

Atmospheric gas plasmas (AGPs) up-regulate intracellular ROS levels and induce apoptosis in melanoma cells. Evidence for TNF-signaling dependence of ASK1-mediated apoptosis suggests possible mechanisms for AGP activation and regulation of apoptosis-signaling pathways in tumor cells.
Abstract
Atmospheric gas plasmas (AGPs) are able to selectively induce apoptosis in cancer cells, offering a promising alternative to conventional therapies that have unwanted side effects such as drug resistance and toxicity. However, the mechanism of AGP-induced cancer cell death is unknown. In this study, AGP is shown to up-regulate intracellular reactive oxygen species (ROS) levels and induce apoptosis in melanoma but not normal melanocyte cells. By screening genes involved in apoptosis, we identify tumor necrosis factor (TNF)–family members as the most differentially expressed cellular genes upon AGP treatment of melanoma cells. TNF receptor 1 (TNFR1) antagonist–neutralizing antibody specifically inhibits AGP-induced apoptosis signal, regulating apoptosis signal–regulating kinase 1 (ASK1) activity and subsequent ASK1-dependent apoptosis. Treatment of cells with intracellular ROS scavenger N-acetyl-l-cysteine also inhibits AGP-induced activation of ASK1, as well as apoptosis. Moreover, depletion of intracellular ASK1 reduces the level of AGP-induced oxidative stress and apoptosis. The evidence for TNF-signaling dependence of ASK1-mediated apoptosis suggests possible mechanisms for AGP activation and regulation of apoptosis-signaling pathways in tumor cells.
INTRODUCTION
In recent decades, there has been significant progress in the development of new therapies to treat human cancers. However, fundamental problems related to chemotherapeutic drug delivery—resistance and toxicity to normal cells—remain. An ideal anticancer treatment should selectively kill cancer cells with limited side effects on normal cells and minimal drug resistance. Selective induction of apoptosis in target cancer cells would be an ideal treatment (Nagata, 1997; Kaelin, 1999).
Two pathways of apoptosis have been described for mammalian cells: the intrinsic mitochondrial pathway that is activated in response to cellular stress, and the extrinsic death receptor pathway that is activated at the cell surface by the binding of tumor necrosis factor (TNF)–family cytokines to their cognate death receptors (TNFR1, Fas/CD95, DR4/5). The extrinsic and the intrinsic pathways converge in a caspase cascade that results in cellular shrinkage and DNA fragmentation, culminating in apoptosis (Micheau and Tschopp, 2003; Kamata et al., 2005).
Cellular oxidative stress can induce apoptosis by initiating the activation of a specialized group of mitogen-activated protein kinase (MAPK) cascades. Apoptosis signal–regulating kinase 1 (ASK1) is a MAPK kinase kinase (MAPKKK)–family member that plays a major role in stress-induced apoptosis. ASK1 is activated by various stress-related stimuli, including oxidative stress, reactive oxygen species (ROS), genotoxic agents, serum withdrawal, endoplasmic reticulum stress, and tumor necrosis factor (TNF; Ichijo et al., 1997; Nishitoh et al., 1998; Liu et al., 2000). The activated ASK1 phosphorylates and activates the downstream kinases MKK4/MKK7 and MKK3/MKK6. These are in turn required to activate c-Jun N-terminal kinase (JNK) and p38 MAPK kinase, respectively, before caspase-3 activation and apoptosis. The knockdown of ASK1 inhibits ROS (H2O2)- and TNF-induced apoptosis (Noguchi et al., 2005, 2008). Overexpression of a constitutively active form of ASK1 in cancer cells induces caspase-3–dependent apoptosis (Ichijo et al., 1997). ROS such as H2O2 activate ASK1 by dissociation of thioredoxin (Trx), a reduction/oxidation (redox) regulatory protein that inhibits the kinase activity of ASK1 (Saitoh et al., 1998). It has also been proposed that ROS generated by cytokines like TNFα or stress may oxidize and consequently dissociate Trx to activate ASK1 and subsequent ASK1-dependent apoptosis-signaling cascades (Saitoh et al., 1998; Liu et al., 2000; Nadeau et al., 2009). These findings demonstrate that ASK1 plays an important role in stress-induced apoptosis.
Recently atmospheric gas plasmas (AGPs) delivered in a plume have shown the potential to be a safe anticancer therapy that can kill selectively a variety of cancer cells, such as melanoma (Keidar et al., 2011; Kim et al., 2011a), neuroblastoma (Walk et al., 2013), glioma (Koeritzer et al., 2013), and colorectal (Vandamme et al., 2012), pancreatic (Partecke et al., 2012), breast (Kalghatgi et al., 2011), liver (Yan et al., 2012), ovarian (Iseki et al., 2012), leukemia (Thiyagarajan et al., 2012), and lung (Huang et al., 2011) cancers. AGP delivered as a plume has also shown significant effects in in vivo animal models by decreasing the size of tumors (Vandamme et al., 2010, 2012; Keidar et al., 2011; Partecke et al., 2012; Walk et al., 2013). It has been proven effective against drug-resistant brain tumor (glioblastoma) in combination with temozolomide (Koeritzer et al., 2013). The AGP plume generated by an electrical discharge in a gas generates reactive oxygen species (e.g., H2O2, O3, OH, NO), ions, and free electrons (Sensenig et al., 2008; Liu et al., 2010; Weltmann et al., 2010; Keidar et al., 2011; Graves, 2012; Ostrikov et al., 2013). AGP leads to the formation of reactive oxygen species, including H2O2, in liquids that simulate cellular fluids (Winter et al., 2013). Despite early promising results obtained using AGP as an antitumor treatment to kill cancer cells, little effort has been made to translate this into clinical applications. This is possibly due to a lack of understanding about the underlying cellular mechanisms and the means by which AGP can selectively kill cancer cells by inducing apoptosis without affecting normal cells.
In this study, we find that AGP treatment enhances the production of intracellular ROS. This elevated level of intracellular ROS induces oxidative stress, leading to apoptosis in melanoma cells without affecting normal control cells. Moreover, AGP treatment activates TNF and ASK1 pathways to increase downstream activity of JNK and p38 kinases and stimulate caspase-3/7–dependent apoptosis. Thus AGP shows selective induction of apoptosis in cancer cells by stimulating the oxidative stress–induced TNF-ASK1-JNK/p38–caspase-3/7 apoptotic pathway.
RESULTS
Selective apoptotic response to AGP is ROS dependent
We quantified the effect of AGP (presented and briefly described in Materials and Methods, Figure 1, and Supplemental Methods) on the cellular ROS level in melanoma cancer cells (Mel007 and Mel-RM) using the redox-sensitive fluorescent probe 2′-,7′-dichlorofluorescein diacetate (CM-H2DCFDA). Treatment with AGP caused a significant increase in ROS level in melanoma cells. AGP did not increase ROS level significantly in normal control cells (primary human epidermal melanocytes and human fetal lung fibroblasts (Figure 2a). Cotreatment with the intracellular ROS inhibitor N-acetyl-l-cysteine (NAC) fully reversed the AGP-induced increase in ROS in melanoma cells (Figure 2b). It has been shown that an increase in ROS production can lead to decrease in reduced glutathione (GSH) levels in cancer cells (Estrela et al., 2006). GSH is an important intracellular antioxidant that protects cells from damage caused by ROS. It is able to remove O2•− and provide electrons for glutathione peroxidase to reduce H2O2 to H2O. Because AGP has been shown to induce ROS production and modulate redox homeostasis (Graves, 2012), we tested whether AGP can lead to a decrease in GSH levels. This was found to occur in melanoma cells. However AGP-mediated GSH depletion was inhibited by addition of the reducing agent NAC (Figure 2c). Parallel counting of viable cells showed that AGP-induced cell death in melanoma cells correlated with decrease in cellular GSH contents (Supplemental Table S1). We also found that nitric oxide was among the ROS species induced by AGP in melanoma cells but not in normal control cells (Figure 2d). Supplemental Figure S1 demonstrates the optical emission spectrum of the AGP plume over the range from 300 to 800 nm, further confirming that excited species of OH, N2, N2+, He, and O exist in the AGP plume.
FIGURE 1:
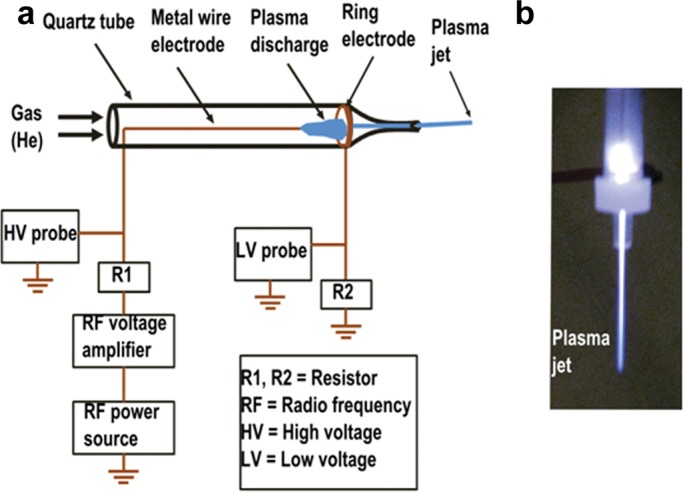
(a) Schematic of the atmospheric gas plasma jet device. (b) Digital photo of the generated gas plasma jet for cell treatment.
FIGURE 2:

AGP enhances ROS accumulation in melanoma cells. (a) AGP enhances ROS levels in melanoma cells (Mel-RM, Mel007) but not normal cells (melanocytes, human fetal lung fibroblasts). Cells were treated with AGP (5, 15, 30 s). ROS level was quantified by the fluorescent dye CM-H2DCFDA and is shown as a fold change relative to control cells treated only with He gas flow. Data were normalized to ROS levels in control melanocyte cells. (b) Mel007 or melanocyte cells were treated with AGP (5, 15, 30s) or pretreated with NAC (1–2 h) followed by AGP treatment (5, 15, 30 s). ROS level was determined as in a. (c) Mel007 or melanocyte cells were treated with AGP or pretreated with NAC (1–2 h), followed by AGP treatment. GSH level was quantified as described in Materials and Methods. (d) Mel007 or melanocyte cells were treated with AGP (5, 15, 30 s) or pretreated with NAC (1–2 h), followed by AGP treatment (5, 15, 30 s). Nitric oxide was quantified after cells were labeled with DAF-FM diacetate. All values are mean ± SD of three independent experiments performed in triplicate. *p ≤ 0.01, **p ≤ 0.001; ANOVA.
We next examined the effect of AGP on the viability of several cultured melanoma cells (Mel-RM, Mel007, and Mel-JD) and normal control cells (primary human epidermal melanocytes and human fetal lung fibroblasts). AGP treatment significantly induced apoptosis in the melanoma cells. When primary cultures of normal cells were treated with AGP, there was little effect on cell growth and proliferation. This indicated that AGP selectively induces cell death in melanoma cells (Figure 3a). As shown earlier (Figure 2, a and d), AGP induced intracellular ROS, so we next tested to see whether the selective cytotoxic effect of AGP on melanoma cells was ROS dependent or independent. Cotreatment of AGP and NAC completely reversed the toxic effects of AGP in melanoma cells (Figure 3b). AGP induced apoptosis and DNA damage in melanoma cells, as observed by increased activity of caspases 3/7 and stress response target protein γ-H2AX in AGP-treated melanoma cells, with no significant activity in normal cells (Figure 3, c and d). Taken together, this different response of cancer and normal cells to AGP treatment indicates that AGP targets cancer cell redox homeostasis, which results in both a stress response and DNA damage, leading to apoptosis in cancer cells. This selective induction of ROS in cancer cells indicates that the AGP-specific apoptotic response in melanoma cells is mediated by perturbation of cellular redox homeostasis.
FIGURE 3:

AGP selectively induces apoptosis in melanoma cells, and this is ROS dependent. (a) AGP treatment induced cell death in melanoma cells but not normal cells. Melanoma cells (Mel-RM, Mel007, Mel-JD), melanocytes (MC), and human fetal lung fibroblasts (MRC5) were cultured in 96-well plates overnight, treated with AGP for 5–30 s, and grown for 18–24 h before analysis. Cell viability was measured by cell titer nonradioactive cell proliferation assay. (b) AGP-induced cell death in melanoma cells was reversed by NAC. Mel007 or Mel-RM cancer cells or melanocytes were treated with AGP (5, 15, 30 s) or pretreated with NAC for 1–2 h, followed by AGP treatment (5, 15, 30 s) for 18–24 h. Cell viability was measured by cell titer nonradioactive cell proliferation assay. (c) Mel007, Mel-RM, and melanocytes were treated with AGP or pretreated with NAC. Caspase 3/7 activity was measured by Caspase-Glo 3/7 assay. (d) The effect of AGP on stress response targets was determined by Western blot analysis of H2AX and γ-H2AX protein in normal (melanocytes) and melanoma cells (Mel007). GAPDH expression was used as a loading control. In all experiments, control cells were mock treated with He gas flow only. All values are mean ± SD of three independent experiments performed in triplicate. *p ≤ 0.01, **p ≤ 0.001; ANOVA.
TNF is involved in AGP-induced apoptosis
Next we examined the mechanism by which AGP specifically induces apoptosis in melanoma cells. To find the specific cellular factors involved in selective AGP-induced apoptosis, we screened >90 genes involved specifically in prosurvival or proapoptotic pathways by using real-time quantitative PCR (qPCR). We found that TNF family members are the cellular factors that most frequently showed differential expression in AGP-treated melanoma cells relative to control untreated cells (Supplemental Figure S2). We confirmed our qPCR gene expression screening data by measuring the gene expression of TNF-receptor family member 1 (TNFR1) in melanoma cells treated with AGP for different time intervals (5, 15, and 30 s) by qPCR and Western blotting. However, this AGP-induced TNFR1 expression was inhibited by the ROS scavenger NAC (Figure 4, a and b). This shows the involvement of ROS in AGP-induced TNF signaling. Moreover, we also observed increase in the production of TNF signaling ligand (TNFα) in AGP-treated melanoma (Mel007) cells but not in normal melanocytes (Figure 4c). Outcomes showed increased activity of stress response target protein γ-H2AX in the AGP-treated melanoma cells but no significant activity in melanoma cells pretreated with anti–TNFR1-neutralizing antibody (Figure 4d). Determination of cell viability and caspase 3/7 activity demonstrated that AGP-induced apoptosis and cytotoxicity was inhibited by cotreatment of AGP with antagonistic anti–TNFR1-neutralizing antibody, the caspase inhibitor Z-VAD-FMK, the inhibitor of nitric oxide synthetase diphenyleneiodonium chloride (DPI), or the H2O2 depleter catalase (Figure 4, e and f). These results indicate that selective AGP-induced apoptosis in melanoma cells is dependent on intracellular ROS production. Moreover, cell death induced by AGP is mediated by TNF-receptor pathways by activating caspase 3/7 activity.
FIGURE 4:

TNF-apoptotic pathway is involved for AGP-induced selective apoptosis in cancer cells. (a, b) Mel007 cells were treated with AGP (5, 15, 30 s), and gene expression for TNFR1 was measured by quantitative real-time PCR and Western blotting. (c) Effect of AGP on the activation of TNF ligand in Mel007 and melanocytes was determined by enzyme-linked immunosorbent assay. Cells were treated with AGP (5, 15, 30 s) for 48 h. The concentration of TNFα was measured in cell culture supernatant. (d) Effect of AGP pretreated with or without TNFR1-neutralizing antibody on stress response targets were determined by Western blot analysis of H2AX and γ-H2AX protein in melanoma cells (Mel007). Mouse IgG1 isotype antibody was used as negative control. GAPDH expression was used as loading control. (e, f) Mel007 cells were treated with AGP or pretreated with anti-TNFR1 antibody, caspase inhibitor zVAD, H2O2-depleting-agent catalase, and nitric oxide synthesis inhibitor DPI, followed by AGP treatment. Cell viability was measured using a cell titer nonradioactive cell proliferation assay, and caspases 3/7 activity was measured by Caspase-Glo 3/7 assay. In all experiments, control cells were mock treated with He gas flow only. All values are mean ± SD of three independent experiments performed in triplicate. *p ≤ 0.01, **p ≤ 0.001; ANOVA.
ASK1 is required for AGP-induced apoptosis
We next sought to explore the downstream signaling factors involved in the AGP-induced TNF apoptosis pathway. It was shown that ROS (e.g., H2O2) induced by the TNF pathway plays a role in programmed cell death signaling mediated by ASK1 (Noguchi et al., 2005). We examined whether ASK1 is involved in AGP-induced apoptosis in melanoma cells. AGP induced ASK1 activity at a similar level to H2O2. However, this ASK1 activation was inhibited by the ROS scavenger NAC (Figure 5a). This shows that AGP-induced ASK1 activation is ROS dependent. We further determined that AGP activated the ASK1-activity–dependent downstream-signaling kinases p38 and JNK at a level similar to H2O2, whereas this activation was strongly reduced by NAC (Figure 5, b and c).
FIGURE 5:

AGP stimulates ASK1-mediated p38, and JNK activation is ROS dependent and reversed with NAC. (a–c) Mel007 cancer cells were treated with AGP or H2O2 with or without pretreatment with NAC. Western blot analysis was performed on cell lysate using different antibodies as indicated (p-ASK1, S83 phosphospecific antibody for ASK1; p-JNK, phosphospecific antibody for JNK; p-p38, phosphospecific antibody for p-38). GAPDH antibody was used as a loading control. (d) Knockdown of ASK1 leads to decreased activation effect of AGP in melanoma cells. Mel007 cancer cells were transfected with small interfering RNA (siRNA) for ASK1 or control siRNA. The cells were analyzed 36–48 h after transfection to determine ASK1 protein levels. GAPDH was used as protein loading control. (e, f) At 24 h after treatment of ASK1 siRNA or control siRNA, Mel007 cells were treated with AGP (30 s) with or without pretreatment with NAC. Caspase 3/7 activity was measured by Caspase-Glo 3/7 assay, and cell viability was measured by cell titer nonradioactive cell proliferation assay. All values are mean ± SD of three independent experiments performed in triplicate. *p ≤ 0.01, **p ≤ 0.001; ANOVA. (g) 2At 4 h posttreatment of ASK1 siRNA or control siRNA, Mel007 cells were treated with AGP (30 s) with or without pretreatment with NAC. The effects of AGP on stress response targets were determined by Western blot analysis of H2AX and γ-H2AX protein levels in cancer cells. GAPDH expression was used as a loading control. In all experiments, control cells were mock treated with He gas flow only.
ASK1 and JNK/p38 kinases play important roles in DNA damage responses by inducing cyclin-dependent kinase inhibitors, and depletion of ASK1 significantly attenuates p38 activation and DNA damage response (Thornton and Rincon, 2009). ASK1 is also required for H2O2 (ROS)-induced DNA fragmentation and apoptosis. It was shown that depletion of the ROS scavenger NAC as well as of ASK1 strongly reduced DNA fragmentation and apoptosis (Noguchi et al., 2008). To determine the specific role of ASK1 in AGP-induced apoptosis, we knocked down the ASK1 gene in melanoma cells by RNA interference (RNAi; Figure 5d). First, we tested the effect of AGP on caspase 3/7 activity in ASK1-knockdown melanoma cells. The results showed that activation of caspases 3/7 induced by AGP was inhibited in ASK1-depleted melanoma cells, as in cells pretreated with intracellular ROS scavenger NAC (Figure 5e). We also observed that pretreatment of melanoma cells with either NAC or knockdown of ASK1 by RNAi inhibited the toxic effects of AGP in melanoma cells (Figure 5f). Our results also showed that AGP-induced activation of stress response protein (H2AX) was inhibited in ASK1-knockdown or NAC-pretreated melanoma cells (Figure 5g). These results show the involvement of ASK1 and downstream kinases p38 and JNK in AGP-induced apoptosis in melanoma cells and clearly reveal that ASK1 plays a critical role in selective AGP-dependent signal transduction and apoptosis in melanoma cells.
TNF pathway involved in AGP-induced activation of ASK1 signaling and apoptosis
From the results already presented, it is plausible to conclude that ASK1 is an AGP effector that activates JNK and p38 kinases. However, the mechanism of ASK1 activation by AGP is still unclear. Previous studies show that TNFR family members are involved in the activation of stress-activated protein kinases (SAPKs) and MAPKs (Natoli et al., 1997). The similarity between AGP- and H2O2-induced changes in the ASK1 signaling complex (Figure 5), together with the findings that TNF-induced activation of ASK1 depends largely on ROS, prompted us to examine whether TNF signaling may also be involved in AGP-induced activation of the ASK1 pathway (Natoli et al., 1997). Tumor necrosis factor 1 (TNFR1) blocking antibody was used to neutralize the TNFR1 or inhibit the TNF signaling pathway (Defer et al., 2007; Moh et al., 2013).
AGP stimulates the production of ROS and TNF, and both ROS and TNF activation of ASK1 can be inhibited on depletion of ROS by using free radical scavenger NAC and the TNFR1-antagonist-neutralizing antibody (Figure 6a). Similarly, AGP-induced activation of p38 and JNK signaling were suppressed in melanoma cells pretreated with NAC or antagonist anti-TNFR1 antibody (Figure 6, b and c). ASK1, p38, and JNK kinases were found in the inactive form in normal melanocytes and melanoma (Mel007) cells at basal level. Moreover, AGP treatment did not activate ASK1, p38, and JNK kinases in melanocytes (Figure 6d)
FIGURE 6:
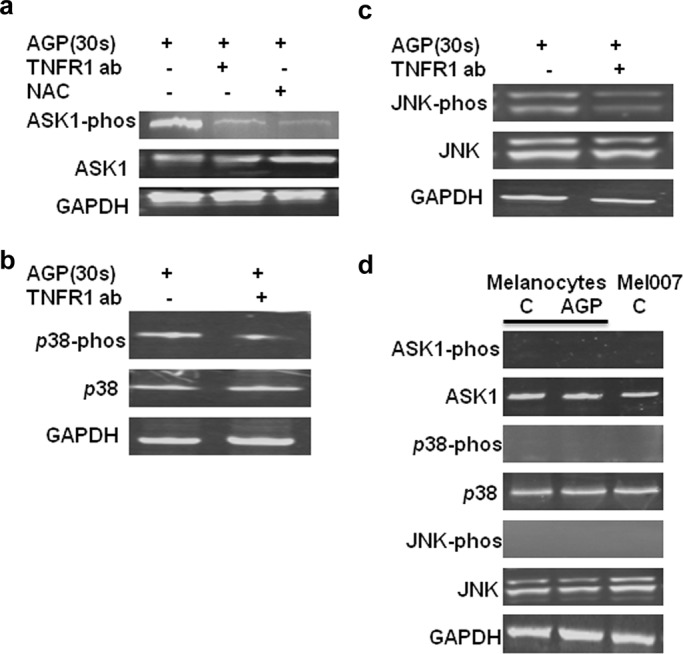
TNF signaling involved in the activation of ASK1, p38, and JNK by AGP. (a–c) Mel007 cancer cells were treated with AGP with or without pretreatment with TNFR1- neutralizing antibody or NAC. Western blot analysis was performed on cell lysate using different antibodies as indicated (p-ASK1, S83 phosphospecific antibody for ASK1; p-JNK, phosphospecific antibody for JNK; p-p38, phosphospecific antibody for p-38). GAPDH antibody was used as a loading control. (d) The basal protein expression levels for ASK1, p38, JNK, ASK1-phos, p38-phos, and JNK-phos were quantified in normal melanocytes and Mel007 cells by Western blot analysis. GAPDH antibody was used as a loading control.
These results suggest that the TNF pathway is required for AGP-induced activation of ASK1 signaling and apoptosis pathways.
DISCUSSION
Recent literature reviews show that AGPs are being studied with great interest for cancer treatment (Graves, 2012; Ishaq et al., 2014). It has been shown that AGPs increase intracellular ROS production (Vandamme et al., 2012), induce senescence (Arndt et al., 2013), cause cell cycle arrest (Volotskova et al., 2012), and induce oxidative stress, DNA damage, and apoptosis (Keidar et al., 2011; Iseki et al., 2012; Kalghatgi et al., 2012). A major obstacle in the development of this tool for clinical application as an anticancer therapeutic is the lack of understanding of the mechanisms underlying the intercellular response to AGP exposure. For cancer treatment, knowledge of the specific intracellular signaling pathway factors involved in tumor sensitivity and resistance is critical to successful therapy. This requires exploration of the intracellular mechanisms involved in the interaction of AGP with the localized cellular environment. This knowledge gap motivated our examination of the intracellular pathways involved in the sensitivity to AGP of cancer cell death, as shown in Figure 7.
FIGURE 7:
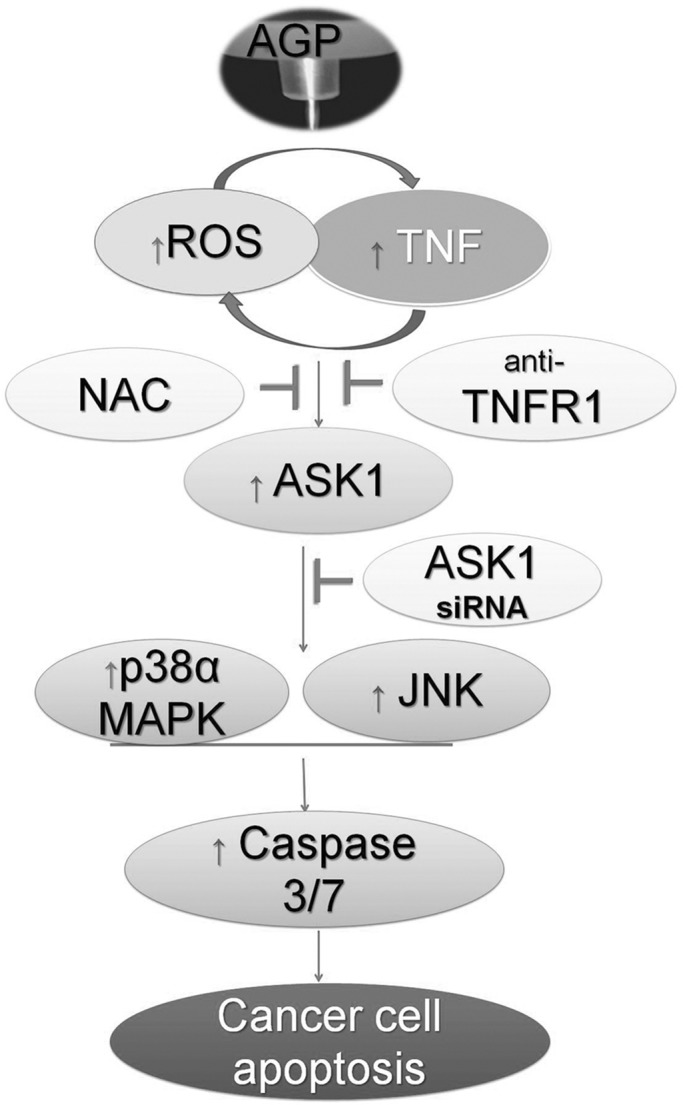
Model for the regulation of TNF-receptor–mediated apoptosis pathway induced by AGP. The induced production of ROS and TNF pathway activation by cross-talk (direct or indirect interaction), which leads to activation of ASK1 and stimulation of downstream p38α MAPK or JNK to induce caspase-3/7–dependent cancer cell apoptosis.
The AGP-produced ROS, such as H2O2 and NO, are the main factors that form a system of oxidizing or nitrosylating species, which activate several signaling pathways. Excessive ROS inside cells results in oxidative stress, which leads to DNA damage and apoptosis by unknown mechanisms (Landino et al., 1996; Kalghatgi et al., 2011; Vandamme et al., 2012). Here we found that AGP selectively induces increased ROS levels in melanoma cells compared with normal control cells, which are blocked by the endogenous ROS scavenger NAC. We also demonstrated that AGP-induced ROS are responsible for selective apoptosis of cancer cells but that this does not occur in normal control cells (Figure 3). This is the first report to show that AGPs induce ROS in cancer cells but not normal cells.
It has been suggested that ROS are involved in apoptosis of cancer cells by the TNF-dependent signaling pathway (Natoli et al., 1997). We also found in our apoptosis-related gene screening study that TNF family members are differentially expressed in AGP-treated melanoma cells. We observed that blocking TNFR1 inhibits the activity of AGP-induced caspases 3/7 and reduces activation of stress response protein H2AX (Figure 4). This indicates the involvement of the TNF-apoptotic pathway in AGP-induced apoptosis in melanoma cells.
Several reports have shown that ROS (H2O2) induced by the TNF pathway participates in the apoptosis of cancer cells in ASK1-dependent signaling (Hatai et al., 2000; Liu et al., 2000; Nishitoh et al., 2002; Kamata et al., 2005). ASK1 is required for ROS-induced activation of JNK and p38. We also found that AGPs, similar to H2O2, activate the ASK1 pathway, which results in downstream activation of JNK and p38 signaling. The AGP-induced activation of endogenous JNK and p38 is lost by blocking with NAC. Moreover, intracellular depletion of ASK1 by RNAi inhibits AGP-induced caspase 3/7 activity, cell viability, and stress response (Figure 5). These results suggest the involvement of ASK1 in the AGP-induced apoptosis pathway.
Stimulation of the TNF pathway and inducement of ROS production for the activation of ASK1 by AGPs are believed to be involved in this selective cell death process. Although TNF is known to stimulate the production of ROS, and activation of the SAPKs can be partially reversed with free-radical scavengers (Natoli et al., 1997), AGP-induced and TNF-mediated production of ROS had not been demonstrated. We found that AGP-induced ROS production and ASK1 activation was mediated by the TNF pathway. Pretreatment with antioxidant NAC and antagonist anti-TNFR1 antibody attenuated AGP-induced ASK1, p38, and JNK phosphorylation (Figure 6). These results reveal that AGP-induced production of cellular ROS is upstream of the ASK1 and p38/JNK signaling pathway.
Our results provide a plausible mechanisms for AGP-induced apoptosis in cancer cells. Extracellular ROS generated by AGP activates TNF signaling and TNF-dependent intracellular ROS generation. This high intracellular level of ROS activates caspases 3/7 via activation of ASK1 and subsequent p38/JNK pathway and eventually leads to apoptosis (Figure 7).
The precise mechanisms by which AGP increases intracellular ROS production and activate caspase-3–induced apoptosis need to be elucidated. It was reported that AGP induced apoptosis via caspase-3–dependent mechanisms (Kim et al., 2011b; Sensenig et al., 2011), but the upstream mechanisms remained unknown. Our results clearly demonstrate that ASK1 is strongly activated in AGP-treated cancer cells, similar to those treated with H2O2, and this may provide a useful model system to understand the AGP-ROS–dependent signaling pathway leading to apoptosis.
In summary, the present work demonstrates that activation of ASK1 through TNF signaling is required in AGP-induced apoptosis. This suggests the importance of TNF signaling and subsequent activation of ASK1 in AGP- and/or ROS-induced programmed cell death of melanoma cells. Further studies are required to elucidate the physiological functions of the AGP-TNF-ASK1 pathway in resistant tumor treatment.
MATERIALS AND METHODS
Atmospheric gas plasma jet device
All cells were treated with atmospheric gas plasma plume, which was generated using a custom-designed atmospheric gas plasma jet device (Figure 1). The device consists of a fused quartz tube equipped with two conducting electrodes. One electrode is a metal wire placed inside the tube along the axis and ∼1.8 cm before the nozzle exit. The second electrode is a metal ring attached to the outer wall of the tube near the nozzle exit. The distance between the two electrodes is ∼1.0 cm. On helium gas flow (2 l/min) through the quartz tube, a nonequilibrium plasma discharge is produced between the two electrodes by an applied AC high voltage. The high voltage was generated by RF power source coupled with RF voltage amplifier. The discharge voltage and operating frequency were kept at 1.1–1.8 kV and 230–270 kHz, respectively, during cell treatment. The plasma discharge produced at the end of the metal wire electrode was located in two different spatial regions. One part of the discharge was located entirely inside the tube, from the end of the metal wire electrode to the ring electrode, and part of the discharge from the ring electrode propagated along with the gas flow and extended out of the tube through the nozzle as a collimated plasma jet. The length of the plasma jet was >2.5 cm. The end diameter of this collimated plasma jet was ∼500 μm and is referred as a cold plasma due to the measured low gas temperature (∼35–40°C). During the discharge process, electrical parameters of the plasma, such as discharge voltage and current, were measured using high- and low-voltage probes, respectively. The discharge current between the metal wire and the ring electrodes was 10 mA. However, only 5–10% of this current is likely to extend out of the discharge tube, which was used for cell treatment (Keidar et al., 2013).
Cell culture and reagents
The human melanoma cell lines Mel-RM, Mel-007, and Mel-JD and control human epidermal melanocytes were a kind gift of Peter Hershey, Melanoma Institute, University of Sydney, Sydney, Australia (Jiang et al., 2010), and were maintained in DMEM (Invitrogen) plus 10% fetal bovine serum (FBS). Melanocytes were grown in special melanocyte growth medium 254 plus with human melanocyte growth supplement (S-002-05; Invitrogen, Grand Island, NY). Human fetal lung fibroblasts (MRC5) were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and grown in MEM plus 10% FBS. H2O2 (216763), caspase inhibitor Z-VAD-FMK (G7231), NAC (antioxidant and ROS scavenger; A0737), DPI (inhibitor of nitric oxide synthetase; D2926), and catalase (H2O2 depleter; C-30) were purchased from Sigma-Aldrich (St. Loius, MO). TNFR1-neutralizing antibody (Defer et al., 2007) and mouse immunoglobulin (Ig) G1 isotype control antibody were purchased from R&D Systems (Minneapolis, MN).
Cell viability assay
All cells were cultured in 96-well plates at 2 × 104 cells/well overnight, treated with AGP for indicated time periods, and incubated for 18–24 h. Cell viability was measured using CellTiter 96 Aqueous Non-Radioactive Cell Proliferation (MTS) Assay (G5421; Promega, Madison, WI) following the manufacturer's protocol. In some experiments the caspase inhibitor zVAD-FMK (50 μM), NAC (3 mM), TNFR1-neutralizing antibody (50 μg/ml), catalase (2000 U/ml), or DPI (10 μM) was added 1–2 h before AGP treatment. Assays were performed in triplicate.
ROS detection assay
Cells were cultured in 96-well plates at 2 × 104 cells/well overnight and treated with AGP for the indicated time periods. ROS generation was detected with CM-H2DCFDA (C6827; Invitrogen, Grand Island, NY) following the manufacturer's instructions. Briefly, 1–2 h after AGP treatment, cells were incubated with 10 μM of CM-H2DCFDA for 30 min at 37°C in the dark. ROS were measured by a FLUOstar Omega fluorescence plate reader at excitation and emission wavelengths of 485 and 530 nm, respectively. Assays were performed in triplicate.
Caspase 3/7 activity assay
Cells were plated in 96-well plates at 2 × 104 cells/well, incubated for 24 h, and then treated with AGP for the indicated time periods. After 4–8 h of AGP treatment, caspase 3/7 activity was quantified by adding Caspase Glo 3/7 reagent (G8091; Promega). In some experiments the caspase inhibitor zVAD-FMK (50 μM), NAC (3 mM), TNFR1-neutralizing antibody (50 μg/ml), catalase (2000 U/ml), or DPI (10 μM) was added 1–2 h before AGP treatment. Triplicate samples were run in standard 96-well plates to quantify the apoptotic response. Luminescence values were then determined using a LUMIstar Omega plate reader.
Total cellular glutathione assay
Cells were cultured in six-well plates overnight and treated with AGP, in some cases after pretreatment with NAC, for the indicated time periods. A total of 1 × 106 cells were collected and lysed in 100 μl of ice-cold lysis buffer for 10 min. At 1–2 h after the AGP treatment, supernatant was collected by centrifugation of lysate for a glutathione assay performed with a glutathione (GSH/GSSG/Total) assay kit (K264-100; BioVision Research Products, Milpitas, CA) following the manufacturer's instructions. The total amount of GSH was measured using a FLUOstar Omega fluorescence plate reader at excitation and emission wavelengths of 340 and 420 nm, respectively. Trypan blue stain was used in parallel, and the viable cells were counted.
Nitric oxide measurement
Cells were cultures in 96-well plates at 2 × 104 cells/well, incubated for 24 h, treated with AGP, and in some cases pretreated with NAC (3 mM), for the indicated time periods. Nitric oxide production was measured with 4-amino-5-methylamino-2′,7′-difluofluorescein diacetate (DAF-FM diacetate) following the manufacturer's protocols (D23844; Invitrogen). At 1–2 h after AGP treatment, cells were labeled with 10 μM DAF-FM diacetate for 30 min at 37°C. Nitric oxide radical was measured using a FLUOstar Omega fluorescence plate reader at excitation and emission wavelengths of 485 and 520 nm, respectively.
Quantitative real-time PCR
Cells were cultured in six-well plates at 5 × 105 cells/well overnight and treated with AGP for the specified time periods. At 18–24 h after AGP treatment, total cell RNA was extracted using TRIzol reagent (15596-026; Invitrogen) and reverse transcribed into single-stranded cDNA as explained previously (Ishaq et al., 2008). Quantitative relative gene expression was determined by using SsoAdvanced SYBR Green SuperMix (172-5265; Bio-Rad, Hercules, CA) with a Roche LightCycler 480 qPCR system. Data were analyzed with LightCycler 480 software. The human apoptosis PCR array primers library (HPA-1; RealTimePCR.com, Elkins Park, PA) was used to quantify relative gene expression of >90 genes involved in intracellular prosurvival and proapoptotic pathways. glyceraldehyde-3-phosphate dehydrogenase (GAPDH), actin-b, GUS-b, B2M, HPRT1, PPIA, and RPL13A were used as internal controls to normalize data. Relative quantification of gene expression after AGP treatment was obtained by using the ∆CT method compared with fold change of untreated (He gas flow only) control cells.
Enzyme-linked immunosorbent assay
Cells were cultured in six-well plates and either untreated or treated with AGP for the specified time period. After 48 h of treatment, cell culture supernatants were harvested, and the concentration of TNFα was measured by using the TNFα human enzyme-linked immunosorbent assay kit (ab46087) from Abcam (Cambridge, United Kingdom) as per the manufacturer's instructions.
RNA interference and Western blot
Cells grown to a density of ∼70% confluence with antibiotic-free media were transfected with the ASK1 short hairpin RNA (shRNA; sc-29748; Santa Cruz Biotechnology, Santa Cruz, CA) and negative control shRNA expression vector by Lipofectamine (Invitrogen) according to the manufacturers’ instructions. After 24 h of shRNA transfection, cells were treated with AGP. Cells were harvested and lysed in RIPA lysis and extraction buffer (89901), halt protease inhibitor cocktail (87786), and halt phosphatase inhibitor cocktail (78420; Thermo Scientific, Pittsburgh, PA) by incubating on ice for 30 min, as described previously (Ishaq et al., 2009). Protein concentration was determined by BCA Protein Assay Kit (23227; Thermo Scientific). After adding 2× SDS loading buffer, the samples were subjected to SDS–PAGE (MP TGX 4–20%; 4561094; Bio-Rad). Protein was then transferred onto immunoblot polyvinylidene fluoride membrane (Millipore, Billerica, MA) and probed with the primary antibodies as specified and horseradish peroxidase (HRP)–conjugated secondary antibodies. The bonded proteins were visualized with a chemiluminescence detection kit (Clarity Western ECL substrate; Bio-Rad, 170-5060) using ImageQuant LAS400 (GE Technology, Little Chalfon, United Kingdom). Antibodies used included phospho-p38 MAPK (Thr-180/Tyr-182; 4511), p38 MAPK (8690), phospho-SAPK/JNK (Thr-183/Tyr-185; 4668), SAPK/JNK (9258), phospho-ASK1 (Thr-845; 3765), ASK1 (8662), H2AX (2595), phospho-histone H2A.X (Ser-139; 2577) and GAPDH from Cell Signaling Technology (Danvers, MA) and goat anti-rabbit IgG HRP (170-6515) and goat anti-mouse IgG HRP (170-6516) from Bio-Rad. In some experiments the antioxidant NAC (3 mM), TNFR1-neutralizing antibody (50 μg/ml), or mouse IgG1 antibody as negative isotype control were added 1–2 h before AGP treatment.
Statistical analysis
All values are presented as mean ± SD of three independent experiments. Statistical differences between controls and treated groups were determined by one-way analysis of variance (ANOVA) or Student's t test where applicable. Differences were considered statistically significant for p ≤ 0.05 (*p ≤ 0.01, **p ≤ 0.001).
Supplementary Material
Acknowledgments
We thank Peter Hershey for providing melanoma cell lines and melanocytes. We also thank Penny Bean, Vijay Vaithilingam, Gail Wertz, Denise Lewy, Bernie Tuch, Peter Molloy, Sumeet Bal, Glenn Brown, and Thu Ho for useful discussions and support for the experimental work and equipment. This study was supported by the CSIRO OCE postdoctoral fellowship program, the CSIRO Science Leadership program, CSIRO Transformational Biology and Advanced Materials Transformational Capability Platforms, and the Australian Research Council.
Abbreviations used:
- AGP
atmospheric gas plasmas
- ASK-1
apoptosis signal–regulating kinase 1
- B2M
beta-2 microglobulin
- BCA
bicinchoninic acid
- CD95
cluster of differentiation 95
- CM-H2DCFDA
2′-,7′-dichlorofluorescein diacetate
- CSIRO
Commonwealth Scientific and Industrial Research Organization
- DAF-FM
4-amino-5-methylamino-2′,7′-difluofluorescein
- DPI
diphenyleneiodonium chloride
- DR4/5
death receptor 4/5
- FAS
TNF receptor superfamily, member 6
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GSH
glutathione
- GSSG
glutathione disulfide
- GUS-b
glucuronidase beta
- H2AX
H2A histone family, member X
- γ-H2AX
gamma-H2AX
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- HRP
horseradish peroxidase
- IgG
immunoglobulin G
- JNK
c-Jun N-terminal kinase
- MC
melanocytes
- MKK3
dual specificity mitogen-activated protein kinase kinase 3
- MKK4
mitogen-activated protein kinase kinase 4
- MKK6
dual specificity mitogen-activated protein kinase kinase 6
- MKK7
dual specificity mitogen-activated protein kinase kinase 7
- MRC5
human fetal lung fibroblasts
- MTS
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
- NAC
N-acetyl-l-cysteine
- OCE
Office of the Chief Executive
- p38 MAPK
p38 mitogen-activated protein kinase
- PMS
phenazine methosulfate
- PPIA
peptidylprolyl isomerase A
- qPCR
quantitative PCR
- RF
radio frequency
- RNAi
RNA interference
- ROS
reactive oxygen species
- RPL13A
ribosomal protein L13a
- SAPK
stress-activated protein kinases
- SYBR
N’,N’-dimethyl-N-[4-[(E)-(3-methyl-1,3-benzothiazol-2-ylidene)methyl]-1-phenylquinolin-1-ium-2-yl]-N-propylpropane-1,3-diamine
- TNF
tumor necrosis factor
- TNFα
tumor necrosis factor α
- TNFR1
tumor necrosis factor receptor 1
- Trx
thioredoxin
- Z-VAD-FMK
3-[2-(2-benzyloxycarbonylamino-3-methyl-butyrylamino)-propionylamino]-4-oxo-pentanoic acid
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-10-0590) on February 26, 2014.
REFERENCES
- Arndt S, Wacker E, Li YF, Shimizu T, Thomas HM, Morfill GE, Karrer S, Zimmermann JL, Bosserhoff AK. Cold atmospheric plasma, a new strategy to induce senescence in melanoma cells. Exp Dermatol. 2013;22:284–289. doi: 10.1111/exd.12127. [DOI] [PubMed] [Google Scholar]
- Defer N, Azroyan A, Pecker F, Pavoine C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J Biol Chem. 2007;282:35564–35573. doi: 10.1074/jbc.M704003200. [DOI] [PubMed] [Google Scholar]
- Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. 2006;43:143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- Graves DB. The emerging role of reactive oxygen and nitrogen species in redox biology and some implications for plasma applications to medicine and biology. J Phys D Appl Phys. 2012;45:263001–263042. [Google Scholar]
- Hatai T, Matsuzawa A, Inoshita S, Mochida Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, Ichijo H, Takeda K. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J Biol Chem. 2000;275:26576–26581. doi: 10.1074/jbc.M003412200. [DOI] [PubMed] [Google Scholar]
- Huang J, Li H, Chen W, Lv G-H, Wang X-Q, Zhang G-P, Ostrikov K, Wang P-Y, Yang S-Z. Dielectric barrier discharge plasma in Ar/O2 promoting apoptosis behavior in A549 cancer cells. Appl Phys Lett. 2011;99:253701. [Google Scholar]
- Ichijo H, Nishida E, Irie K, tenDijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Iseki S, Nakamura K, Hayashi M, Tanaka H, Kondo H, Kajiyama H, Kano H, Kikkawa F, Hori M. Selective killing of ovarian cancer cells through induction of apoptosis by nonequilibrium atmospheric pressure plasma. Appl Phys Lett. 2012;100:113702. [Google Scholar]
- Ishaq M, Evans MM, Ostrikov KK. Effect of atmospheric gas plasmas on cancer cell signaling. Int J Cancer. 2014;134:1517–1528. doi: 10.1002/ijc.28323. [DOI] [PubMed] [Google Scholar]
- Ishaq M, Hu J, Wu X, Fu Q, Yang Y, Liu Q, Guo D. Knockdown of cellular RNA helicase DDX3 by short hairpin RNAs suppresses HIV-1 viral replication without inducing apoptosis. Mol Biotechnol. 2008;39:231–238. doi: 10.1007/s12033-008-9040-0. [DOI] [PubMed] [Google Scholar]
- Ishaq M, Ma L, Wu X, Mu Y, Pan J, Hu J, Hu T, Fu Q, Guo D. The DEAD-box RNA helicase DDX1 interacts with RelA and enhances nuclear factor kappaB-mediated transcription. J Cell Biochem. 2009;106:296–305. doi: 10.1002/jcb.22004. [DOI] [PubMed] [Google Scholar]
- Jiang CC, et al. Apoptosis of human melanoma cells induced by inhibition of B-RAF(V600E) involves preferential splicing of bim(S) Cell Death Dis. 2010;1:e69. doi: 10.1038/cddis.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG. Choosing anticancer drug targets in the postgenomic era. J Clin Invest. 1999;104:1503–1506. doi: 10.1172/JCI8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalghatgi S, Fridman A, Azizkhan-Clifford J, Friedman G. DNA damage in mammalian cells by non-thermal atmospheric pressure microsecond pulsed dielectric barrier discharge plasma is not mediated by ozone. Plasma Processes Polymers. 2012;9:726–732. [Google Scholar]
- Kalghatgi S, Kelly CM, Cerchar E, Torabi B, Alekseev O, Fridman A, Friedman G, Azizkhan-Clifford J. Effects of non-thermal plasma on mammalian cells. PLoS One. 2011;6:e16270. doi: 10.1371/journal.pone.0016270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang LF, Hirata H, Karin M. Reactive oxygen species promote TNF alpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Keidar M, Shashurin A, Volotskova O, Stepp MA, Srinivasan P, Sandler A, Trink B. Cold atmospheric plasma in cancer therapy. Phys Plasmas. 2013;20:057101. [Google Scholar]
- Keidar M, Walk R, Shashurin A, Srinivasan P, Sandler A, Dasgupta S, Ravi R, Guerrero-Preston R, Trink B. Cold plasma selectivity and the possibility of a paradigm shift in cancer therapy. Br J Cancer. 2011;105:1295–1301. doi: 10.1038/bjc.2011.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Choi JD, Hong YC, Kim G, Noh EJ, Lee J-S, Yang SS. Atmospheric-pressure plasma-jet from micronozzle array and its biological effects on living cells for cancer therapy. Appl Phys Lett. 2011b;98:073701. [Google Scholar]
- Kim JY, Wei Y, Li J, Foy P, Hawkins T, Ballato J, Kim S-O. Single-cell-level microplasma cancer therapy. Small. 2011a;7:2291–2295. doi: 10.1002/smll.201100456. [DOI] [PubMed] [Google Scholar]
- Koeritzer J, et al. Restoration of sensitivity in chemo-resistant glioma cells by cold atmospheric plasma. PLoS One. 2013;8:e64498. doi: 10.1371/journal.pone.0064498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc Natl Acad Sci USA. 1996;93:15069–15074. doi: 10.1073/pnas.93.26.15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DX, Bruggeman P, Iza F, Rong MZ, Kong MG. Global model of low-temperature atmospheric-pressure He + H2O plasmas. Plasma Sources Sci Technol. 2010;19:025018. [Google Scholar]
- Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Moh MC, Lorenzini PA, Gullo C, Schwarz H. Tumor necrosis factor receptor 1 associates with CD137 ligand and mediates its reverse signaling. FASEB J. 2013;27:2957–2966. doi: 10.1096/fj.12-225250. [DOI] [PubMed] [Google Scholar]
- Nadeau PJ, Charette SJ, Landry J. REDOX reaction at ASK1-Cys250 is essential for activation of JNK and induction of apoptosis. Mol Biol Cell. 2009;20:3628–3637. doi: 10.1091/mbc.E09-03-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C, Levrero M. Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science. 1997;275:200–203. doi: 10.1126/science.275.5297.200. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Gene Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Ishii K, Fukutomi H, Naguro I, Matsuzawa A, Takeda K, Ichijo H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J Biol Chem. 2008;283:7657–7665. doi: 10.1074/jbc.M708402200. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Takeda K, Matsuzawa A, Saegusa K, Nakano H, Gohda J, Inoue J, Ichijo H. Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J Biol Chem. 2005;280:37033–37040. doi: 10.1074/jbc.M506771200. [DOI] [PubMed] [Google Scholar]
- Ostrikov K, Neyts EC, Meyyappan M. Plasma nanoscience: from nano-solids in plasmas to nano-plasmas in solids. Adv Phys. 2013;62:113–224. [Google Scholar]
- Partecke LI, et al. Tissue tolerable plasma (TTP) induces apoptosis in pancreatic cancer cells in vitro and in vivo. BMC Cancer. 2012;12:473. doi: 10.1186/1471-2407-12-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensenig R, et al. Non-thermal plasma induces apoptosis in melanoma cells via production of intracellular reactive oxygen species. Ann Biomed Eng. 2011;39:674–687. doi: 10.1007/s10439-010-0197-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sensenig R, Kalghatgi S, Goldstein A, Friedman G, Friedman G, Brooks A. Induction of apoptosis in melanoma cells by non-thermal atmospheric plasma discharge. Ann Surg Oncol. 2008;15:65–65. [Google Scholar]
- Thiyagarajan M, Waldbeser L, Whitmill A. THP-1 leukemia cancer treatment using a portable plasma device. Stud Health Technol Inform. 2012;173:515–517. [PubMed] [Google Scholar]
- Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009;5:44–51. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandamme M, et al. ROS implication in a new antitumor strategy based on non-thermal plasma. Int J Cancer. 2012;130:2185–2194. doi: 10.1002/ijc.26252. [DOI] [PubMed] [Google Scholar]
- Vandamme M, Robert E, Pesnel S, Barbosa E, Dozias S, Sobilo J, Lerondel S, Le Pape A, Pouvesle JM. Antitumor effect of plasma treatment on U87 glioma xenografts: preliminary results. Plasma Processes Polymers. 2010;7:264–273. [Google Scholar]
- Volotskova O, Hawley TS, Stepp MA, Keidar M. Targeting the cancer cell cycle by cold atmospheric plasma. Sci Rep. 2012;2:636–610. doi: 10.1038/srep00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walk RM, Snyder JA, Srinivasan P, Kirsch J, Diaz SO, Blanco FC, Shashurin A, Keidar M, Sandler AD. Cold atmospheric plasma for the ablative treatment of neuroblastoma. J Pediatr Surg. 2013;48:67–73. doi: 10.1016/j.jpedsurg.2012.10.020. [DOI] [PubMed] [Google Scholar]
- Weltmann KD, Kindel E, von Woedtke T, Hahnel M, Stieber M, Brandenburg R. Atmospheric-pressure plasma sources: prospective tools for plasma medicine. Pure Appl Chem. 2010;82:1223–1237. [Google Scholar]
- Winter J, Wende K, Masur K, Iseni S, Dunnbier M, Hammer MU, Tresp H, Weltmann KD, Reuter S. Feed gas humidity: a vital parameter affecting a cold atmospheric-pressure plasma jet and plasma-treated human skin cells. J Phys D Appl Phys. 2013;46:295401. [Google Scholar]
- Yan X, Xiong Z, Zou F, Zhao S, Lu X, Yang G, He G, Ostrikov K. Plasma-induced death of HepG2 cancer cells: intracellular effects of reactive species. Plasma Processes Polymers. 2012;9:59–66. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.