

Abstract
Sex and age-matched wild-type and TCR transgenic mice were infected with cytomegalovirus (CMV) at 6 months of age and followed for 12 additional months to examine aging of the immune system. It was found that viral infection of C57Bl/6 mice resulted in accelerated aging of the immune system as shown by a loss of CD8+28+ cells and an accumulation of KLRG1+ T cells. CMV infection of OT-1 transgenic mice had no influence on immune aging of these mice which nonetheless demonstrated an accumulation of CD8+28− and KLRG1+ T cells with time. CD4+ T cells were unaffected in either strain of mice. Thus, immunological aging was found to be due to both cell-intrinsic and cell-extrinsic factors. Persistent viral infections may accelerate immunological aging but consideration must be given to individual variation in the aging process.
Keywords: aging, immunity, OT-1, mice, MCMV
INTRODUCTION
Aging of the human immune system is characterized by a gradual decrease in immune function and a skewing of hematopoiesis toward the myeloid lineage, with a reduction in the lymphocytic lineage, and progressive increases in senescent memory T cells at the expense of naïve T cells. Both the innate and the adaptive branches of the immune system are affected, including neutrophils (PMN), macrophages, dendritic cells (DC) and lymphocytes. The hematopoietic stem cell (HSC) population is also detrimentally affected by aging as reflected by its inability to maintain both hematopoiesis and lymphopoiesis (Kovacs et al, 2010; DiCarlo et al, 2009).
Aging of the immune system (and the subsequent loss of function) has been attributed both to “time after birth” (i.e., how old an individual is) and ongoing immune responses to endogenous viral (e.g., herpetic) infections, which focus the immune response on these pathogens at the detriment of other responses (Goronzy et al, 2012; Smithey et al, 2012; Pawelec et al, 2004; Mekker et al, 2012). Which variable (chronological age [intrinsic factor] or persistent viral infection [extrinsic factor]) is more important is not known. However, it is consistently reported that immune aging changes seem to be more pronounced in the cytotoxic T cells (CD8+) subpopulation of lymphocytes, which could reflect the significant impact of persistent viral infections (Smithey et al, 2012; Mekker et al, 2012). Fortuitously, T cell aging can easily be followed phenotypically as demonstrated by the loss of CD28 molecules concomitant with the increased expression of the KLRG1 molecule (Vallejo, 2005; Hensen and Akbar, 2009; Fagnoni et al, 2000). However, there is controversy as to whether these phenotypic changes occur in both humans and mice (Ohteki and MacDonald, 1993; Ortiz-Suarez et al, 2002; Ku et al, 2001; Connoy et al, 2006; Effros et al, 1994; Boucher et al, 1998; Castle, 2000).
To address this question we utilized 6 month old control and virally-infected (cytomegalovirus; CMV) “wild-type” C57Bl/6 (B6) and TCR transgenic OT-1 mice. OT-1 mice express a TCR specific for the ovalbumin (OVA) peptide, and thus are unable to “see” and respond to CMV infections (Hogquist et al, 1994). We hypothesized that if immunological aging was due to recognition of persistent endogenous viruses then aging should only be observed in B6 mice. If aging was also due to “time after birth” then immune system aging should be observed in both strains of mice. Infected and control mice were followed until 18 months of age.
We observed that immunological aging was influenced by both cell intrinsic and extrinsic factors, that CMV infection could accelerate this process, but that immunological aging may differ significantly between strains of mice.
MATERIALS AND METHODS
Mice
All (female) mice were obtained from Jackson Laboratories (Bar Harbor, ME) and used according to an IACUC approved protocol. All care and handling of mice was in accordance with the AAALAC guidelines. The C57Bl/6 and the OT-1 strains of mice were utilized. While the B6 mouse is the “wild type” counterpart, the OT-1 mice contain transgenic inserts for mouse Tcra-V2 and Tcrb-V5 genes, a transgenic T cell receptor (TCR) that is designed to recognize ovalbumin residues 257–264 in the context of H2Kb and used to study the response of CD8+ T cells to antigen (Hogquist et al, 1994). The OT-1 TCR transgenic mice were congenic to the C57Bl/6 background.
Virus Infections
Mouse cytomegalovirus (MCMV, strain smith MSGV) was purchased from American Type Culture Collection (ATTC, VR-1399). Mice were inoculated by the intraperitoneal route with 1×104 plaque forming units (pfu) of MCMV. Both B6 and OT-1 mice were infected at 6 months of age, and followed over the next 12 months. MCMV was measured in peripheral blood and occasionally in tissues by real time PCR (Vliegen et al, 2004; Gold et al, 2004). Briefly genomic DNA was extracted from blood followed by outer PCR amplification using primers, primer A 5’TTCGTTCGGACCATGGCCG (+) and primer B 5’ TCGCCGTTCGTGCAGTCCAA (-) followed by inner PCR amplification using primers, primer C 5’TCGCCCATCGTTTCGAGA (+) and primer D 5’TCTCGTAGGTCCACTGACGGA (-). The outer PCR was performed at 95C (30 sec), 55C (30 sec) and 72C (2 min) for 35 cycles and inner PCR at 95C (30 sec), 60C (30 sec) and 72C (2 min) for 35 cycles. The inner PCR yielded a 105bp band visualized on 2% agarose gel.
Fluorescence Activated Cell Sorter (FACS) Analyses
Approximately 20µl of peripheral blood was collected from the cheek pouch and lysed in ACK lysis buffer for 5 minutes at room temperature to remove red blood cells. Cells were washed once and resuspended in 100µl of PBS-2% FBS. All Fc receptors were blocked using 10µl (0.01mg/ml) of mouse IgG for 5 minutes to reduce spurious antibody binding. Cell suspensions were then stained using the following antibody-fluorochrome conjugates: CD3-Brilliant Violet 421, CD4-PE, CD8-FITC, CD28-PE/Cy7, B220-PerCP/Cy5.5, KLRG1-APC. Cells were stained with all antibodies simultaneously. After staining cells were fixed in 1% paraformaldehyde. Cells were analyzed on an LSRII flow cytometer using FacsDiva software.
RESULTS
6-month old B6 and OT-1 mice were infected with murine CMV and followed over the next 12 months. As shown in Table 1 both strains of mice displayed persistent viral infections as MCMV could be detected at multiple time points for up to 1 year post-infection. At no time did the animals become overtly viremic or unhealthy.
Table 1.
Expression of MCMV in Host Tissues.
| Strain/animal# | 1 month after infection |
|---|---|
| B6 (n=4) Control (uninfected) | − |
| OT1 (n=5) Control (uninfected) | − |
| B6-34B | + |
| B6-34L | + |
| B6-34N | + |
| B6-41R | + |
| B6-43R | + |
| OT1-41L | + |
| OT1-46B | + |
| OT1-46L | + |
| OT1-46N | + |
| OT-46R | + |
The indicated strains of mice were infected with MCMV as described in Methods. At the indicated times the expression of MCMV was analyzed by PCR. Positive expression is indicated by (+) while lack of expression is indicated by (−).
Mice were analyzed monthly for almost 1 year after infection for any phenotypic changes associated with persistent viral infection. As shown in Figure 1, there was a small but significant change in total T cell numbers between control and MCMV infected B6 mice. However, there was no significant different in total T cell numbers in OT-1 mice as a result of MCMV infection.
Figure 1. Effects of Age and Viral Infection on Total T cells.

B6 and OT-1 mice were infected with MCMV at 6 months of age as described and followed over the next 12 months. At the indicated times the numbers of total T cells in the peripheral blood was determined. Data is presented as the mean +/− standard deviation for 6 mice for each time point and condition. * indicates significant difference at p<0.05 by Student’s t-test for the B6 mice only.
When CD8+ cytotoxic T cells were analyzed there was a significant increase over time in CD8+ KLRG1+ T cells in the MCMV-infected B6 mice only. The levels of such cells in OT-1 mice did not change (Figure 2).
Figure 2. Effects of Age and Viral Infection on KLRG1 Expression by Cytotoxic T Cells.
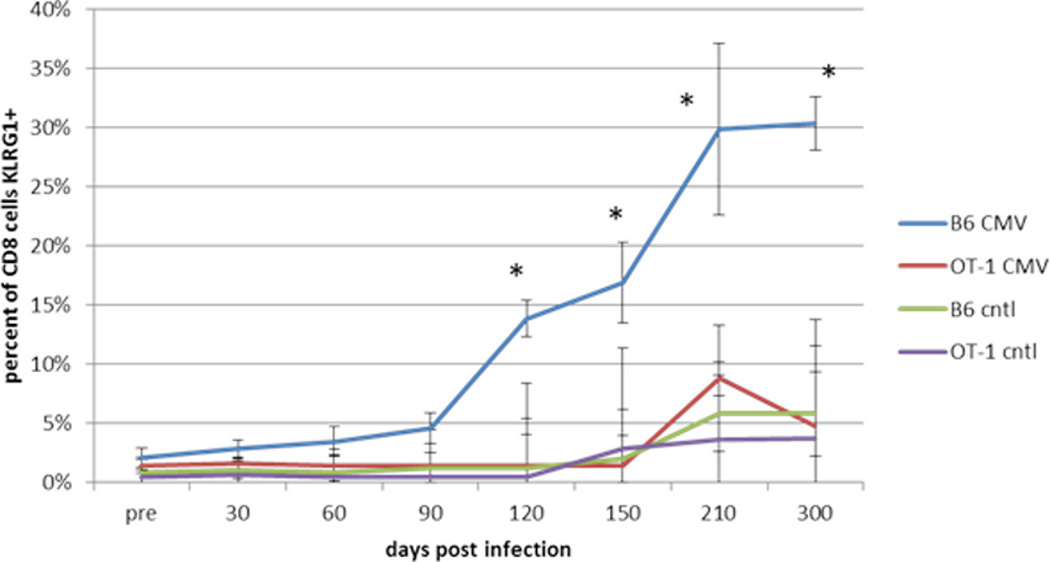
B6 and OT-1 mice were infected with MCMV at 6 months of age as described and followed over the next 12 months. At the indicated times the percentage of KLRG1+ CD8+ T cells in the peripheral blood was determined. Data is presented as the mean +/− standard deviation for 6 mice for each time point and condition. * indicates significant difference at p<0.05 by Student’s t-test for the B6 mice only.
Concurrently, as shown in Figure 3, there was a loss of CD8+CD28+ T cells over time in these MCMV-infected B6 mice that was significantly different that the uninfected control B6 mice. Unexpectedly, OT-1 mice also displayed a loss of CD8+CD28+ T cells with increasing age which was observed in both control and virus-infected animals. Only at very late time points did viral infection accelerate the loss these cells in B6 mice. These observations were in contrast to some reports in the literature for CD28 expression in aging mice (Weng et al, 2004; Ortiz-Suarez et al, 2002).
Figure 3. Effects of Age and Viral Infection on CD28 Expression by Cytotoxic T Cells.
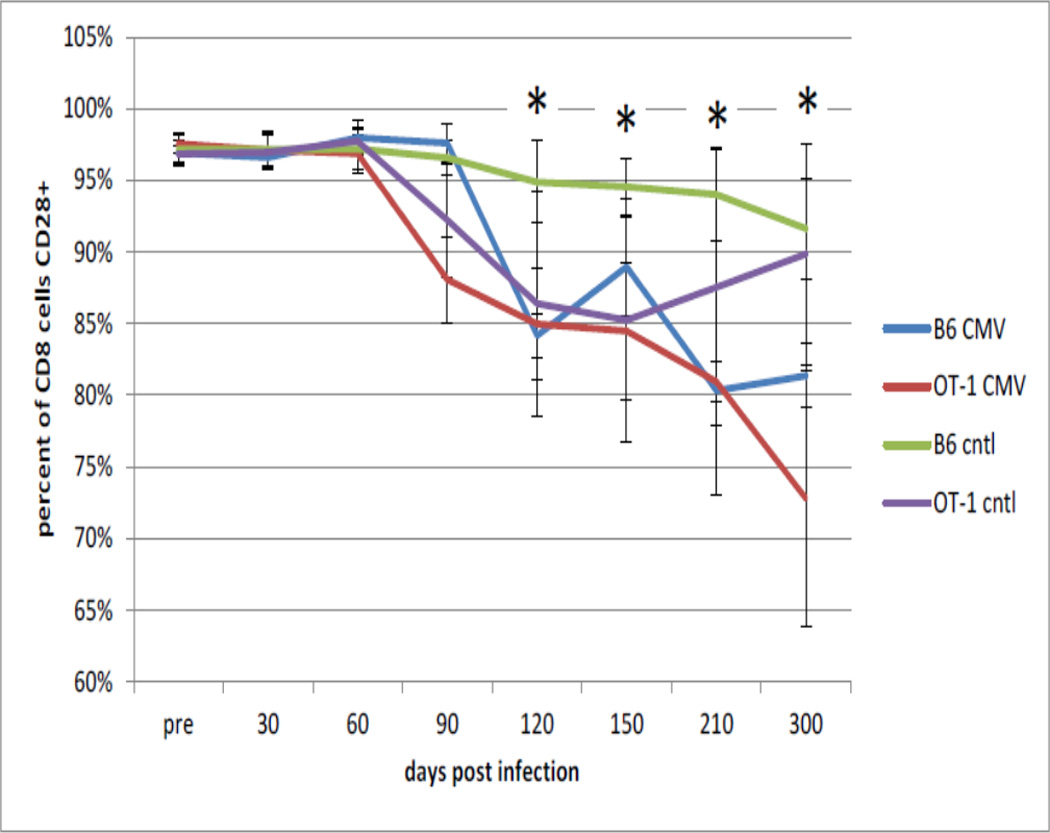
B6 and OT-1 mice were infected with MCMV at 6 months of age as described and followed over the next 12 months. At the indicated times the percentage of CD28+ CD8+ T cells in the peripheral blood was determined. Data is presented as the mean +/− standard deviation for 6 mice for each time point and condition. * indicates significant difference at p<0.05 by Student’s t-test between the B6 mice only.
Interestingly, and as expected from the literature, there was no significant change in helper (CD4+) T cells in either strain of mice, whether MCMV infected or not, in terms of CD4+KLRG1+ T cells (Figure 4). Similarly, the percentage of CD4+ T cells co-expressing CD28 also did not change with age or viral infection (Figure 5) in either strain of mice.
Figure 4. Effects of Age and Viral Infection on KLRG1 Expression by Helper T Cells.

B6 and OT-1 mice were infected with MCMV at 6 months of age as described and followed over the next 12 months. At the indicated times the percentage of KLRG1+ CD4+ T cells in the peripheral blood was determined. Data is presented as the mean +/− standard deviation for 6 mice for each time point and condition. No significant differences were observed between control and infected mice.
Figure 5. Effects of Age and Viral Infection on CD28 Expression by Helper T Cells.
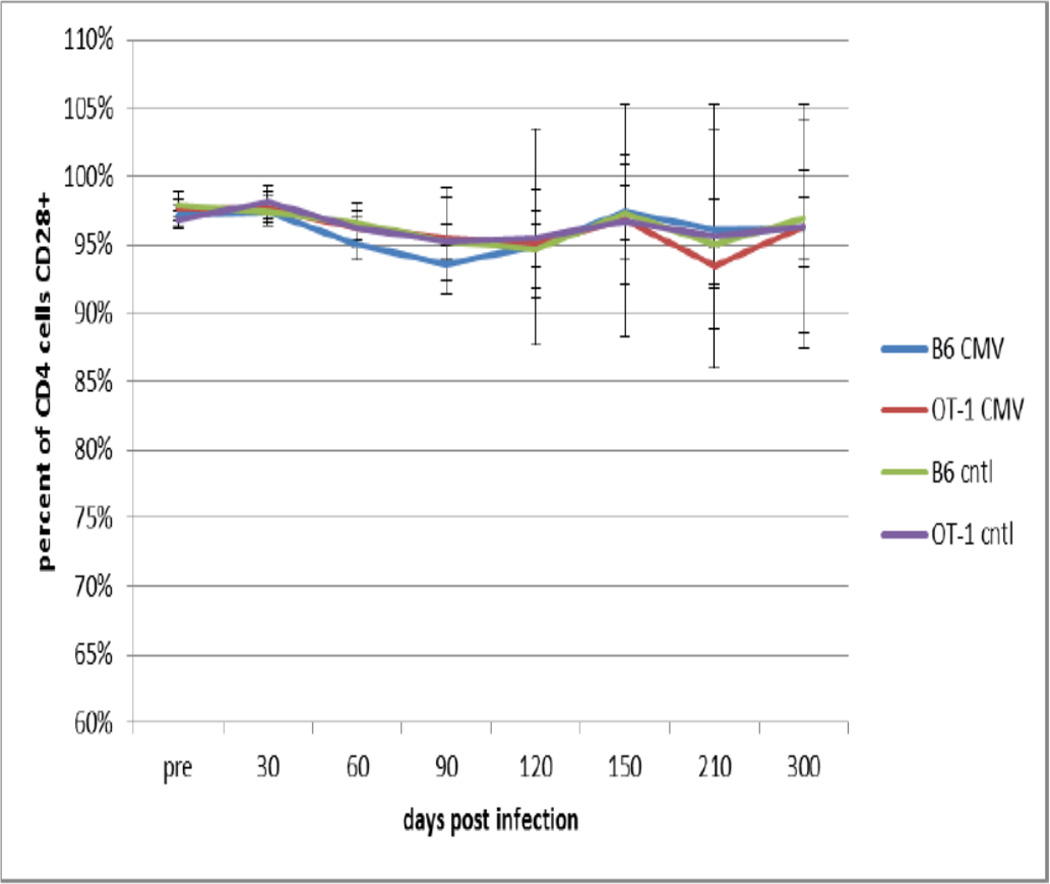
B6 and OT-1 mice were infected with MCMV at 6 months of age as described and followed over the next 12 months. At the indicated times the percentage of CD28+ CD4+ T cells in the peripheral blood was determined. Data is presented as the mean +/− standard deviation for 6 mice for each time point and condition. No significant differences were observed between control and infected mice.
DISCUSSION
In this study we determined the contributions of chronological age and persistent viral infection on T cell aging in mice. To that end we examined normal B6 and TCR transgenic OT-1 mice in the presence or absence of persistent MCMV infection over a period of 18 months. As expected B6 mice infected with MCMV displayed increasing numbers of CD8+ KLRG1+ cells over time that was not reflected in the CD4+ T cell subpopulation, thought to be indicative of ongoing cytotoxic responses to the persistent viral infection. KLRG1 is the “Killer cell Lectin-like Receptor subfamily G member 1” which is known to be expressed on activated T cells. No changes in CD8+KLRG1+ or CD4+KLRG1+ cells were seen in OT-1 mice, indicating that these TCR transgenic mice were “blind” to the viral infection. Unexpectedly, we found that CD8+ T cells in both B6 and OT-1 mice displayed a loss of the CD28 molecule with age, contrary to what some others have reported in the literature (Ortiz-Suarez et al, 2002; Ku et al, 2001; Connoy et al, 2006; Weng et al, 2009). Even more surprising the OT-1 mice displayed a loss of CD8+CD28+ T cells even in the absence of MCMV infection. Even though the decreases in CD8+CD28+ T cells were small (20% for B6 mice and 25% for OT-1 mice) in comparison to changes often seen in the human immune system (70–90% presence of CD8+CD28− T cells; Vallejo, 2005) it was significant, and in contrast to literature reports of either no change or even increases in CD8+CD28+ T cells in aged mice (Ortiz-Suarex et al, 2002; Ku et al, 2001; Connoy et al, 2006; Effros et al, 1994; Boucher et al, 1998; Castle, 2000). Reasons for this apparent discrepancy are unknown.
It has generally been assumed that an unequivocal biomarker of immune aging is the accumulation of CD8+CD28−KLRG-1+ T cells, and the assumption that this phenotype represents cells that are "senescent". However, our data showed clearly that this effect is mostly dependent on MCMV infection and that a late-differentiated T cell phenotype emerged with no indication of senescence or any other dysfunctional attributes that might be considered to contribute to senescence or aging. The "age-effect" observed in the TCR transgenic OT-1 mice is likely to be due to other differentiation events of a similar nature over the animal’s lifespan, plus indirect effects of CMV via other cell types. This effect could be referred to as a "remodeling" of immunity, rather than aging, and for some unclear reason is strongly associated with MCMV infection.
It is likely that in the absence of ova stimulation, peripheral T cell selection with increasing age may have shifted the immune repertoire observed in the transgenic TCR OT-1 mice. This hypothesis may explain some of the phenotypic shifts seen in the transgenic mice. However, as the mice were maintained in the absence of any antigenic stimulation (ova or otherwise) we cannot confirm that any peripheral selection has occurred that is not due to the mere maintenance of the naïve T cells due to stimulation with cross-reactive, self-antigens. One of the limitations of the current study is that the frequency of MCMV-specific T cells (in either strain of mice) was not determined. It is therefore unclear whether what we described as “immune aging” in the C57Bl/6 mice is actually accelerated T cell aging or merely an accumulation of MCMV-specific cells.
It is unclear as to why no obvious changes occur in the CD4+ T cell population, especially in the B6 strain of mice. As effective CD8+ T cell responses require CD4+ T cell help, which would imply recognition of the MCMV virus in the B6 strain, why significant increases in KLRG1+ CD4+ T cells is not observed in this subpopulation and why this population does not appear to age is unclear. We and others have hypothesized that it is simply a numbers game and much fewer CD4+ T cells are responding (i.e., lesser clonal expansion) to the infection for a limited time period and thus below the limit of detection.
Based on the current observations it appears that “aging” of the immune system is due to a combination of chronological age and immune responses to persistent viral infection; at least for the CD8+ T cell population. MCMV infection appears to be able to accelerate the aging process but at least in some strains of mice these changes are independent of an immune response. The caveat to this conclusion is that different strains of mice may age differently (similar to what would be observed in humans) in a chronological sense; or that persistent infections may affect different strains of mice differently.
ACKNOWLEDGEMENTS
This work was supported in part by grant #5 R01 AG038021-03 from the National Institute of Aging, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure Statement: None of the authors have a financial interest with any company that might be concerned with the topic of this work.
REFERENCES
- Boucher N, Dufeu-Duchesne T, Vicaut E, Farge D, Effros RB, Schachter F. CD28 expression in T cell aging and human longevity. Exp. Gerontol. 1998;33:267–282. doi: 10.1016/s0531-5565(97)00132-0. [DOI] [PubMed] [Google Scholar]
- Castle SC. Clinical relevance of age-related immune dysfunction. Clin. Infect. Dis. 2000;31:578–585. doi: 10.1086/313947. [DOI] [PubMed] [Google Scholar]
- Connoy AC, Trader M, High KP. Age-related changes in cell surface and senescence markers in the spleen of DBA/2 mice: A flow cytometric analysis. Experimental Gerontology. 2006;41:225–229. doi: 10.1016/j.exger.2005.11.003. [DOI] [PubMed] [Google Scholar]
- DiCarlo AL, Fuldner R, Kaminiski J, Hodes R. Aging in the context of immunological architecture, function and disease outcomes. Trends in Immunology. 2009;30:293–294. doi: 10.1016/j.it.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Effros RB, Boucher N, Porter V, Zhu X, Spaulding C, Walford RL, Kronenberg M, Cohen D, Schachter F. Decline in CD28+T cells in centenarians and in long-term T cell cultures: a possible cause for both in vivo and in vitro immunosenescence. Exp. Gerontol. 1994;29:601–609. doi: 10.1016/0531-5565(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Fagnoni FF, Vescovini R, Passeri G, Bologna G, Pedrazzoni M, Lavagetto G, Casti A, Franceschi C, Passeri P, Paolo S. Shortage of circulating naive CD8+ T cells provides new insights on immunodeficiency in aging. Blood. 2000;95:2860–2868. [PubMed] [Google Scholar]
- Gold MC, Munks MW, Wagner M, Mcmahon CW, Kelly A, Kavanagh DG, Slifka MK, Koszinowski UH, Raulet DH, Hill AB. Murine cytomegalovirus interference with antigen presentation has little effect on the size or the effector memory phenotype of the CD8 T cell response. J of Immunology. 2004;172:6944–6953. doi: 10.4049/jimmunol.172.11.6944. [DOI] [PubMed] [Google Scholar]
- Goronzy JJ, G Li, M Yu, Weyand CM. Signaling pathways in aged T cells: a reflection of T cell differentiation, cell senescence and host environment. Semin. Immunol. 2012;24:365–72. doi: 10.1016/j.smim.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensen SM, Akbar AN. KLRG1: more than a marker for T cell senescence. AGE. 2009;31:285–291. doi: 10.1007/s11357-009-9100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Kovacs EJ, Palmer JL, Fortin CF, Fulop T, Goldstein DR, Linton P-J. Aging and innate immunity in the mouse: impact of intrinsic and extrinsic factors. Trends in Immunology. 2010;30:319–324. doi: 10.1016/j.it.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CC, Kappler J, Marrack P. The growth of the very large CD8+ T cell clones in older mice is controlled by cytokines. J. Immunol. 2001;166:2186–2193. doi: 10.4049/jimmunol.166.4.2186. [DOI] [PubMed] [Google Scholar]
- Mekker A, Tchang VS, Haeberli L, Oxenius A, Trkola A, Karrer U. Immune senescence: relative contributions of age and cytomegalovirus infection. PLoS Patholog. 2012;8:e1002850. doi: 10.1371/journal.ppat.1002850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohteki TV, MacDonald RH. Expression of the CD28 costimulatory molecule on subsets of murine intestinal intraepithelial lymphocytes correlates with lineage and responsiveness. Eur. J. Immunol. 1993;23:1251–1255. doi: 10.1002/eji.1830230609. [DOI] [PubMed] [Google Scholar]
- Ortiz-Suarez A, Miller RA. A Subset of CD8 Memory T Cells from Old Mice Have High Levels of CD28 and Produce IFN-gamma. Clinical Immunology. 2002;Vol. 104:282–292. doi: 10.1006/clim.2002.5221. [DOI] [PubMed] [Google Scholar]
- Pawelec G, Akbar A, Caruso C, Effros R, Grubeck-Loebenstein B, Wikby A. Is immunosenescence infectious? Trends Immunol. 2004;25:406–410. doi: 10.1016/j.it.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Smithey MJ, Li G, Venturi v, MP Davenport MP, Nikolich-Zugich J. Lifelong persistent viral infection alters the naïve T cell pool, impairing CD8 T cell immunity in late life. J. Immunol. 2012;189:5356–5366. doi: 10.4049/jimmunol.1201867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T cell senescence. Immunol. Rev. 2005;205:158–169. doi: 10.1111/j.0105-2896.2005.00256.x. [DOI] [PubMed] [Google Scholar]
- Vliegen I, Duijvestijn A, Stassen F, Bruggeman C. Murine cytomegalovirus infection directs macrophage differentiation into proinflammatory immune phenotype: implications for atherogenesis. Microbes and Infection. 2004;6:1056–1062. doi: 10.1016/j.micinf.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Weng N-P, Akbar AN, Goronzy J. CD28(−) T cells: their role in the age-associated decline of immune function. Trends in Immunology. 2009;30:306–312. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]