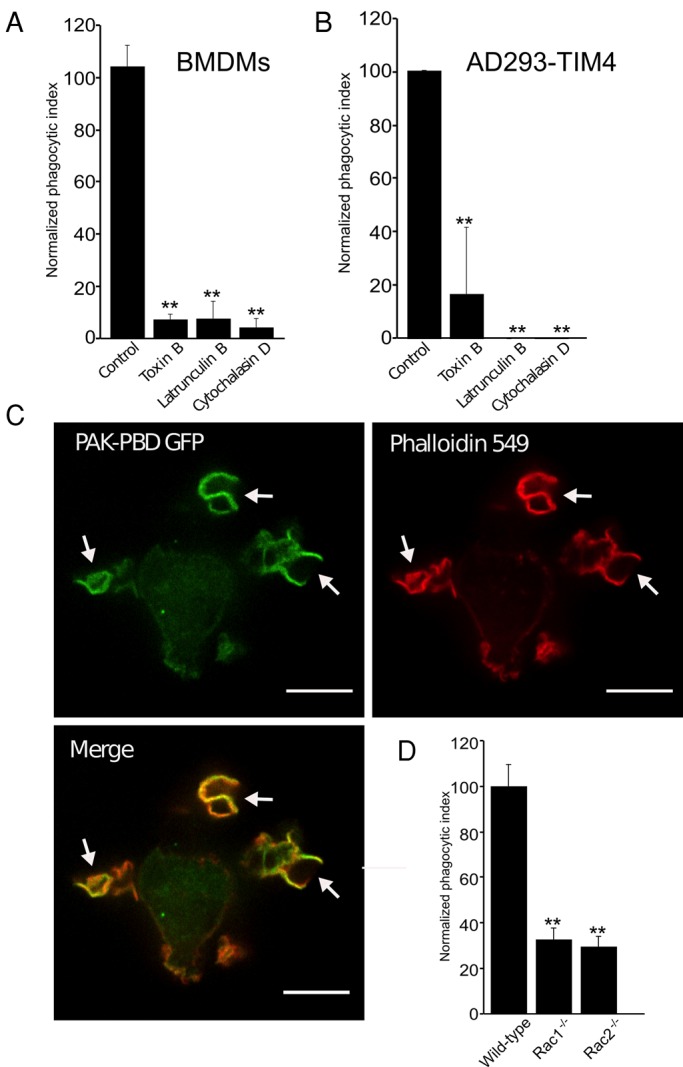
TIM4-dependent phagocytosis is actin dependent and requires Rac1 and Rac2. (A, B) The effect of the actin-disrupting drugs latrunculin B and cytochalasin D on TIM4-driven phagocytosis in wild-type BMDMs (A) and AD293 TIM4 transfectants (B), expressed as percentage of the vehicle control. Data are means ± SEM from three independent experiments. (C) Distribution of the active Rac/Cdc42 biosensor mCherry-PAK-PBD and of actin, visualized by phalloidin staining, in wild-type BMDMs exposed to targets coated with PtdSer. Images are representative XY fluorescence micrographs obtained by spinning disk confocal microscopy. White arrows indicate mCherry-PAK-PBD and actin accumulation beneath the PtdSer-bearing phagocytic target. Scale bar, 13 μm. (D) Wild-type, Rac1−/−, and Rac2−/− BMDMs were exposed to targets coated with PtdSer, and the phagocytic index was determined. Data are the means ± SEM from three independent experiments. **p ≤ 0.01.
Because PBD-PAK is mostly responsive to Rac GTPases (Srinivasan et al., 2003), we tested the requirement for Rac1 and Rac2 in TIM4-mediated phagocytosis using BMDMs isolated from mice in which Rac1 was conditionally deleted in cells of the monocyte lineage and from mice with complete deletion of Rac2 (Koh et al., 2005). Deficiency of either Rac1 or Rac2 resulted in >60% reduction in particle uptake (Figure 7D). These observations imply that both Rac1 and Rac2 are involved in TIM4-initiated phagocytosis and that their actions are not merely additive, but synergistic.
The participation of other Rho-family candidates was tested by treating the cells with the C3 toxin of Clostridium botulinum. This transferase catalyzes the ADP-ribosylation and functional inactivation of RhoA (Jank et al., 2007). When pretreated with cell-permeant TAT-C3 (10 μg/ml), both wild-type BMDMs and TIM4-transfected AD293 cells retained the ability to bind PtdSer-coated beads, but internalization was reduced by ≥60 and ≥75%, respectively (Figure 8, A and B). To assess more specifically the role of RhoA, we used siRNA. Immunoblot analysis revealed that, under the conditions used, we achieved on average ∼60% reduction of RhoA protein expression relative to control cells treated with scrambled siRNA (Figure 8, C and D). Such partial silencing of RhoA produced commensurate inhibition of the phagocytic efficiency (Figure 8E). Jointly these observations indicate that particle internalization triggered by TIM4 requires the cooperative action of Rac1, Rac2, and RhoA.
FIGURE 8:
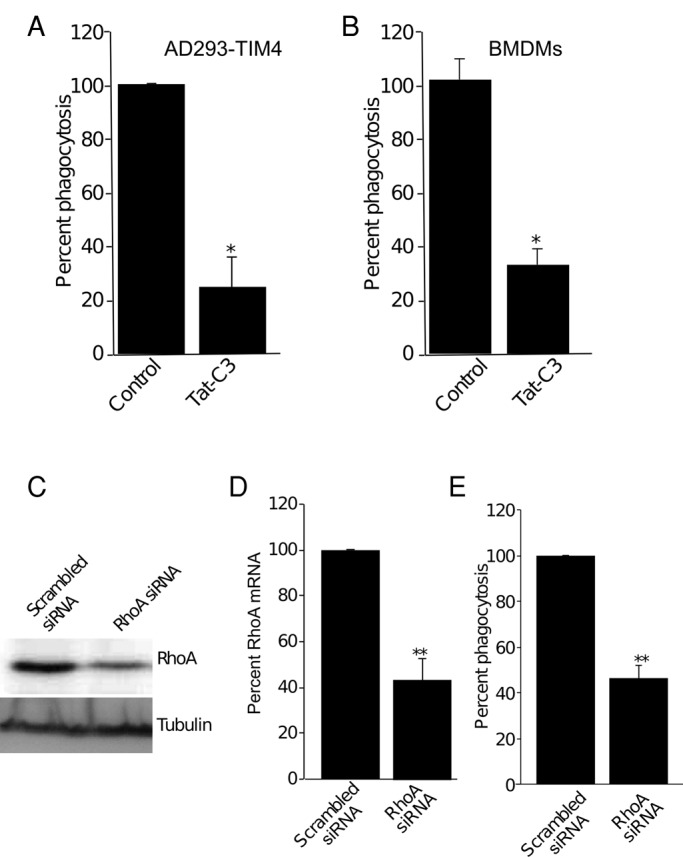
RhoA is required for TIM4-dependent phagocytosis. (A, B) Effect of the cell-permeant Rho inhibitor TAT-C3 transferase on phagocytosis of PtdSer-coated beads by TIM4-transfected AD293 cells (A) and wild-type BMDMs (B). The phagocytic index normalized to vehicle control. Data are means ± SEM of three independent experiments. (C) Immunoblot showing the effect of RhoA-specific and scrambled siRNA on RhoA protein expression (top). Tubulin was used as a loading control (bottom). Blots are representative of three similar experiments. (D) Quantitation of the effect of RhoA-specific and scrambled siRNA on RhoA protein expression, from experiments like that in C and quantified by densitometry of immunoblots. Data are mean ± SEM of three experiments. (E) Effect of RhoA-specific and scrambled siRNA on TIM4-mediated phagocytosis of PtdSer beads. Data were normalized to scrambled siRNA-treated cells and are the mean ± SEM of three independent experiments. *p < 0.05 and **p ≤ 0.01.
Vav3 is required for TIM4-dependent phagocytosis
The identity of the guanine nucleotide exchange factor(s) (GEFs) involved in the activation of Rac1, Rac2, and RhoA during TIM4-mediated phagocytosis was investigated next. Vav3, a RhoA and Rac GEF broadly expressed in multiple tissues and cell types, was an attractive candidate. We therefore investigated its distribution during the course of TIM4-driven phagocytosis. TIM4-mCherry and Vav3-citrine were coexpressed in AD293 cells and their localization monitored by spinning disk confocal microscopy. As shown in Figure 9A, Vav3 was consistently found adjacent to TIM4 during the development of the phagosomal cup.
FIGURE 9:
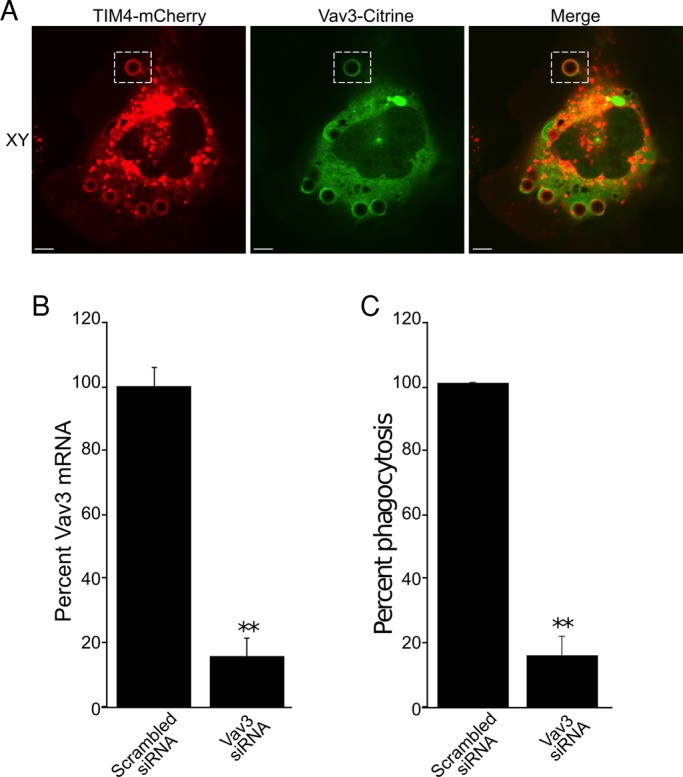
Vav3 is recruited to TIM4-positive phagocytic cups and is required for TIM4-dependent phagocytosis. (A) Confocal fluorescence micrographs of an AD293 cell expressing both TIM4-mCherry and Vav3-citrine that was exposed to PtdSer beads; XY slices are depicted. The hatched box demarcates the phagocytic cup that is presented in the YZ reconstruction, and the white arrow points to accumulated citrine fluorescence in the phagocytic cup. Scale bars, 6.8 μm. (B) AD293 cells were treated with either scrambled siRNA or Vav3-specific siRNA, and knockdown was assessed by real-time PCR. Data were normalized to scrambled siRNA–treated cells and are the mean ± SEM of three independent experiments. (C) Effect of Vav3-specific and scrambled siRNA on TIM4-mediated phagocytosis of PtdSer beads. Data were normalized to scrambled siRNA-treated cells and are the mean ± SEM of three independent experiments. **p ≤ 0.01.
That Vav3 can in fact activate RhoA in AD293 cells was suggested by experiments in which the GEF was deliberately overexpressed. In such cells Vav3 induced spreading and the formation of prominent phalloidin-positive stress fibers (unpublished data), as described for other cells (Movilla and Bustelo, 1999). To assess more directly the involvement of Vav3 in TIM4-mediated phagocytosis, we quantified particle internalization in cells that were treated with siRNA directed to human Vav3. Because no antibodies were available to detect endogenous Vav3, we ensured the effectiveness of the silencing procedure by quantitative PCR. siRNA treatment resulted in >80% decrease in Vav3 mRNA (Figure 9B). Of importance, the ability of TIM4-expressing cells to engulf PtdSer-coated beads was also reduced (by 72%) when treated with Vav3 siRNA relative to scrambled siRNA–treated control cells (Figure 9C).
DISCUSSION
The mode of TIM4 function has been the epicenter of controversy despite its relatively recent discovery. Conflicting data relating to its molecular function have emerged, with some researchers reporting that the cytosolic and transmembrane domains of the receptor are dispensable for its phagocytic function and others suggesting that this is not the case (Park et al., 2009; Wong et al., 2010). In fact, it has been argued that TIM4 is not itself a phagocytic receptor but merely a tether that ensnares and immobilizes particles for other receptors to subsequently signal engulfment (Park et al., 2009; Toda et al., 2012). Our results using truncated forms of TIM4 indicate that the cytosolic domain is not required for effective phagocytosis. Nevertheless, we find that TIM4 its not a passive, remote molecular tether but is in fact an active participant in target engulfment. This important conclusion is borne out by the observation that a surrogate, even more effective transmembrane tether failed to compensate for the absence TIM4 in the phagocytosis of PtdSer-coated targets (Figure 3).
Investigation of the underlying molecular mechanism revealed that integrin expression is also required for TIM4-dependent engulfment of PtdSer beads. The β3/β5 integrins were previously characterized as phagocytic receptors for apoptotic cells, using lactadherin as a bridging element (Andersen et al., 2000; Hanayama et al., 2002; Akakura et al., 2004). Lactadherin, a secreted serum protein, carries an N-terminal, PtdSer-binding domain and a C-terminal, RGD integrin-binding motif; it can consequently bridge PtdSer-bearing targets to integrins to trigger phagocytosis. Of note, we found that TIM4 engulfment occurs in a lactadherin-independent manner, as the removal or addition of lactadherin-containing serum had no effect on engulfment, nor did it obviate the absolute requirement for TIM4 expression for phagocytosis of PtdSer-coated beads (Figure 3E). How then can TIM4 elicit phagocytosis of PtdSer targets in an integrin-dependent manner? One possibility is that integrins interact directly with the RGD motif present in the N-terminal exofacial domain of TIM4. This possibility is lent credence by perusal of the crystal structure of TIM4: its RGD motif resides in a loop predicted to be readily accessible from the surrounding milieu yet removed from the PtdSer-binding pocket. In this manner integrins could engage the receptor without interfering with its ability to bind PtdSer. In this model TIM4 would serve to couple PtdSer-bearing targets to integrins, which would in turn serve to transduce phagocytic signals. The notion of direct interaction between these molecules is strengthened by colocalization with surface-expressed β1 integrins observed upon clustering of TIM4. Conversely, clustering of integrins also caused increased colocalization with TIM4. Inability to detect significant colocalization of β1 integrins and TIM4 before cross-linking suggests that these molecules may form short-lived metastable complexes that become stabilized upon receptor aggregation. This predicted weak constitutive association would also explain why attempts to coimmunoprecipitate integrin via TIM4, or vice versa, were unsuccessful.
We also report that TIM4-mediated phagocytosis depends on the activity of Src-family and FAK kinases and is accompanied by accumulation of talin at the sites of receptor engagement, all hallmarks of integrin activation. In addition, PtdIns(3,4,5)P3 is found to accumulate at the phagocytic cup and be essential for particle engulfment. Activation of PI3K is another well-documented consequence of integrin activation. Taken together, our data support a model in which TIM4 uses integrins as coreceptors to effect the engulfment of surface-bound prey. This model is entirely consistent with the observation that the transmembrane and cytoplasmic domains of TIM4 are dispensable for its function in phagocytosis (Park et al., 2009).
Other PtdSer receptors, such as BaiI and stabilin, converge on the small GTPase Rac to effect the actin rearrangements that drive particle engulfment (Kinchen et al., 2005; Park et al., 2007; Kim et al., 2012). In sharp contrast, Park et al. (2009) found that TIM4-dependent phagocytosis was virtually independent of Rac. Contrary to their findings, we report here that RhoA, as well as Rac1 and Rac 2, is required for the actin changes that accompany TIM4-mediated phagocytosis. This finding, too, is consistent with involvement of the integrins. Indeed, uptake of complement-opsonized particles—an integrin-mediated event—is believed to require the activation of RhoA, Rac1, and Rac2 (Caron and Hall, 1998; Olazabal et al., 2002; Hall et al., 2006). In good accordance with the observation that RhoA, Rac1, and Rac2 contribute to TIM4-dependent phagocytosis we find that Vav3, a Rho/Rac GEF (Movilla and Bustelo, 1999; Colomba et al., 2008), accumulates at sites of TIM4 engagement and is required for target engulfment. Vav3 is a modular protein that, in addition to its DBL-homology domain, which mediates nucleotide exchange, possesses a Src-homology 2 domain and a PH domain, which bind phosphorylated tyrosine residues and phosphoinositides, respectively. It is therefore predisposed to be recruited by the phosphotyrosine residues generated by Src-family kinases and by the PtdIns(3,4,5)P3 synthesized in response to integrin activation.
In summary, we demonstrate that TIM4, a PtdSer receptor expressed on professional phagocytes, is a direct, active participant in the phagocytosis of PtdSer-bearing targets. Furthermore, we show that TIM4 uses integrins as coreceptors to effect target engulfment via a FAK-, PI3K-, and RhoA/Rac1/Rac2-dependent pathway.
MATERIALS AND METHODS
Reagents
Rabbit polyclonal anti-HA and mouse anti-HA (12CA5) antibodies were purchased from Rockland (Boyertown, PA) and the Sunnybrook Health Sciences Centre Hybridoma core facility (Toronto, Canada), respectively. The rat anti-human CD29 antibody (clone AIIB2) and the mouse anti-human CD29 antibody (clone P5D2) were purchased from the Iowa Developmental Studies Hybridoma Bank (Iowa City, IA). Mouse anti-RhoA (clone 67B9) was from Cell Signaling Technology (Beverly, MA). Mouse anti-biotin antibody, goat anti-rabbit Fab2 antibody, and all fluorophore-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). The actin-disrupting agents latrunculin B and cytochalasin D were from Sigma-Aldrich (St. Louis, MO). The Src inhibitor PP1 and the FAK inhibitor PF 573228 were purchased from Tocris (Bristol, United Kingdom). The PI3K inhibitors wortmannin and LY294002 were from EMD Biosciences (Billerica, MA). Clostridium difficile toxin B and cell-permeant C3-transferases were purchased from Techlab (Blacksburg, VA) and Cytoskeleton Inc. (Denver, CO), respectively. Fluorescent avidin and phalloidin were purchased from Invitrogen Molecular Probes (Carlsbad, CA). The RGD-containing peptide Gly-Arg-Gly-Asp-Ser was purchased from Sigma-Aldrich. The murine M-CSF was purchased from Peprotech (Rocky Hill, NJ).
Tissue culture, transfection, and plasmids
AD293, a semiadherent HEK-derived cell line, was purchased from Stratagene. AD293 cells were cultured in DMEM containing 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) in the presence of 5% CO2. For live-cell imaging or incubations in the absence of CO2, AD293 cells were cultured in bicarbonate-free medium RPMI 1640 containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) without serum. cDNA was transfected into AD293 cells using FuGENE 6 transfection reagent (Promega) according to the manufacturer's instructions. After plating and overnight incubation, the cells were routinely transfected with 1 μg of cDNA and incubated at least 16–24 h before experimentation. To isolate BMDMs, mice were killed and the femora and tibiae removed, and bone marrow was flushed with cold phosphate-buffered saline (PBS) using a 21G needle. The marrow was homogenized by repeated passage through a 21G needle and plated in RPMI 1640 supplemented with 10% heat-inactivated FBS, 25 ng/ml macrophage colony-stimulating factor (M-CSF), and penicillin/streptomycin. The M-CSF was replaced every 2–3 d, and BMDMs were used on days 7–8 after isolation. Transfection of BMDMs was carried out using the Neon transfection system. In short, BMDMs were resuspended in electroporation buffer and loaded into the Neon transfection system for electroporation at 1400 V for 30 m. Macrophages were then replated in RPMI 1640 supplemented with 10% heat-inactivated FBS and incubated for 12–16 h before use. The parental plasmid encoding human TIM4 (MCG clone 4184237) was purchased from Invitrogen. To generate TIM4-GFP and TIM4-mCherry, the TIM4 coding sequence was PCR amplified using MCG clone 4184237 as a template and cloned into pEGFP-N1 (Clontech) and p-mCherry-N1 (Clontech) as a BamHI/XhoI fragment. TIM4(RGE)-GFP was created using the site-directed mutagenesis primers CTCAAACACCAATCGAGGTGAAAGTGGGGTGTACTGC and GCAGTACACCCCACTTTCACCTCGATTGGTGTTTGAG and primers derived from the kanamycin resistance cassette in pEGFP-N1 to generate PCR products that were fused using the In-Fusion HD Cloning System (Clontech). To make the plasmid mCherry-PBD-PAK, the PBD-PAK was excised from yPET-PBD-PAK as a BamHI/EcoRI fragment and cloned into pmCherry-C1 digested similarly. FcγRIIA-GFP and the signaling mutant FcγRIIA(Y2Y3F)-GFP, called FcγRIIAmut-GFP, were a gift of A. Schreiber (University of Pennsylvania, Philadelphia, PA). TIM4-HA was kindly provided by K. Ravichandran and was generated as previously described (Meyers et al., 2005). The plasmid encoding human Vav3-citrine was provided by R. Bagshaw (Lunenfeld Research Institute, Toronto, Canada). The plasmids PM-RFP, PM-GFP, and PH-Akt-RFP are described elsewhere (Teruel et al., 1999; Haugh et al., 2000).
Small interfering RNA
siRNA directed to human RhoA, Vav3, integrin β1, integrin β3, and integrin β5 was purchased from Dharmacon. To effect knockdown, 20 nM siRNA was transfected into AD293 cells using RNAiMAX Oligofectamine transfection reagent (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after siRNA transfection, the medium was replaced, and the cells were transfected as described with cDNA encoding TIM4 as appropriate. RhoA knockdown was verified by Western blot analysis after 15% SDS–PAGE. To evaluate knockdown of integrin β1, endogenous protein was detected by immunofluorescence. Briefly, cells were fixed with 4% (vol/vol) paraformaldehyde (PFA) and blocked with 5% (wt/vol) skim milk powder in PBS. Integrin β1 was detected using a mouse anti-human β1 integrin antibody (clone P5D2) at 1:200 dilution, followed by staining with a secondary fluorophore-conjugated antibody. Fluorescence intensity measurements of integrin β1 knockdown were performed on images acquired with identical acquisition parameters using ImageJ (National Institutes of Health, Bethesda, MD) after background correction. The knockdown of integrin β3, integrin β5, and Vav3 was evaluated by real-time PCR using TaqMan Gene Expression Assays (Applied Biosystems by Life Technologies).
Pharmacological treatments
AD293 cells were treated with 10 μM cytochalasin D for 30 min or 2 μM latrunculin B for 10 min before the addition of phagocytic targets. PP1 and LY294002 were added to cells 20 min before the addition of phagocytic targets at a final concentration of 10 and 100 μM, respectively. PF 573228 was used at 30 μM and incubated with AD293 cells for 30 min before addition of phagocytic targets. Cell-permeant C3-transferase was used at a final concentration of 10 μg/ml and was added to the cells for 2 h before phagocytosis. C. difficile toxin B was used at a final concentration of 100 ng/ml, pretreating the cells for ∼1.5 h. The pharmacological agents were present throughout the measurements.
Preparation of phagocytic targets
To generate lipid-coated phagocytic targets, 5-μm glass beads (Bangs Laboratories) were mixed with 76.2 mol% PtdCho, 23 mol% PtdSer, and 0.8 mol% biotinylated PtdEth or rhodamine-labeled PtdEth (Avanti Polar Lipids) dissolved in 1:1 (vol/vol) chloroform/methanol and then dried under constant N2 flow. Once dried, the beads were washed and resuspended in PBS. Beads without PtdSer were generated as described, except that PtdSer was omitted and 99.2 mol% PtdCho was used instead. To opsonize lipid-coated beads with IgG, beads bearing biotinylated PtdEth were incubated with a subagglutinating concentration of mouse anti-biotin IgG in PBS for 15 min with constant mixing.
Phagocytosis assays
Before the addition of targets, AD293 cells cultured on 18-mm coverslips were rinsed with PBS and incubated in serum-free medium RPMI 1640 (bicarbonate free) containing 25 mM HEPES at 37°C in the absence of CO2 for 10 min. Phagocytic targets were then added to AD293 cells at a ratio of ∼10 beads/cell, and target binding was synchronized by a 1-min centrifugation at 277 × g. Phagocytosis was allowed to proceed at 37°C for 30 min. To evaluate the specificity of binding, cells were gently rinsed after an initial 2-min incubation at 37°C and then incubated in medium RPMI 1640 (bicarbonate free) with 25 mM HEPES without serum for a further 28 min. For experiments in which FBS was included during phagocytosis, it was used at 10% (vol/vol), and the cells were maintained in DMEM in the presence of 5% CO2. To distinguish internalized targets from those that remained accessible to the fluid phase, coverslips were gently rinsed with cold PBS and incubated with PBS containing 2 μg/ml streptavidin conjugated to Alexa Fluor 647 for 2–3 min before fixation with 4% (vol/vol) PFA or ice-cold methanol as necessary.
Fluorescence microscopy
Fluorescence images were acquired using spinning disk confocal microscopy. The spinning disk confocal system (Quorum Technologies) used in our laboratory is based on a Zeiss Axiovert 200M microscope (Carl Zeiss) with 63× (numerical aperture [NA] 1.4) or 100× (NA 1.45) oil immersion objective, equipped with diode-pumped solid-state lasers (405, 440, 491, 561, and 655 nm; Spectral Applied Research) and a motorized XY stage (Applied Scientific Instruments). Images were acquired using a back-thinned, electron-multiplied, cooled, charge-coupled device camera (C9100-13 ImagEM; Hamamatsu Photonics) controlled by the Volocity software (PerkinElmer).
Use of optical tweezers to evaluate TIM4 dependence of phagocytic cup formation
Lipid-coated beads bearing PtdCho/PtdSer/rhodamine-PtdEth were generated as described and added to coverslips with adherent AD293 cells expressing either PM-GFP or TIM4-GFP. Individual lipid-coated beads were captured in an optical trap using a Molecular Machines and Industries laser tweezer system (30W Nd:YAG infrared laser) installed on a Zeiss Axiovert 200M spinning disk confocal microscope with a 63× objective (NA 1.4). Individually trapped beads were pressed using the laser tweezer against the surface of transfected AD293 cells, and the accumulation of GFP-positive membrane at sites of forced particle binding was monitored by time-lapse spinning disk confocal microscopy.
Single-particle labeling, detection, and spatial apposition analysis
The colocalization of TIM4 and integrin β1 was assessed by microscopy at the single-particle level. AD293 cells were transfected with plasmids encoding wild-type TIM4 with an N-terminal HA epitope. TIM4 and integrin β1 were detected by labeling with 22 ng/ml anti-HA (clone 12CA5) and 2.9 μg/ml rat anti-human CD29 (clone AIIB2) for 10 min at 10°C. After a gentle wash with cold PBS, the cells were incubated for 8 min at 10°C with a rabbit anti-mouse Fab conjugated to DyLight549 and a donkey anti-rat Fab conjugated to DyLight488. To induce cross-linking, an unlabeled goat anti-Rabbit Fab2 was used at a final concentration of 6 μg/ml for 5 min at 10°C. Cells were fixed immediately after labeling using 4% (vol/vol) PFA for 30 min, followed by imaging. In instances in which integrin β1 was cross-linked, a rabbit anti-rat Fab DyLight488 was used as a secondary antibody and donkey anti-mouse Cy3 Fab was used to detect mouse anti-HA, followed by goat anti-rabbit Fab2 and then fixed as described.
Individual features were visualized by epifluorescence microscopy using a Zeiss Axiovert 200M microscope equipped with a custom 2.5× lens, a 100× (NA, 1.45) objective, and an electron-multiplied, cooled, charge-coupled device camera (C9100-13 ImagEM). Volocity was used to acquire single images for each channel, which were then exported as OME TIFFs for analysis. The resulting images contained individual resolvable point spread functions for each fluorescence channel, which were detected and localized as previously described (Jaqaman et al., 2008; Flannagan et al., 2010). Briefly, the position and intensity of each fluorescently labeled TIM4-HA or integrin β1 molecule were determined by identifying local intensity maxima for each feature. Next a Gaussian kernel approximating the microscope's point spread function was fit over each local maximum to approximate the position of the point emitter to a positional precision (σ) of ∼22 nm.
The colocalization between TIM4-HA and integrin β1 molecules was determined by determining the spatial relationship between fluorescently labeled proteins employing a nearest-neighbor approach similar to that of Dunne et al. (2009), as described in detail in Heit et al. (2013). Briefly, the positional precision was calculated individually for each detected feature in each image. The predicted positional precision for colocalized features was determined by calculating the root mean square of the positional precision for each fluorescence feature in each channel. The colocalization distance cutoff (CDC) was established by multiplying the positional precision of colocalized features by a 90% probability cutoff. The image registration accuracy (determined to be 105 nm) was then factored into the CDC. Fluorescent features below the CDC were considered colocalized. The CDC for our microscope is calculated to be 165–175 nm, and a conservative 180 nm CDC was used for image analysis. Using a Monte Carlo simulation, we determined whether the degree of observed colocalization was significantly different from the fortuitous colocalization of features having a random distribution (Metropolis and Ulam, 1949). To generate relevant random distributions, the dimensions of the image being analyzed, as well as the number of fluorescence features in each channel, were determined. Using this information, a virtual image of the same dimensions and number of features in each channel was created at random, for which the degree of colocalization was determined as described. This simulation was repeated 2000 times for every image analyzed to give a frequency of colocalization for nonassociated molecules with a random distribution. Finally, a t test was used to determine whether a statistically significant difference in the degree of colocalization exists between the actual observed molecules and the randomized virtual molecules.
Statistical analysis
All t tests were performed using Prism software (GraphPad). All mathematical functions for single-particle detection, localization, and colocalization analysis were performed using Matlab software (MathWorks). Data are presented as mean ± SE, unless noted otherwise. *p ≤ 0.05, **p ≤ 0.01.
Supplementary Material
Acknowledgments
This work was supported by Grants MOP7075, MOP93634, and TBO-122068 from the Canadian Institutes of Health Research (to S.G. and M.G.). R.S.F. was supported by a Restracomp Fellowship from the Hospital for Sick Children Research Training Center. TIM4−/− bones were kindly provided by John Brumell (Hospital for Sick Children, Toronto, Canada).
Abbreviations used:
- BMDM
bone marrow–derived macrophage
- EDTA
ethylenediaminetetraacetic acid
- FAK
focal adhesion kinase
- HEK293
human embryonic kidney 293 cells
- PBD-PAK
p21-binding domain of p21-activated kinase
- PH
pleckstrin homology domain
- PI3K
class I phosphoinositide 3-kinase
- PP1
1-(1,1-dimethylethyl)-1-(4-methylphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- PtdCho
phosphatidylcholine
- PtdEth
phosphatidylethanolamine
- PtdIns(3,4)P2
phosphatidylinositol 3,4-bisphosphate
- PtdIns(3,4,5)P3
phosphatidylinositol 3,4,5-trisphosphate
- PtdSer
phosphatidylserine
- RGD
arginine, glycine, and aspartic acid–containing peptide
- TAT
trans-activator of transcription
- TIM4
T-cell immunoglobulin mucin protein 4
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-04-0212) on March 12, 2014.
*These authors contributed equally.
REFERENCES
- Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akakura S, Singh S, Spataro M, Akakura R, Kim J-I, Albert ML, Birge RB. The opsonin MFG-E8 is a ligand for the alphavbeta5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res. 2004;292:403–416. doi: 10.1016/j.yexcr.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT. Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry. 2000;39:6200–6206. doi: 10.1021/bi992221r. [DOI] [PubMed] [Google Scholar]
- Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- Colomba A, Courilleau D, Ramel D, Billadeau DD, Espinos E, Delsol G, Payrastre B, Gaits-Iacovoni F. Activation of Rac1 and the exchange factor Vav3 are involved in NPM-ALK signaling in anaplastic large cell lymphomas. Oncogene. 2008;27:2728–2736. doi: 10.1038/sj.onc.1210921. [DOI] [PubMed] [Google Scholar]
- Dunne PD, Fernandes RA, McColl J, Yoon JW, James JR, Davis SJ, Klenerman D. DySCo: quantitating associations of membrane proteins using two-color single-molecule tracking. Biophys J. 2009;97:L5–L7. doi: 10.1016/j.bpj.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista V, et al. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood. 2007;109:2461–2469. doi: 10.1182/blood-2006-06-029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Harrison RE, Yip CM, Jaqaman K, Grinstein S. Dynamic macrophage “probing” is required for the efficient capture of phagocytic targets. J Cell Biol. 2010;191:1205–1218. doi: 10.1083/jcb.201007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaipl US., et al. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–121. doi: 10.1016/j.jaut.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Hall AB, Gakidis MAM, Glogauer M, Wilsbacher JL, Gao S, Swat W, Brugge JS. Requirements for Vav guanine nucleotide exchange factors and Rho GTPases in FcgammaR- and complement-mediated phagocytosis. Immunity. 2006;24:305–316. doi: 10.1016/j.immuni.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- Haugh JM, Codazzi F, Teruel M, Meyer T. Spatial sensing in fibroblasts mediated by 3’ phosphoinositides. J Cell Biol. 2000;151:1269–1280. doi: 10.1083/jcb.151.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B, Kim H, Cosío G, Castaño D, Collins R, Lowell CA, Kain KC, Trimble WS, Grinstein S. Multimolecular signaling complexes enable Syk-mediated signaling of CD36 internalization. Dev Cell. 2013;24:372–383. doi: 10.1016/j.devcel.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell. 2004;15:3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jank T, Giesemann T, Aktories K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology. 2007;17:15R–22R. doi: 10.1093/glycob/cwm004. [DOI] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Park SY, Kim SY, Bae DJ, Pyo JH, Hong M, Kim IS. Cross talk between engulfment receptors stabilin-2 and integrin αvβ5 orchestrates engulfment of phosphatidylserine-exposed erythrocytes. Mol Cell Biol. 2012;32:2698–2708. doi: 10.1128/MCB.06743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, Schnabel H, Schnabel R, Hengartner MO. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature. 2005;434:93–99. doi: 10.1038/nature03263. [DOI] [PubMed] [Google Scholar]
- Koh AL, Sun CX, Zhu F, Glogauer M. The role of Rac1 and Rac2 in bacterial killing. Cell Immunol. 2005;235:92–97. doi: 10.1016/j.cellimm.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Korns D, Bratton DL. Modulation of macrophage efferocytosis in inflammation. Front Inflamm. 2011;2:57. doi: 10.3389/fimmu.2011.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Massol P, Montcourrier P, Guillemot JC, Chavrier P. Fc receptor-mediated phagocytosis requires CDC42 and Rac1. EMBO J. 1998;17:6219–6229. doi: 10.1093/emboj/17.21.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metropolis N, Ulam S. The Monte Carlo method. J Am Stat Assoc. 1949;44:335–341. doi: 10.1080/01621459.1949.10483310. [DOI] [PubMed] [Google Scholar]
- Meyers JH, et al. TIM-4 is the ligand for TIM-1, and the TIM-1–TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005;6:455–464. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- Movilla N, Bustelo XR. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol Cell Biol. 1999;19:7870–7885. doi: 10.1128/mcb.19.11.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olazabal IM, Caron E, May RC, Schilling K, Knecht DA, Machesky LM. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcgammaR, phagocytosis. Curr Biol. 2002;12:1413–1418. doi: 10.1016/s0960-9822(02)01069-2. [DOI] [PubMed] [Google Scholar]
- Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19:346–351. doi: 10.1016/j.cub.2009.01.042. [DOI] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont A-C, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- Santiago C, Ballesteros A, Martínez-Muñoz L, Mellado M, Kaplan GG, Freeman GJ, Casasnovas JM. Structures of T cell immunoglobulin mucin protein 4 show a metal-ion-dependent ligand binding site where phosphatidylserine binds. Immunity. 2007;27:941–951. doi: 10.1016/j.immuni.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, Kalman D, Bourne HR. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol. 2003;160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruel MN, Blanpied TA, Shen K, Augustine GJ, Meyer T. A versatile microporation technique for the transfection of cultured CNS neurons. J Neurosci Methods. 1999;93:37–48. doi: 10.1016/s0165-0270(99)00112-0. [DOI] [PubMed] [Google Scholar]
- Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–1428. doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda S, Hanayama R, Nagata S. Two-step engulfment of apoptotic cells. Mol Cell Biol. 2012;32:118–125. doi: 10.1128/MCB.05993-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utomo A, Cullere X, Glogauer M, Swat W, Mayadas TN. Vav proteins in neutrophils are required for FcgammaR-mediated signaling to Rac GTPases and nicotinamide adenine dinucleotide phosphate oxidase component p40(phox) J Immunol. 2006;177:6388–6397. doi: 10.4049/jimmunol.177.9.6388. [DOI] [PubMed] [Google Scholar]
- Wong K, Valdez PA, Tan C, Yeh S, Hongo J-A, Ouyang W. Phosphatidylserine receptor Tim-4 is essential for the maintenance of the homeostatic state of resident peritoneal macrophages. Proc Natl Acad Sci USA. 2010;107:8712–8717. doi: 10.1073/pnas.0910929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.