

Abstract
Once a backwater in medical sciences, aging research has emerged and now threatens to take the forefront. This dramatic change of stature is driven from three major events. First and foremost, the world is rapidly getting old. Never before have we lived in a demographic environment like today and the trends will continue such that 20% percent of the global population of 9 billion will be over the age of 60 by 2050. Given current trends of sharply increasing chronic disease incidence, economic disaster from the impending silver tsunami may be ahead. A second major driver on the rise is the dramatic progress that aging research has made using invertebrate models such as worms, flies and yeast. Genetic approaches using these organisms have led to hundreds of aging genes and, perhaps surprisingly, strong evidence of evolutionary conservation among longevity pathways between disparate species, including mammals. Current studies suggest that this conservation may extend to humans. Finally, small molecules such as rapamycin and resveratrol have been identified that slow aging in model organisms, although only rapamycin to date impacts longevity in mice. The potential now exists to delay human aging, whether it is through known classes of small molecules or a plethora of emerging ones. But how can a drug that slows aging become approved and make it to market when aging is not defined as a disease. Here, we discuss the strategies to translate discoveries from aging research into drugs. Will aging research lead to novel therapies toward chronic disease, prevention of disease or be targeted directly at extending lifespan?
Introduction
While the quest for immortality goes back thousands of years, critical thinking about why we age begins, it can be argued, with Darwin and natural selection. If only the fittest survive, why would an organism age, lose functional capacity and die. Yet aging occurs in almost every species (1). Alfred Rüssel Wallace proposed an early version of group selection, where it would be beneficial for older individuals to be eliminated so that the reproducing younger individuals can have access to a larger allotment of available resources.
The evidence for group selection with respect to aging is limited, but the question was right and the apparent answer at least in part comes from data on life expectancy. Essentially, average life expectancy among humans was under 25 and very few people died from age-related diseases (2). More prevalent causes of mortality were infectious disease, childbirth and malnutrition. There was no selective pressure to extend reproductive capacity or life expectancy since very few people lived long enough for it to matter. While beyond the scope of this review, evolutionary theories of aging have continued to evolve, with elegant hypotheses such as antagonistic pleiotropy and the disposable soma theory emerging (3, 4). Readers are encouraged to seek out the following reviews (5-7).
What is worth considering here is whether the aging process is fixed or malleable? All data collected from the wild and from experimental organisms indicates the latter: life expectancy and the intrinsic aging process are relatively easily altered. For instance, similar species in the wild can have widely divergent life expectancies based on requirements imposed by evolutionary life history traits and other environmental factors (8). Moreover, hundreds of genetic mutants have been identified with longer lifespan and often longer healthspan, the disease free and highly functional period of life (9).
Against this backdrop, a dramatic change in demography has occurred within the last two centuries. Never before have humans been so old. Life expectancy has surpassed 80 in many countries and, coupled with a declining birthrate in many of those same countries, the percentage of seniors is skyrocketing. In the near future, up to 40% of the Japanese and Korean populations will be over 65 and the rest of the developed world will not be far behind (http://www.who.int/ageing/publications/global_health.pdf). Using current projections, two of the nine billion people on the planet in 2050 will have lived at least six decades.
Older people offer experience and wisdom, but are also increasingly beset with chronic disease. Aging itself is the biggest risk factor for most of the leading causes of disease burden and mortality, including cardiovascular and neurodegenerative disease, metabolic syndromes and most forms of cancer. Add to that societal changes leading to over nutrition, lack of exercise and stress and the result is that most people over 65 in the US have 1-3 chronic diseases (http://www.who.int/ageing/publications/global_health.pdf). This reduces their productivity and dramatically escalates health care costs. In summary, the silver tsunami threatens to leave wrecked economies in its wake.
One partial solution is to keep people healthy longer and aging research may have much to offer. By delaying aging, it may be possible to prevent the onset of chronic diseases and increase healthspan. Healthy seniors could work longer at high rates of productivity and would certainly reduce the burden of healthcare. But aligning medical research on aging with prevention has its challenges. How do drugs that slow aging make it to market when aging is a slow process and not even recognized as a disease by the FDA. Prevention trials for chronic diseases are also slow and often expensive. Will drugs that slow aging be effective for treatment of chronic diseases? It is not obvious that a drug which slows aging will have any impact after an age-related disease is already creating havoc.
In this review, we cover briefly the progress in studies on the genetics of aging and then turn to small molecules that modulate aging. The two best studied, rapamycin and resveratrol will be discussed in some detail. Finally, we return to the question of the potential utility of aging drugs, making the argument that they will be effective therapeutic agents for disease states, but maybe not for the reasons that are most obvious.
The Genetics of Aging
Before active pursuit of aging in invertebrates began in earnest, much of our understanding of the molecular and genetic events driving aging was based on correlative studies of young and old animals. However, starting in the 1960s, genetic studies of aging Drosophila melanogaster and Caenorhabditis elegans, simpler and cheaper model organisms, began to yield insights (10-14). Studies in yeast replicative aging, the number of times one mother cell can divide and produce a daughter (15, 16), date back to 1959 and turned to genetics as well (17, 18). Yeast chronological aging, the survival of yeast cells in a post-replicative environment (15, 19), has also proven highly informative. It was clear from these studies that extending lifespan was possible through methods such as mutagenesis and screening (10, 12, 20), or through selection, for instance, by isolating flies that maintain late reproductive capacity over many generations (13, 21).
The first longevity mutants began to be isolated in C. elegans and for yeast replicative aging in the 1990s and this field of research has gained steam in dramatic fashion ever since (11, 20). Worms and yeast have led the way in part because they are amenable to whole genome screening for longevity and hundreds of genes whose reduced expression lead to lifespan extension in both organisms have been identified (22-30). Critically, in many cases ortholog families have been identified that modulate aging across multiple species (31, 32). In addition, quantitative evidence has been generated that longevity pathways are conserved between C. elegans and yeast (replicative aging) (33). These results provide a demonstration that the aging process has significant overlap between disparate species, although there will certainly be unique features as well. By studying the conserved ones, it is likely possible to gain major insights into the human aging process.
Indeed research is starting to bear out this assertion. Four of the major pathways known to influence aging are insulin/IGF signaling (IIS) (11, 34-36), Target of Rapamycin (TOR) (22, 23, 37-40), the protein kinase A (PKA) pathway (41, 42) and the protein deacetylase SIR2 (43-45). All four of these were defined first in invertebrates but evidence for the first three affecting aging has since followed in mice and even humans. The case for SIR2 and its orthologs is more complex, but also promising (see below). Mice with reduced IIS, mTOR or PKA signaling all have extended lifespan and a majority of evidence suggests they are healthier and protected from many age-related diseases (46-51).
For humans, most evidence comes from genetic studies of centenarians. Mutations in the FOXO3A gene, encoding a transcription factor downstream of IIS signaling have been associated with enhanced longevity (52-57). Further linking the IIS pathway to human longevity, centenarians are more likely to have IGF receptor mutations (58). In this case, these mutations are known to be hypomorphic, IGF signaling is reduced when they are introduced into cells in culture. With respect to the PKA pathway, Zhao et al. identified hypomorphic mutations in the β-adrenergic receptor, ADRB2, that are predicted to lead to reduced PKA signaling (59). All of these findings are consistent with the theory that longevity pathways are conserved among eukaryotes, implying that interventions to slow aging in animal models may have a similar effect in humans. In the next sections, we examine the two primary pharmacological interventions that affect aging and age-related disease in animal models.
Rapamycin and the Tor Pathway
The story of rapamycin starts with a wide-ranging scientific expedition by a group of Canadians to Easter Island in the 1960s and a soil sample that proved to have an activity capable of killing eukaryotic cells. That activity was later attributed to a small molecule, rapamycin, which was produced by bacteria (60). Since its discovery, rapamycin has been the focus of intense research both from academics and pharmaceutical companies. Clinical trials have been performed with rapamycin and derivatives (rapalogs) in a wide range of disease conditions and while side effects of treatment are significant, the class of drugs has been approved for several disease indications (61, 62).
A major discovery that advanced the field involved the identification of the Target Of Rapamycin (TOR) kinase and the vast biology that emerged from this line of investigation. Here we will not cover TOR signaling in depth and readers are directed to several recent reviews (63, 64). The finding that reduced TOR signaling enhanced longevity in yeast, worms and flies suggested that reduced TOR signaling might have a similar effect in mammals (22, 23, 37-40). The NIA Intervention Testing Program tested the small molecule during mouse aging and rapamycin turned out to be the first robust hit, extending lifespan by 15% in females and 10% in males, even when administered relatively late in the lifespan of a mouse (20 months) (47). Rapamycin has also been reported to extend lifespan in another mouse strain (48), as has deletion of the downstream TOR target, S6 kinase 1 (S6K1) (65). Moreover, in physiological aging studies, rapamycin is reported to delay a subset of age-associated pathologies, including neurodegenerative diseases, age-related cardiac hypertrophy and others (66-71). It is not a panacea for chronic disease, however as chronic administration does not affect phenotypes including kyphosis and may accelerate others such as cataracts (66, 70).
There are two TOR complexes, and the majority of evidence indicates that it is reduced TORC1, and not TORC2, activity that promotes longevity (61, 63). Rapamycin primarily inhibits TORC1 but upon chronic administration can inhibit TORC2 signaling as well (72). TORC1 is ideally suited to modulate aging processes. Among upstream regulatory pathways that control TORC1 activation are extracellular nutrient levels including carbohydrates (mediated through the IIS pathway) and amino acids (63, 64, 73). High nutrients and activated TORC1 translates to rapid cell growth and proliferation. A rapamycin-mediated reduction in TORC1 signaling may thus phenocopy dietary restriction, known to enhance longevity in a wide range of organisms for many decades. The jury remains out on this hypothesis and perhaps the best interpretation of current data is that dietary restriction and rapamycin-mediated TORC1 inhibition extend lifespan through overlapping but non-identical effects (74, 75). Other inputs controlling TORC1 activation include stress sensors such as MAP kinase and AMP kinase signaling (76, 77).
Downstream of TORC1 are a range of pathways that permit control of cell growth and proliferation, as well as stress response pathways. For instance, activated TORC1 leads to enhanced protein translation and cell cycle entry, whereas reduced TORC1 activity instead enhances autophagy and proteasome-mediated turnover (63, 64, 73). Thus, TORC1 is poised to integrate environmental signals and modulate downstream responses accordingly. Although likely something of an oversimplification, high TORC1 equates to rapid growth, reproduction and aging whereas reduced TORC1 signaling delays growth and enhances cellular stress response pathways leading to enhanced longevity.
Rapamycin and derivatives have been and continue to be tested in a wide range of clinical trials for numerous chronic disease indications, and the drugs have already been approved for uses in cancers including renal carcinomas and to inhibit restenosis after implantation of stents during angioplasty (61). It is also used to prevent organ transplant rejection in combination with other potent immunosuppressants such as cyclosporine A (78). In addition, chronic rapalog administration results in a range of side effects such as hyperglycemia and dyslipidemia. Interestingly, at least some of the side effects appear to be through inhibition of hepatic TORC2 (79),. Whether rapalogs prove effective to slow aging in humans remains to be determined, but the results establish proof-in-principle that it is possible to slow aging and, when achieved, delay the onset or progression of at least a subset of age-related chronic diseases.
Resveratrol, Stacs and Sirtuins
Overexpression of SIR2 enhances replicative lifespan in yeast (43). Sir2 is the founding member of a class of protein deacetylases termed Sirtuins. Eukaryotic species have multiple Sirtuins (80), but it is SIR2 orthologs that have been linked to aging in worms and flies, albeit controversially. Some studies report that overexpression of Sir.2-1 extends lifespan in C. elegans, but others have failed to replicate this finding (45, 81, 82). Similar findings have been reported with Sir2 in flies (81, 83, 84). The reasons for these discrepancies remain to be fully elucidated. In mice, the closest ortholog by sequence homology is SIRT1, however constitutive expression failed to extend lifespan in mice (85). Recently, it was reported that overexpression of SIRT1 in the brain enhances longevity likely through enhanced hypothalamic function with age (86). Overexpression of another Sirtuin, SIRT6, also extends lifespan, but only in male mice (87).
The functions of Sirtuins linked to enhanced longevity remain to be elucidated. In yeast, enhanced Sir2 activity suppresses rDNA recombination (43), reducing the number of extrachromosomal rDNA circles, which accelerate aging through still poorly defined mechanisms (88). However, there are other functions of yeast Sir2 that have been linked to longevity, including possible effects on gene expression near telomeres and on replication at ARS elements in rDNA repeats (89, 90). In worms, Sir.2-1 overexpression may lead to enhanced function of the FOXO ortholog, DAF-16, possibly linking the Sirtuin pathway to IIS signaling (45). In flies, the mechanisms also remain poorly defined but involve interaction with another deacteylase, Rpd3 (44).
In mice, the mechanisms underlying the longevity effects of SIRT1 overexpression in the brain likely involve enhanced hypothalamic neural activity, which protects and maintains mitochondrial function in aging skeletal muscle (86). It also remains possible that tissue-specific SIRT1 overexpression may affect aging directly. Also of note, deletion of SIR2 orthologs have pronounced metabolic effects and may be required for lifespan extension by dietary restriction, depending on the DR regimen that is chosen and other unidentified factors (91, 92).
In a screen for SIRT1 activators, the molecule resveratrol was identified to enhance the SIRT1 activity in an in vitro assay using acetylated non-native substrates (93). The substrate was engineered for high throughput screening and involved the release of a fluorescent moiety triggered by the deacetylase reaction. A series of other molecules, named Sirtuin Activating Compounds (STACs), were also identified that could activate SIRT1. These molecules have triggered a wave of studies reporting potentially beneficial effects in chronic disease assays, and several clinical trials have been initiated. See recent reviews for a more complete description of these studies (80, 94).
The activity of resveratrol and STACs toward SIRT1 has also been the subject of great controversy, starting with the discovery that these molecules generally fail to activate SIRT1 toward native peptides in vitro (95, 96). Follow up studies suggested that in the original screen, the non-native fluorescent moiety interfered with SIRT1 deacetylation and that resveratrol and STACs alleviate that interference. In vivo, however, these same molecules could promote deacetylation of SIRT1 substrates and their physiologic properties were often, but not always, found to be SIRT1-dependent (97). A recent report may have resolved this controversy, finding that a subset of native substrates have amino acids that act similarly to the fluorescent moiety in the screen (98). Therefore, resveratrol and STACs appear to demonstrate selective SIRT1 activation.
To date, resveratrol and STACs have been reported not to extend mouse lifespan (99), with data in non-vertebrate organisms conflicting (100). Specifically, a large study by the NIA Intervention Testing Program was unable to detect enhanced longevity in mice (99, 101). Given the recent report that brain-specific SIRT1 expression extends mouse lifespan, it is possible that other STACs will have a longevity effect, but further studies will be needed to address this question in detail. Interestingly, resveratrol does extend lifespan in one fish model, the annual fish Nothobranchius (102, 103).Regardless of their impact on lifespan, the Sirtuin activating compounds represent another intriguing class of small molecules derived from aging studies that may have significant clinical applications.
Other Drugs Linked to Aging
Within the next few years, several more drugs likely will be reported to extend mouse lifespan. However, two already widely used drugs are worth discussing. One, metformin, has recently been reported to increase male mouse median and maximum lifespan by approximately 5% (104). This is the latest and most definitive of several studies examining the effects of metformin on lifespan, with a variety of results observed (105). It is particularly relevant since metformin is a widely used drugs for type II diabetes and relatively safe for human administration. Also consistent with the possibility that metformin modulates aging, it has been reported to be beneficial in more than one age-related disease. Reports from clinical studies indicate that the drug also reduces risk of cardiovascular disease and cancer (106, 107). Although other activities have been reported, metformin is primarily thought of as an activator of AMP kinase, which responds to cellular energy deficits to mediate starvation responses in the cell (108). Therefore, as proposed for both rapamycin and resveratrol, metformin can be considered a dietary restriction mimetic.
The case for statins as an aging drug can also be proposed. Statins inhibit HMG-CoA reductase, leading to reduced levels of low density lipoprotein (LDL) associated cholesterol and are widely used with efficacy in cardiovascular disease states (109, 110). Statins may also prevent cellular senescence, possibly through reducing levels of reactive oxygen species and/or stabilizing telomere structures (111). Although the NIA Intervention Testing Program was unable to detect any longevity benefit with simvastatin (99), statins have been reported in human clinical trials to protect in some cases against other age-related diseases including dementias and forms of cancer. It has also been suggested that these drugs should be administered in a widespread fashion to individuals above 50. However, there are several notes of caution (109, 110). First, there are significant although manageable side effects associated with statins in a subset of patients. In addition, the protective effects of statins against diseases other than CVD are not always seen in clinical studies and finally, it has been debated whether statins are beneficial for CVD in individuals over the age of 80 (109). While further studies are certainly warranted, it seems premature to classify statins as anti-aging drugs at this juncture.
How Does Aging Research Translate to Clinical Applications?
The last two decades of research have led to the following hypotheses: (1) aging can be delayed in animal models (including mammals) with genetic interventions and small molecules; (2) the pathways modulating aging are at least partially conserved in eukaryotes; and (3) aging is a common cause of many if not most chronic diseases that are the leading contributors to morbidity and mortality. The latter assertion is based on findings that interventions delaying aging in animal models protect against chronic diseases. Put this together and the potential is obvious: develop interventions to slow human aging as a means of preventing disease and extending healthspan. This approach could improve quality-of-life while simultaneously lowering healthcare costs. Whether aging drugs will also serve as treatment for diseases is another issue, addressed below.
But this approach does not blend well with current drug development strategies, which are primarily aimed at treatment strategies. Many of these treatments are accompanied by high costs and some fail to significantly improve quality-of-life. In fact, approval of drugs aimed at chronic diseases has occurred at a slow pace. Approval of drugs aimed at prevention is feasible, but these studies are often shunned to extended trial times and high costs. Finally, even if a drug existed that extended lifespan without side effects, it would be hard to get it approved as aging is not a disease indication. Thus the conundrum - there is a disconnect between the likely benefits of drugs that slow aging and likely strategies that would get them approved for human use (Figure 2).
Figure 2. The translational promise of aging research is mis-aligned with current approaches to drug development.
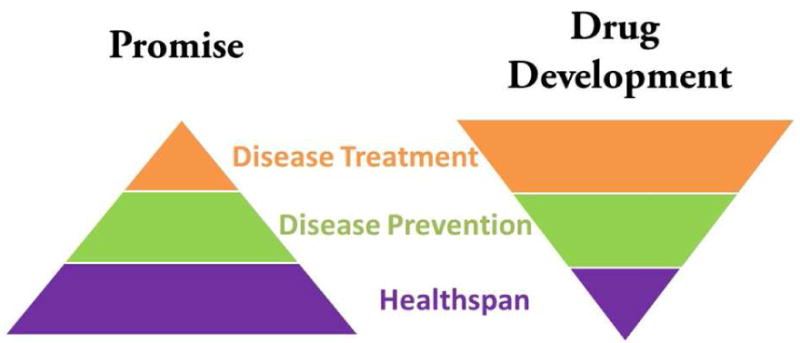
By manipulating the pathways that underlie and promote aging, researchers hope to extend healthspan, the disease free and highly functional period of life (purple). This approach will likely lead to interventions that prevent the onset of age-associated diseases (green). Less clear is whether treatments for disease will be generated (orange). However, most drug development is focused on disease treatment, with a smaller emphasis on prevention in part due to the high cost of often lengthy trials. The concept of providing drugs to healthy people to keep them healthy longer has yet to emerge as a mainstream approach.
This has led the small number of researchers focused on aging to take hits and direct them at treatment of chronic diseases. But why should this work? Aging may indeed enable disease, but why should the specific molecular pathology that accompanies chronic diseases be mitigated by methodologies slowing aging. Early evidence, however, suggests that this strategy might work. In this section, we lay out a hypothesis for why drugs that slow aging may be viable treatment options for chronic diseases and it comes with a twist - it may not be the diseases of aging for which small molecules extending lifespan show the most efficacy.
As an example, we describe a recent study we reported examining a mouse model of Emery-Dreifuss Muscular Dystrophy (EDMD2/3) and Dilated Cardiomyopathy (CDM1A) (112). These diseases are caused by mis-sense mutations in the LMNA gene, which encodes the nuclear intermediate filaments A-type lamins (113, 114). Interestingly, there are over a dozen diseases associated with mutations in LMNA, including multiple progeroid syndromes such as Hutchinson-Gilford Progeria Syndrome (HGPS). The motivation for the study was to test rapamycin in a mouse progeria model, reasoning that a drug that affects normal aging may be highly efficacious in progeria. As a control, with the intent of showing specificity for rapamycin toward HGPS, we also tested the Lmna−/− mice (115), which succumb to conduction defects due to the dilated cardiomyopathy after 6-8 weeks (116). The surprise, rapamycin was ineffective in the progeria model (unpublished observation), but highly effective in extending survival as well as improving cardiac function in the Lmna−/− mice (112, 117). Why should rapamycin work for this disease, which is not associated with aging? The likely answer, the TORC1 pathway is aberrantly upregulated in the Lmna−/− mice (112), and in another Lmna mutant mouse model as well as tissue from human patients (117). The connection between A-type lamin and TORC1 is still being elucidated, but the reason rapamycin works is readily apparent - it suppresses elevated TORC1 activity in pathologic tissues.
Interestingly, TORC1 activity may also be upregulated in a variety of aging cells and tissues, including the mouse liver, hematopoietic stem cells and others (48, 118). In the initial NIA Intervention Testing Program study, rapamycin was only administered at 20 months of age (47), leading many to predict even bigger benefits associated with earlier administration. Yet this proved largely incorrect. Rapamycin administration starting at 9 months only led to lifespans slightly longer than when given at 20 months (99). From these findings, it may be that the benefits of rapamycin come not from reduced steady-state TORC1 signaling in young animals, but rather from suppression of age-associated aberrant upregulation. While more studies are needed to confirm this hypothesis, it presents an intriguing picture of the genetic pathways identified to modulate aging.
While TORC1 becomes elevated at least in a subset of aging tissues, Sirtuin activity declines. For instance, Sir2 degradation is enhanced during yeast replicative aging, leading to reduced activity and accompanying phenotypes (89). Similarly, SIRT1 function declines with age in at least a subset of mouse tissues (119, 120). Interestingly, while it has been relatively easy to identify conserved longevity pathways, determining why they affect aging and how they interface with mechanistic hypothesis of aging such as oxidative damage, altered proteostasis, telomere shortening etc. has been very challenging. One possibility is that Sirtuin and TOR dysfunction are not the primary events that drive aging but rather the secondary responses to those events. In other words, these are the pathways that go wrong when things go wrong, and moreover, that once aberrantly regulated they further contribute to the pathology of aging. Suppressing this dysfunction therefore enhances lifespan and healthspan.
If this is the case, there may be significant overlap between aging and disease states - the same pathways may be mis-regulated. This may be the case at least with both TOR and Sirtuins, whose altered function has been linked to pathology in a wide range of disease conditions. Of course, the concept will require testing with other longevity pathways as they emerge, but if true, it leads to a reasonable explanation of why small molecules that slow aging will be effective therapeutic agents, as well as a path to approval. Moreover, it suggests that screening for delayed aging may yield a wide range of new pathways and chemical classes for chronic diseases. Finally, it implies that the molecules slowing aging may be as likely to have efficacy for non-aging diseases as ones associated with aging.
Ultimately, the focus has to turn to healthspan, either tested directly or through disease prevention studies. Avoiding disease rather than slowing progression is an economic and a quality-of-life winning ticket. This will require a change of thinking in drug development, including an increased tolerance for prevention trials and acceptance of new parameters of aging, such as frailty and altered biomarkers, as disease indications. This latter issue remains a major challenge for aging and longevity specifically. While many efforts involving both unbiased large-scale and targeted hypothesis-driven studies have been performed, there remains a dearth of solid biomarkers that are predictive of longevity in mice or humans. Studies of miRNA levels in serum may prove to be a useful approach to generate these critically needed biomarkers. In hand, it would be feasible to examine whether anti-aging drugs reduce biomarkers of aging in human trials.
Aging research has unique features. Not only does it touch upon the major chronic disease states that are currently plaguing humanity, but it offers the potential of aligning medical research with disease prevention. After all, at least in the cases studied to date, slowing aging is coupled with extending healthspan. The best way to treat a major debilitating disease is to not get it in the first place and aging research has the potential to help achieve that goal. With new small molecule interventions that slow aging emerging, the opportunity is there for aging research to revolutionize medicine. Rapamycin derivatives and STACs may or may not be appropriate to extend healthspan in humans, but as more and more candidates emerge, the likelihood dramatically increases that one will work in humans.
Methods
This paper conforms to the relevant ethical guidelines for animal research.
Figure 1. Possibilities for lifespan extension.
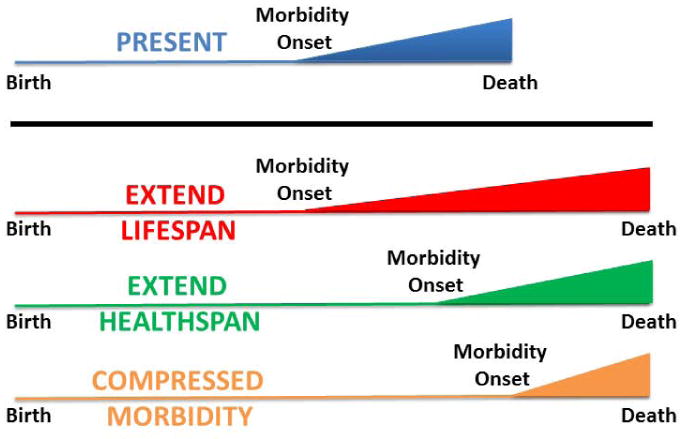
In the diagram (Blue - normal lifespan, the total lifespan is depicted as the length of the line, whereas morbidity onset associated with chronic diseases occurs at the inflection point. There are three ways in which lifespan might theoretically be extended. In red, lifespan extended with no alteration in the onset of morbidity. This is the most non-preferable option since it creates a prolonged period of high healthcare costs and low quality of life. In green, lifespan extension is accompanied by a coordinate delay in the onset of morbidity. Thus, healthspan is enhanced relative to morbidity. Many animal models of enhanced longevity appear to fit this paradigm. Finally, in orange, lifespan is extended with a reduction in the period of chronic disease, a condition termed compressed morbidity. While the most preferable, this result has not been consistently achieved in animal models to date.
Figure 3. What goes wrong when things go wrong.
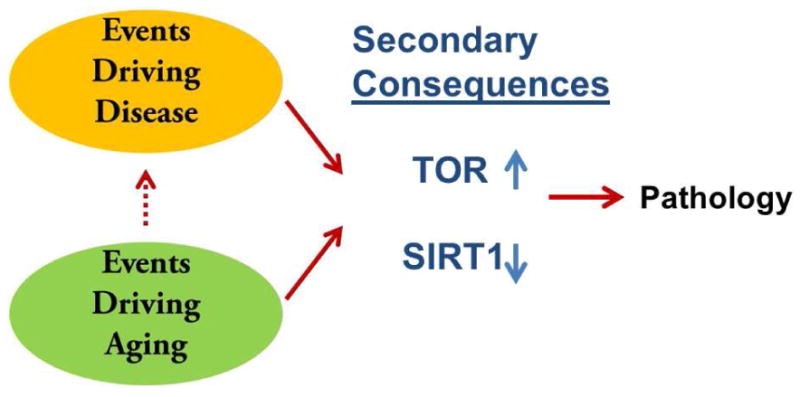
From recent studies, we put forward the hypothesis that major aging pathways that have been discovered, TOR signaling and SIRT1-dependent protein deacetylation, may be linked not only to normal aging processes but also over-represented in disease states. With mammalian aging, it is emerging that TOR signaling is elevated aberrantly and SIRT1 activity is reduced. These may occur in response to the primary molecular events that underlie aging, but appear not to be beneficial compensations. Restoring SIRT1 activity of suppressing elevated TOR signaling suppresses aging pathology, extends lifespan or both. Primary aging molecular events almost certainly promote many disease states as well. Interestingly, however, aberrant TOR and SIRT1 function may be an enriched occurrence in a range of diseases, even ones not associated with aging. If true and if the trend continues, it suggests that a new approach to therapy may be possible. Aging pathways may emerge as targets for a wide range of disease indications, not all of which age-related. Thus it may be possible to identify small molecules that target aging pathways, such as rapamycin and resveratrol, and then use them to probe disease indications for efficacious outcomes. If so, aging research will yield therapeutic strategies, but not in the way expected.
Acknowledgments
All authors have read the journal's policy on conflicts of interest and have none to declare. No editorial support was used in the preparation of this manuscript.
Abbreviations
- FDA
Food and Drug Administration
- IGF
Insulin-like Growth Factor
- SIR2
Silent Information Regulator Two
- IIS
Insulin/IGF Signaling
- PKA
Protein Kinase A
- mTOR
Mammalian Target Of Rapamycin
- TORC1
Target of Rapamycin Complex 1
- TORC2
Target of Rapamycin Complex 2
- ADRB2
Beta-Adrenergic Receptor
- NIA
National Institute on Aging
- S6K1
S6 Kinase 1
- MAP Kinase
Mitogen-activated Protein Kinase
- AMP Kinase
5′ Adenosine Monophosphate-activated Protein kinase
- STACs
Sirtuin Activating Compounds
- DR
Dietary Restriction
- LMNA
Lamin A/C
- HGPA
Hutchinson-Gilford Progeria Syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Finch CE. Longevity, Senescence and the Genome. Chicago, IL: Univ. Chicago Press; 1990. [Google Scholar]
- 2.Finch CE. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci U S A. 2010 Jan 26;107(Suppl 1):1718–24. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 4.Kirkwood TB. Evolution of ageing. Nature. 1977;270:301–4. doi: 10.1038/270301a0. [DOI] [PubMed] [Google Scholar]
- 5.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–47. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 6.Kirkwood TB, Austad SN. Why do we age? Nature. 2000 Nov 9;408(6809):233–8. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 7.Ljubuncic P, Reznick AZ. The evolutionary theories of aging revisited--a mini-review. Gerontology. 2009;55(2):205–16. doi: 10.1159/000200772. [DOI] [PubMed] [Google Scholar]
- 8.Austad SN. Diverse aging rates in metazoans: targets for functional genomics. Mech Ageing Dev. 2005 Jan;126(1):43–9. doi: 10.1016/j.mad.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 9.Kennedy BK. The genetics of ageing: insight from genome-wide approaches in invertebrate model organisms. J Intern Med. 2008 Feb;263(2):142–52. doi: 10.1111/j.1365-2796.2007.01903.x. [DOI] [PubMed] [Google Scholar]
- 10.Klass MR. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech Ageing Dev. 1983 Jul-Aug;22(3-4):279–86. doi: 10.1016/0047-6374(83)90082-9. [DOI] [PubMed] [Google Scholar]
- 11.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366(6454):461–4. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 12.Friedman DB, Johnson TE. Three mutants that extend both mean and maximum life span of the nematode, Caenorhabditis elegans, define the age-1 gene. J Gerontol. 1988;43:B102–B9. doi: 10.1093/geronj/43.4.b102. [DOI] [PubMed] [Google Scholar]
- 13.Rose MR, Charlesworth B. Genetics of life history in Drosophila melanogaster. II. Exploratory selection experiments. Genetics. 1981 Jan;97(1):187–96. doi: 10.1093/genetics/97.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark AM, Gould AB. Genetic control of adult life span in Drosophila melanogaster. Exp Gerontol. 1970 Jul;5(2):157–62. doi: 10.1016/0531-5565(70)90004-5. [DOI] [PubMed] [Google Scholar]
- 15.Longo VD, Shadel GS, Kaeberlein M, Kennedy B. Replicative and Chronological Aging in Saccharomyces cerevisiae. Cell Metab. 2012 Jul 3;16(1):18–31. doi: 10.1016/j.cmet.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaeberlein M. Lessons on longevity from budding yeast. Nature. 2010 Mar 25;464(7288):513–9. doi: 10.1038/nature08981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mortimer RK, Johnston JR. Life span of individual yeast cells. Nature. 1959;183:1751–2. doi: 10.1038/1831751a0. [DOI] [PubMed] [Google Scholar]
- 18.Sun J, Kale SP, Childress AM, Pinswasdi C, Jazwinski SM. Divergent roles for RAS1 and RAS2 in yeast longevity. J Biol Chem. 1994;269:18638–45. [PubMed] [Google Scholar]
- 19.Longo VD, Gralla EB, Valentine JS. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J Biol Chem. 1996 May 24;271(21):12275–80. doi: 10.1074/jbc.271.21.12275. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy BK, Austriaco NR, Zhang J, Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80(3):485–96. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- 21.Luckinbill LS, Clare MJ. Selection for life span in Drosophila melanogaster. Heredity (Edinb) 1985 Aug;55(Pt 1):9–18. doi: 10.1038/hdy.1985.66. [DOI] [PubMed] [Google Scholar]
- 22.Kaeberlein M, Powers RW, 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005 Nov 18;310(5751):1193–6. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 23.Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006 Jan 15;20(2):174–84. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamilton B, Dong Y, Shindo M, et al. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005 Jul 1;19(13):1544–55. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003 Jan;33(1):40–8. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 26.Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 2005 Jul;1(1):119–28. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dillin A, Hsu AL, Arantes-Oliveira N, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002 Dec 20;298(5602):2398–401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 28.Chen D, Pan KZ, Palter JE, Kapahi P. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell. 2007 Aug;6(4):525–33. doi: 10.1111/j.1474-9726.2007.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007 Apr 6;3(4):e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim Y, Sun H. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell. 2007;6:489–503. doi: 10.1111/j.1474-9726.2007.00302.x. [DOI] [PubMed] [Google Scholar]
- 31.Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010 Apr 16;328(5976):321–6. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenyon CJ. The genetics of ageing. Nature. 2010 Mar 25;464(7288):504–12. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 33.Smith ED, Tsuchiya M, Fox LA, et al. Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res. 2008 Apr;18(4):564–70. doi: 10.1101/gr.074724.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris JZ, Tissenbaum HA, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382(6591):536–9. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- 35.Clancy DJ, Gems D, Harshman LG, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001 Apr 6;292(5514):104–6. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 36.Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292(5514):107–10. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- 37.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 38.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 40.Meissner B, Boll M, Daniel H, Baumeister R. Deletion of the intestinal peptide transporter affects insulin and TOR signaling in Caenorhabditis elegans. J Biol Chem. 2004 Aug 27;279(35):36739–45. doi: 10.1074/jbc.M403415200. [DOI] [PubMed] [Google Scholar]
- 41.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289(5487):2126–8. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 42.Gendron CM, Minois N, Longo VD, Pletcher SD, Vaupel JW. Biodemographic trajectories of age-specific reproliferation from stationary phase in the yeast Saccharomyces cerevisiae seem multiphasic. Mech Ageing Dev. 2003;124:1059–63. doi: 10.1016/j.mad.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 43.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–80. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci, USA. 2004;101:15998–6003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410(6825):227–30. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 46.Enns LC, Morton JF, Treuting PR, et al. Disruption of protein kinase A in mice enhances healthy aging. PLoS One. 2009;4(6):e5963. doi: 10.1371/journal.pone.0005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009 Jul 16;460(7253):392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2(98):ra75. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan L, Vatner DE, O'Connor JP, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007 Jul 27;130(2):247–58. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 50.Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384(6604):33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 51.Bluher M, Kahn BB, Kahn RC. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–74. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Wang WJ, Cao H, et al. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum Mol Genet. 2009 Dec 15;18(24):4897–904. doi: 10.1093/hmg/ddp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soerensen M, Dato S, Christensen K, et al. Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell. 2010 Dec;9(6):1010–7. doi: 10.1111/j.1474-9726.2010.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pawlikowska L, Hu D, Huntsman S, et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell. 2009 Aug;8(4):460–72. doi: 10.1111/j.1474-9726.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anselmi CV, Malovini A, Roncarati R, et al. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009 Apr;12(2):95–104. doi: 10.1089/rej.2008.0827. [DOI] [PubMed] [Google Scholar]
- 56.Flachsbart F, Caliebe A, Kleindorp R, et al. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2700–5. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willcox BJ, Donlon TA, He Q, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008 Sep 16;105(37):13987–92. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suh Y, Atzmon G, Cho MO, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A. 2008 Mar 4;105(9):3438–42. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao L, Yang F, Xu K, et al. Common genetic variants of the beta2-adrenergic receptor affect its translational efficiency and are associated with human longevity. Aging Cell. 2012 Dec;11(6):1094–101. doi: 10.1111/acel.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975 Oct;28(10):721–6. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 61.Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009 Oct;1790(10):1067–74. doi: 10.1016/j.bbagen.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013 Mar 1;123(3):980–9. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013 Jan 17;493(7432):338–45. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012 Apr 13;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Selman C, Tullet JM, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009 Oct 2;326(5949):140–4. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flynn JM, O'Leary MN, Zambataro CA, et al. Late life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013 Jun 4; doi: 10.1111/acel.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009 Jan;16(1):46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 68.Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5(4):e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010 Jan 20;30(3):1166–75. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilkinson JE, Burmeister L, Brooks SV, et al. Rapamycin slows aging in mice. Aging Cell. 2012 Aug;11(4):675–82. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y, Bokov A, Gelfond J, et al. Rapamycin Extends Life and Health in C57BL/6 Mice. J Gerontol A Biol Sci Med Sci. 2013 May 16; doi: 10.1093/gerona/glt056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006 Apr 21;22(2):159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 73.Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011 Dec;189(4):1177–201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gallinetti J, Harputlugil E, Mitchell JR. Amino acid sensing in dietary-restriction-mediated longevity: roles of signal-transducing kinases GCN2 and TOR. Biochem J. 2013 Jan 1;449(1):1–10. doi: 10.1042/BJ20121098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blagosklonny MV. Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans) Cell Cycle. 2010 Feb 15;9(4):683–8. doi: 10.4161/cc.9.4.10766. [DOI] [PubMed] [Google Scholar]
- 76.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002 Jul 5;277(27):23977–80. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 77.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011 Jun;36(6):320–8. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Touzot M, Soulillou JP, Dantal J. Mechanistic target of rapamycin inhibitors in solid organ transplantation: from benchside to clinical use. Curr Opin Organ Transplant. 2012 Dec;17(6):626–33. doi: 10.1097/MOT.0b013e32835a4be2. [DOI] [PubMed] [Google Scholar]
- 79.Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012 Mar 30;335(6076):1638–43. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family's role in aging and age-associated pathologies. J Clin Invest. 2013 Mar 1;123(3):973–9. doi: 10.1172/JCI64094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burnett C, Valentini S, Cabreiro F, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011 Sep 22;477(7365):482–5. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rizki G, Iwata TN, Li J, et al. The evolutionarily conserved longevity determinants HCF-1 and SIR-2.1/SIRT1 collaborate to regulate DAF-16/FOXO. PLoS Genet. 2011 Sep;7(9):e1002235. doi: 10.1371/journal.pgen.1002235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rogina B, Helfand SL, Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science. 2002 Nov 29;298(5599):1745. doi: 10.1126/science.1078986. [DOI] [PubMed] [Google Scholar]
- 84.Banerjee KK, Ayyub C, Ali SZ, Mandot V, Prasad NG, Kolthur-Seetharam U. dSir2 in the adult fat body, but not in muscles, regulates life span in a diet-dependent manner. Cell Rep. 2012 Dec 27;2(6):1485–91. doi: 10.1016/j.celrep.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 85.Herranz D, Munoz-Martin M, Canamero M, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Satoh A, Brace CS, Rensing N, et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013;18:416–30. doi: 10.1016/j.cmet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kanfi Y, Naiman S, Amir G, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012 Mar 8;483(7388):218–21. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 88.Sinclair DA, Guarente L. Extrachromosomal rDNA circles-a cause of aging in yeast. Cell. 1997;91:1033–42. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 89.Dang W, Steffen KK, Perry R, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009 Jun 11;459(7248):802–7. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kwan EX, Foss EJ, Tsuchiyama S, et al. A Natural Polymorphism in rDNA Replication Origins Links Origin Activation with Calorie Restriction and Lifespan. PLoS Genet. 2013 Mar;9(3):e1003329. doi: 10.1371/journal.pgen.1003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaeberlein M. The ongoing saga of sirtuins and aging. Cell Metab. 2008 Jul;8(1):4–5. doi: 10.1016/j.cmet.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 92.Baur JA, Chen D, Chini EN, et al. Dietary restriction: standing up for sirtuins. Science. 2010 Aug 27;329(5995):1012–3. doi: 10.1126/science.329.5995.1012. author reply 3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–6. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 94.Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012 Jun;11(6):443–61. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kaeberlein M, McDonagh T, Heltweg B, et al. Substrate specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–45. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- 96.Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005 Apr 29;280(17):17187–95. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- 97.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006 Jun;5(6):493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 98.Hubbard BP, Gomes AP, Dai H, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013 Mar 8;339(6124):1216–9. doi: 10.1126/science.1231097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Miller RA, Harrison DE, Astle CM, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011 Feb;66(2):191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hector KL, Lagisz M, Nakagawa S. The effect of resveratrol on longevity across species: a metaanalysis. Biol Lett. 2012 Oct 23;8(5):790–3. doi: 10.1098/rsbl.2012.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Strong R, Miller RA, Astle CM, et al. Evaluation of resveratrol, green tea extract, curcumin, oxaloacetic acid, and medium-chain triglyceride oil on life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2013 Jan;68(1):6–16. doi: 10.1093/gerona/gls070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu X, Li G. Effects of resveratrol on longevity, cognitive ability and aging-related histological markers in the annual fish Nothobranchius guentheri. Exp Gerontol. 2012 Dec;47(12):940–9. doi: 10.1016/j.exger.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 103.Genade T, Lang DM. Resveratrol extends lifespan and preserves glia but not neurons of the Nothobranchius guentheri optic tectum. Exp Gerontol. 2013 Feb;48(2):202–12. doi: 10.1016/j.exger.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 104.Martin-Montalvo A, Mercken EM, Mitchell SJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013 Jul 31;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY) 2010 Nov;2(11):760–74. doi: 10.18632/aging.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012 Sep;2(9):778–90. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 107.Ovalle F. Cardiovascular implications of antihyperglycemic therapies for type 2 diabetes. Clin Ther. 2011 Apr;33(4):393–407. doi: 10.1016/j.clinthera.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 108.Hardie DG, Ross FA, Hawley SA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol. 2012 Oct 26;19(10):1222–36. doi: 10.1016/j.chembiol.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schiattarella GG, Perrino C, Magliulo F, et al. Statins and the elderly: recent evidence and current indications. Aging Clin Exp Res. 2012 Jun;24(3 Suppl):47–55. [PubMed] [Google Scholar]
- 110.Kolovou G, Kolovou V, Vasiliadis I, Wierzbicki AS, Mikhailidis DP. Ideal lipid profile and genes for an extended life span. Curr Opin Cardiol. 2011 Jul;26(4):348–55. doi: 10.1097/HCO.0b013e32834659d4. [DOI] [PubMed] [Google Scholar]
- 111.Olivieri F, Mazzanti I, Abbatecola AM, et al. Telomere/Telomerase system: a new target of statins pleiotropic effect? Curr Vasc Pharmacol. 2012 Mar;10(2):216–24. doi: 10.2174/157016112799305076. [DOI] [PubMed] [Google Scholar]
- 112.Ramos FJ, Chen SC, Garelick MG, et al. Rapamycin Reverses Elevated mTORC1 Signaling in Lamin A/C-Deficient Mice, Rescues Cardiac and Skeletal Muscle Function, and Extends Survival. Sci Transl Med. 2012 Jul 25;4(144):144ra03. doi: 10.1126/scitranslmed.3003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013 Mar 14;152(6):1365–75. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Butin-Israeli V, Adam SA, Goldman AE, Goldman RD. Nuclear lamin functions and disease. Trends Genet. 2012 Sep;28(9):464–71. doi: 10.1016/j.tig.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sullivan T, Escalante-Alcalde D, Bhatt H, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–20. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nikolova V, Leimena C, McMahon AC, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004 Feb;113(3):357–69. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Choi JC, Muchir A, Wu W, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012 Jul 25;4(144):144ra02. doi: 10.1126/scitranslmed.3003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010 Dec 23;468(7327):1100–4. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- 119.Lafontaine-Lacasse M, Richard D, Picard F. Effects of age and gender on Sirt 1 mRNA expressions in the hypothalamus of the mouse. Neurosci Lett. 2010 Aug 9;480(1):1–3. doi: 10.1016/j.neulet.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 120.Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008 Jan;7(1):78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]