

Abstract
Broad-spectrum anticonvulsants are of considerable interest as antiepileptic drugs, especially because of their potential for treating refractory patients. Such “neurostabilizers” have also been used to treat other neurological disorders, including migraine, bipolar disorder, and neuropathic pain. We synthesized a series of sulfamide derivatives (4–9, 10a–i, 11a, 11b, 12) and evaluated their anticonvulsant activity. Thus, we identified promising sulfamide 4 (JNJ-26489112) and explored its pharmacological properties. Compound 4 exhibited excellent anticonvulsant activity in rodents against audiogenic, electrically-induced, and chemically-induced seizures. Mechanistically, 4 inhibited voltage-gated Na+ channels and N-type Ca2+ channels, and was effective as a K+ channel opener. The anticonvulsant profile of 4 suggests that it may be useful for treating multiple forms of epilepsy (generalized tonic-clonic, complex partial, absence seizures), including refractory (or pharmacoresistant) epilepsy, at dose levels that confer a good safety margin. On the basis of its pharmacology and other favorable characteristics, 4 was advanced into human clinical studies.
INTRODUCTION
Epilepsy is a chronic neurological condition that affects at least 50 million people worldwide.1 However, with current medications up to 30% of epileptic patients are not adequately treated and 20% suffer from intractable seizures.1 In the search for next-generation antiepileptic drugs, an important factor has been broad-spectrum anticonvulsant activity, so as to treat multiple seizure types effectively. Besides improved efficacy against refractory epilepsy, an important requirement for new drugs in this area is good safety and tolerability. Interestingly, broad-spectrum anticonvulsant drugs have also proven to be useful for treating other neurological disorders, including neuropathic pain, bipolar disorder/depression, migraine headache, and substance abuse.2
The broad-spectrum anticonvulsant topiramate (1)2f,3 is available worldwide for treating epilepsy and migraine,4 and has several other therapeutic applications (Chart 1).5 Since our discovery of topiramate (1),2f,3 we have been keenly interested in identifying next-generation anticonvulsant drugs with a broad-spectrum pharmacological profile. Some time ago, we reported on cyclic sulfate analogue 2 (RWJ-37947),6 and more recently we described sulfamide-based anticonvulsant 3 (JNJ-26990990), which advanced into human clinical studies (Chart 1).7 Preclinically, 3 demonstrated broad-spectrum anticonvulsant activity in rodents vs. audiogenic, electrically-induced, and chemically-induced seizures. Because of its ability to limit seizure spread and elevate seizure threshold, 3 has an enhanced, preclinical antiepileptic profile relative to topiramate. Therefore, 3 showed promise for treating multiple forms of epilepsy, as well as refractory epilepsy, in humans.
Chart 1.
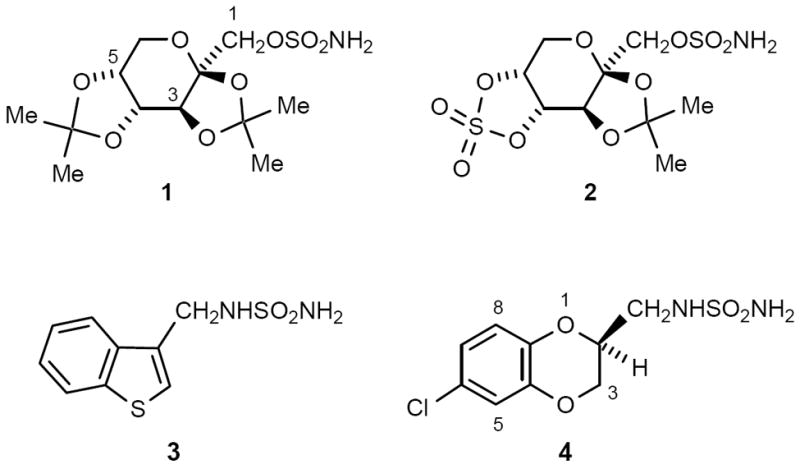
Sulfamate and Sulfamide Anticonvulsants (with Standard Numbering System)
We have continued to investigate sulfamide compounds to find additional anticonvulsants of merit, as potential development candidates. From this research, we identified JNJ-26489112 (4) as a broad-spectrum anticonvulsant and advanced it into human clinical studies (Chart 1). In this paper, we present details on our studies with this novel sulfamide series, focusing on the preclinical pharmacology of 4.8,9 The anticonvulsant profile of 4 suggests that it may be useful for treating multiple forms of epilepsy (generalized tonic-clonic, complex partial, and absence seizures), including refractory (or pharmacoresistant) epilepsy, with a favorable margin of safety.
RESULTS AND DISCUSSION
A specific biological testing protocol was employed to evaluate newly prepared sulfamides, as described previously.7 New compounds (4–9, 10a–i, 11a, 11b, 12) were examined for the virtual absence of carbonic anhydrase-II (CA-II) inhibition (CO2 hydration assay), and compounds with IC50 values of greater than 10 μM were then tested for central nervous system (CNS) behavioral effects in mice and anticonvulsant activity in the mouse maximal electroshock seizure (MES) test. This sequence of events supported the discovery of broad-spectrum anticonvulsants without inhibition of CA-II and with good CNS tolerability. For notable sulfamide compounds, we followed up with advanced anticonvulsant tests to see if the compounds possessed a pharmacological profile superior to that of currently known antiepileptic drugs, including topiramate. This process was assisted by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health (NIH) via their expertise with in vivo models relating to pharmacology and neurotoxicity. Work on the synthesis and testing of various sulfamide derivatives (Table 1) ultimately led to the selection of anticonvulsant 4 for human clinical development.
Table 1.
Sulfamide Compounds and Biological Testing Dataa
| ||||||
|---|---|---|---|---|---|---|
| cmpd | G | synth methb | MES, ipc t (h), effect | MES, pod t (h), effect | CA-II inhe IC50, μM (N) | |
| 4 | A | 2, ED50 = 111 mg/kgf | 0.5, 0/10; 2, 2/10 | 35 | ||
| 2, 5/5g | ||||||
| 5 | h | 0.5, 3/3 | ND | 130 (3) | ||
| (S)-5i | A | 0.5, 3/3 | 0.5, 9/10; 2, 2/10g | 97 (2) | ||
| (R)-5 | A | 0.5, 3/3 | 2, 1/5g | 77 (5) | ||
| 6 | h | 0.5, 3/3; 2, 1/3g | 85 (2) | |||
| 7 | h | 0.5, 4/5; 2, 0/5g | >1000 | |||
| 8 | B | 0.5, 1/5; 2, 0/5 | 130 | |||
| 0.5, 3/3; 2, 2/3g | ||||||
| 9 | h | 0.5, 0/5; 2, 0/5 | 25 | |||
| 0.5, 5/5; 2, 0/5g | ||||||
| 10a | 5-Cl | A | 0.5, 0/5; 2, 1/5 | 220 | ||
| 10b | 7-Cl | A | 0.5, 0/5; 2, 4/5 | 36 (3) | ||
| 10c | 8-Cl | A | 0.5, 1/5; 2, 0/5 | 56 | ||
| 10d | 5-F | A | 0.5, 2/5; 2, 0/5 | 70 | ||
| 0.5, 4/5; 2, 5/5g | ||||||
| 10e | 6-F | A | 0.5, 4/5; 2, 5/5g | 75 | ||
| 10f | 6-Br | A | 0.5, 0/5; 2, 5/5g | 36 | ||
| 10g | 8-OMe | A | 0.5, 0/5; 2, 0/5j | 300 | ||
| 10h | 6,7-diCl | A | 0.5, 0/5; 2, 3/5 | 30 | ||
| 10i | 7-Me | A | 0.5, 0/5; 2, 0/5g | 29 | ||
| 11a | H | B | 0.5, 3/3 | 0.5, 5/5; 2, 0/5 | 71 | |
| 11b | Cl | B | 0.5, 0/5; 2, 0/5 | ND | ||
| 12 | h | 0.5, 2/3 | ND | 17 | ||
| 3k | 0.5, 1/5; 2, 3/5 | 110 | ||||
| 1 | 0.5, 2/5; 2, 4/5l | 0.50m | ||||
Compounds 4 and 10a–i are single enantiomers with the S absolute configuration. ND = not determined.
Synthetic method used (A, B, or other).
Single testing dose was 300 mg/kg, ip. The effect is presented as the number of animals responding out of the total number of animals tested, except for any ED50 values.
Oral dose of 100 mg/kg, unless otherwise noted. The effect is presented as the number of animals responding out of the total number of animals tested.
IC50 values for the inhibition of human CA-II were determined in house by using a CO2 hydration assay (ref 10a–d), unless otherwise noted. The number of independent experiments (N) is one unless indicated otherwise; N > 1 is given in parentheses.
95% confidence interval: 86.6–139.
Oral dose of 300 mg/kg.
See text for information on this other (non-A/non-B) method.
Structure is equivalent to 10 with G = H.
1/5 at 4 h.
Reported in ref 7.
From ref 3a.
From ref 10a.
Chemical Synthesis
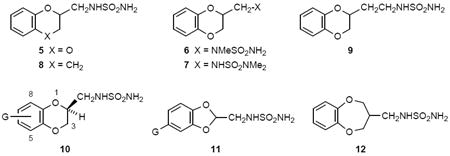
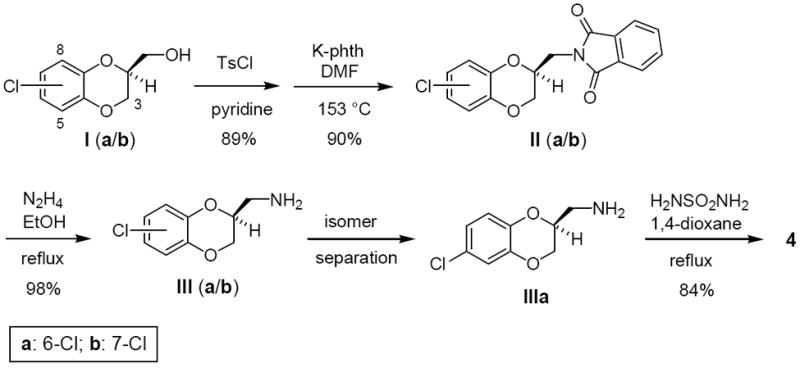
Synthetic routes to sulfamides were reported in our previous papers.10 Several target compounds were obtained from the corresponding alcohol by forming a reactive sulfonate ester,11 displacing the sulfonate with potassium phthalimide, deprotecting the phthalimide with hydrazine, and heating the primary amine with sulfamide (Method A; Scheme 1).10a Derivatives of type 10 were prepared by this route, which is exemplified for the synthesis of 4 in Scheme 1.12 In the case of 5, commercially available racemic (2,3-dihydro-1,4-benzodioxin-2-yl)methylamine was reacted with sulfamide. However, the enantiomers (S)-5 and (R)-5 were synthesized independently as above11 from the known chiral alcohol intermediates (Method A).12 For some derivatives of type 10 a regioselective synthesis was employed,13 possibly with procedural modifications, to afford the single regioisomeric alcohol intermediates required. Some target compounds, such as 8, 11a, and 11b,10a were obtained from the corresponding carboxylic acid by forming a primary amide, reducing the amide to an amine with LiAlH4, and heating the resultant amine with sulfamide (Method B). Compounds 6 and 7 were obtained from commercial (2,3-dihydro-1,4-benzodioxin-2-yl)methylamine: 6 involved formylation with ethyl formate, reduction with LiAlH4 to the N-methylamine, and reaction with sulfamide; 7 involved direct reaction with N,N-dimethylsulfamoyl chloride. Compound 9 was prepared from (2,3-dihydro-1,4-benzodioxin-2-yl)methyl bromide by KCN displacement, borane-THF reduction to the primary amine, and condensation with sulfamide. Compound 12 was obtained starting with catechol (IV) according to the route outlined in Scheme 2. Cyclization with 2-chloromethyl-3-chloro-1-propene yielded alkene V, which was subjected to hydroboration/amination to give requisite primary amine VI.
Scheme 1.
Synthesis of 4.a
a. Alcohols Ia and Ib were prepared as a mixture of regioisomers (~3:1) by following a reported enantioselective synthesis (ref 12). The ratio of IIIa:IIIb was ~3:1. Amine llla was purified as the HCl salt. Ts, p-toluenesulfonyl; phth, phthalimide anion.
Scheme 2.
Synthesis of 12.
Anticonvulsant Screening
Sulfamides with a virtual absence of inhibition of human CA-II10a-c (IC50 > 10 μM) were tested for activity in the mouse MES test (Table 1),3a,14 which involves applying an electrical current to induce tonic extension of the hind limbs. An anticonvulsant compound will inhibit this tonic-extensor response. We analyzed the MES results in terms of the number of responders relative to the number of animals within each group. Early in the project, test compounds were administered intraperitoneally, before we had established oral bioavailability for the series. Later on, compounds were tested by oral administration. Prototype sulfamide 5 (racemate)10a exhibited anticonvulsant activity in the mouse MES test, protecting three out of three mice at 0.5 h after an i.p. dose of 300 mg/kg. Evaluation with i.p. dosing at the NINDS indicated an ED50 at 0.25 h post-dosing of 123 mg/kg. In the rat MES test with oral dosing, 5 had an ED50 at 0.5 h post-dosing of 73 mg/kg. The peak effect in rats with 5 occurred early in the time course and anticonvulsant activity was largely dissipated at the 4-h time point. From these results, it was evident that we had useful anticonvulsant activity, but needed to find an analogue with a longer duration of action.
By way of follow-up, we prepared the individual enantiomers of 5 for MES evaluation. While each of these compounds displayed anticonvulsant activity, (R)-5 was accompanied by ataxia in the rat Irwin test.15 The results for (S)-5 on oral dosing showed good anticonvulsant activity at 0.5 h, but with a decline at 2 h. Since the short duration of action might be attributed to in vivo oxidative metabolism on the electron-rich benzene ring, we pursued the (S) enantiomeric series further but with the introduction of halogen or other substituents, as in compounds 4 and 10a–i. Subsequently, we also looked into eliminating the stereogenicity by symmetrization, as with 11a, 11b, and 12. Our earlier work with aromatic heterocycles, such as 3 and its analogues, actually constitutes removal of stereogenicity by planarization of the ring system.7
Because of the slower in vivo uptake by the oral route of administration, we decided to compare the mouse MES anticonvulsant activity for various test compounds at a 2-h time point (Table 1). Reasonable oral anticonvulsant activity was identified for 4 (6-Cl), 10b (7-Cl), 10d (5-F), 10e (6-F), 10f (6-Br), and 10h (6,7-diCl) in the saturated heterocycle series (encompassing 4 and 10a–i). To build a better basis for comparison, additional oral mouse MES ED50 data were collected for certain derivatives (Table 2). Compounds 4, 10a, 10b, and 11a were relatively close in potency (100–130 mg/kg); 10e and (S)-5 were somewhat less potent (160–200 mg/kg); and 10d, 10h, and 12 were weaker still (230–345 mg/kg). Although the SAR was not systematically examined, we note that minor changes of substituents on the benzene ring can impact potency in the MES assay in an unpredictable manner. Compounds (S)-5, 10a, 10d, 10e, 10h, 11a, and 12 were subjected to a mouse rotorod neurotoxicity screen to gain a sense of tolerability (Table 2). While these data were not obtained at the time of peak potency, we found that the MES ED50 values for (S)-5, 10b, and 11a were considerably to be lower than the doses that produced frank neurotoxicity.
Table 2.
Oral Mouse MES ED50 Data for Selected Compoundsa
| cmpd | ED50 (mg/kg, po) | 95% CI | neurotoxicityb |
|---|---|---|---|
| 4 | 120c | -- | d |
| (S)-5 | 199 | 183–222 | 0/8 (250) |
| 10a | 124 | 85–173 | 6/8 (300) |
| 10b | 104 | 91–128 | 0/8 (150) |
| 10d | 230 | 171–281 | 2/8 (350) |
| 10e | 161 | 138–192 | 3/8 (300) |
| 10h | 296 | 202–473 | 3/8 (600) |
| 11a | 131 | 120–141 | 1/8 (150) |
| 12 | 267 | 232–299 | 4/8 (300) |
| 3 | 119c | 112–126 | d |
| phenytoin | 9.0e | TD50 = 86.7e | |
| 1 | 43.8f | TD50 = 389g |
Maximal electroshock seizure model, at 1 h post dosing, performed at NeuroAdjuvants. CI, 95% confidence interval.
Mouse rotorod toxicity, at the dose in mg/kg given in parentheses. The effect is presented as the number of animals responding out of the total number of animals tested.
Performed in house; result at 3 h post dosing (time of peak effect).
See text for rat data.
At 2 h; ref 3a.
At 1 h; ref 6.
At 1 h; ref 3a.
Some anticonvulsant agents containing a sulfamide functionality have been reported.8 Although several of the described compounds possess N,N- or N,N’-disubstitution, or N,N,N’-trisubstitution, primary sulfamides PhCH2NHSO2NH2 and BuNHSO2NH2 were also examined.8a,b These primary sulfamides are interesting relative to the compounds that we present in this paper, as well as our earlier publication.7 PhCH2NHSO2NH2 was reported to be markedly active (3/3) in the mouse MES test at 0.5 h at 100 and 300 mg/kg, and at 4 h at 300 mg/kg; the ED50 at 0.5 h was of 440 mg/kg. By contrast, analogue BuNHSO2NH2 was not active in the MES test.8b There are some amino acid-derived sulfamides that exhibit anticonvulsant activity of moderate potency.8c
Inhibition of Carbonic Anhydrase-II
Various sulfamide derivatives were examined for inhibition of human CA-II by using a carbon dioxide hydration assay10a-d (Table 1). It is apparent that for a wide diversity of sulfamide structures the inhibition of CA-II is relatively weak, with IC50 values ranging from 17 to >1000 μM. The weakest compound is 7 (IC50 > 1000 μM), which would be expected since it lacks a terminal SO2NH2 group for coordination to Zn(II) within the active site of CA-II. For comparison, the sulfamate anticonvulsant topiramate (1) has Ki values for CA-II inhibition in the range of 0.3–0.6 μM.10a,b Our observation of rather weak CA-II inhibition among this collection of sulfamides is consistent with our prior reports on the behavior of other sulfamides in such a capacity.10 Indeed, we have now examined numerous sulfamides of diverse structure for inhibition of CA-II and have found them to exhibit, in general, weak to very weak CA-II inhibition potency. Moreover, we have found that diverse sulfamides are generally much weaker inhibitors of CA-II than their cognate sulfamates.10
In the CO2 hydration assay (at 0–5 °C), 4 is a very weak inhibitor of CA-II, with an IC50 value of 35 μM (Ki = 8.8 μM); 4 is also just a weak inhibitor of CA-I (IC50 = 18 μM; Ki = 2.2 μM).10a-d Despite our repeated observations that sulfamide derivatives are rather weak inhibitors of CA-II, a controversy has surrounded this issue.10 Consequently, we submitted 4 to an external laboratory, Cerep, for independent assessment.16 Evaluation of the compound with human erythrocyte CA-II via CO2 hydration at 22 °C with fluorimetric detection17 gave a IC50 value of 5 μM, which is within the realm of the values that we have experienced for 4 (not in a double-digit nanomolar range).10
Advanced Pharmacological Assessment of 4
We next sought to study the pharmacological profile of 4, especially with a detailed evaluation of its anticonvulsant properties. This compound was chosen for adnvanced study based on four key attributes: (1) very weak inhibition of CA-II, (2) oral efficacy in the mouse MES test, (3) low neurotoxicity in rodents, and (4) good pharmacokinetics in rats and dogs (high oral bioavailability; long duration of action).18 In MES tests in mice and rats, the oral ED50 values for 4 at the time of peak effect were 120 mg/kg (3 h) and 43 mg/kg (4 h), respectively. The corresponding (R)-enantiomer of 4 (ent-4) was active in the rat MES test with an oral ED50 of 50 mg/kg (at 4 h). Intraperitoneal administration of 4 effectively blocked chemically-induced, forelimb clonic seizures in mice that were caused by subcutaneous bicuculine (Bic), picrotoxin (Pic), or pentylenetetrazol (PTZ), with 1-h ED50 values of 197, 189, or 109 mg/kg, respectively. In the realm of such chemically-induced seizures, the potency demonstrated by 4 is better than that of topiramate (ED50 > 500 mg/kg), valproic acid (ED50 > 200 mg/kg), and levetiracetam (ED50 > 540 mg/kg).7 The oral ED50 values for sulfamide 3 in the mouse and rat MES tests at the time of peak effect were 119 mg/kg (3 h) and 34 mg/kg (2 h), respectively,7 which are analogous to the results with 4. Intraperitoneal administration of 3 blocked forelimb clonic seizures in mice induced by subcutaneous Bic, Pic, and PTZ, with 0.25-h ED50 values of 156, 106, and 161 mg/kg, respectively.7 Clearly, the time to peak effect for 4 was later than that for 3, but these two compounds had similar potencies in blocking chemically induced seizures. Thus, when 4 was evaluated in electrically-induced and chemically-induced seizures in rodents, it was found to exhibit broad-spectrum anticonvulsant activity.
We continued to study 4 in a PTZ seizure-threshold test,7,18 a sensitive method to determine the seizure threshold for a compound. In this protocol activity is judged by the delay of seizure appearance, as opposed to the prevention of seizure occurrence. Of particular value with the i.v. PTZ test, it is possible to characterize a compound’s ability to increase or decrease seizure threshold, that is, to manifest anticonvulsant or proconvulsant behavior, respectively. We used two doses of the test compound, the first of which corresponded to the ED50 for protecting against MES seizures and the second of which corresponded to the median neurotoxic dose (TD50), obtained by the rotorod impairment study.19 We administered i.p. a solution of 4 or vehicle (0.5% aqueous methylcellulose) to mice (N = 9 per dose group) and, after 15 min, we infused a saline solution of PTZ into the tail vein, while recording the time from start of infusion to appearance of the first twitch and the onset of clonus. The test compound would be designated as an anticonvulsant if it increased seizure threshold relative to an increase in the dose of PTZ needed to produce a first twitch or clonic seizure. Compound 4 at its ED50 (111 mg/kg) and TD50 (355 mg/kg) levels (i.p.) markedly increased the seizure threshold for twitch and clonus: at ED50, +39% and +52%; at TD50, +80% and +232%, respectively. Compound 5 also increased the seizure threshold for both twitch and clonus (at ED50 of 107 mg/kg, p.o., +29% and +47%; at TD50 of 182 mg/kg, p.o., +41% and +59%, respectively). Topiramate was somewhat different in that it had no effect on seizure threshold at the ED50 and was actually proconvulsant at the TD50.7
We examined 4 for its effect on audiogenic seizures in mice, which are induced by sound and cause a loss of righting reflex with forelimb and hindlimb tonic extension. An anticonvulsant will be deemed protective in this protocol when treated mice do not experience hindlimb tonic extension.19 One hour before applying the sound, we administered i.p. a solution of 4 to mice (N = 8 per dose group), with at least of four doses being used to determine an ED50 (at time to peak effect). Compound 4 was effective in this model with an ED50 value of 21 (14.1–30.5) mg/kg, although its potency is 5-fold less than topiramate (ED50 = 4.2 mg/kg).
We sought to gauge the potential for treating pharmacoresistant limbic epilepsy by using a rat kindling model, which involves complex partial seizures along with secondarily generalized seizures,19 in a collaboration with the NINDS.20 In this antiepileptic model, the key readouts for a test compound are its effect on seizure score (severity of spread) and after-discharge duration (excitability) of the generalized seizures is gauged, in terms of an ED50 value. For example, a reduction of the seizure score from 5 to 3, with no affect on after-discharge duration, would suggest effectiveness for secondarily generalized seizures; whereas, a reduction of the seizure score from 5 to less than 1, concomitant with reduction of the after-discharge duration, would suggest effectiveness against focal seizures. In this model, i.p. administration of a solution of 4 yielded an ED50 of 68.5 ± 1.3 mg/kg, with a decrease in seizure score at 45 min and peak activity at 165 min, which reflects noteworthy anticonvulsant activity. The seizure score was markedly reduced from 5 to 1 in four out of eight rats (p = 0.0003), with a mean seizure score of 2.1 ± 0.5. There was no statistically significant effect on the after-discharge duration (p = 0.07). Related sulfamide 3 was highly effective on i.p. administration, but it reduced the after-discharge duration [ED50 = 38.9 mg/kg (peak activity at 15 min; sustained at 1 h); seizure score markedly reduced from 5 to 0 in six out of eight rats (p = 0.0001); mean seizure score of 1.6 ± 0.7/0.9 ± 0.6; statistically significant reduction of after-discharge duration (64%; p < 0.01)].7
A comparison of responses for different agents in this rat kindling model at maximally effective dose levels and at the time of peak effect is presented in Figure 1. With 4, there was a significant reduction in global seizure activity (score < 3) in six out of eight rats. Similar protection was achieved with carbamazepine, but only at doses >26 mg/kg, which reached the range of neurotoxic effects. After treatment with sulfamide 3,7 six out of eight rats showed a complete absence of seizure activity. Similar protection was observed with valproic acid, but only at doses >300 mg/kg, which were in the range of neurotoxic effects. With topiramate, only one out of eight treated rats showed complete protection at a maximally effective dose. Besdies topiramate, ethosuximide was not effective in this model; however, phenytoin, carbamazepine and valproic acid significantly suppressed seizure activity, although at dose levels associated with neurotoxicity (Figure 1). Thus, in this antiepileptic model, sulfamide 4 exhibited good antiseizure activity compared with various marketed antiepileptic drugs, while sulfamide 3 exhibited superior antiseizure activity.
Figure 1.
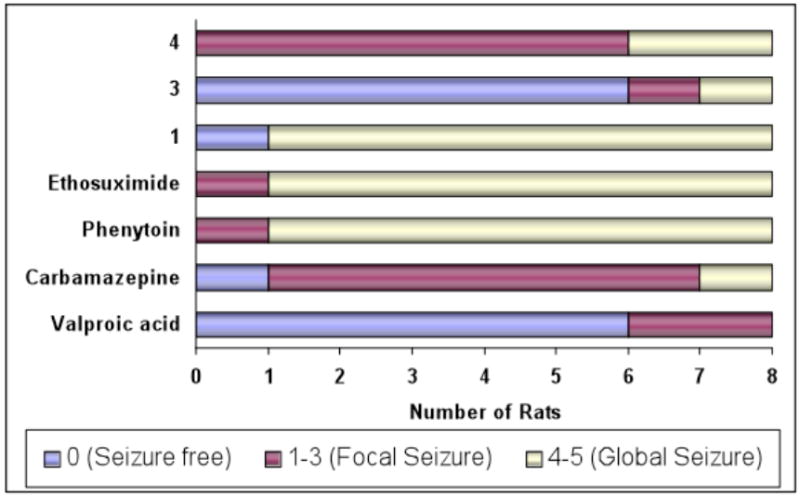
Frequency distribution analysis for individual rats (N = 8 animals) in the hippocampal kindling model after treatment with various anticonvulsants (4, 3, 1, ethosuximide, phenytoin, carbamazepine, and valproic acid; vehicle: 0.5% aqueous methylcellulose). The color code represents the number of rats out of eight that evinced the particular seizure response noted (seizure free, focal seizure, global seizure). Seizure scores were assessed at the time of peak effect and at the maximal effective dose.
The neurotoxicity that we have referred to is acute neurotoxicity, which was evaluated in rats by the NINDS in its anticonvulsant screening process.19 Thus, three other tests were utilized to judge abnormal neurological status: positional sense test, muscle tone test, and gait-and-stance test. A description of these parameters was already reported by us.7 In these tests 4 exhibited no adverse effects at oral doses in rats up to 500 mg/kg. Also, there was a pronounced safety margin between behavioral efficacy (rat oral MES ED50 = 43 mg/kg) and neurotoxicity (rat TD50 > 500 mg/kg) for a protective index (rat oral TD50/MES ED50) of >12-fold. Under the same conditions, the oral protective index for topiramate in rats is 25. The opposite (R)-enantiomer of 4 had a rat oral TD50 of <250 mg/kg, for a protective index of <5-fold, making it much less attractive than 4.
Mechanistic Studies with 4
Anticonvulsant drugs, such as phenytoin, lamotrigine, and topiramate, are known to inhibit voltage-gated Na+ channels.21 Additionally, in human epilepsy the levels of brain Na+ channel mRNA are elevated.22 Consequently, we studied the influence of 4 on Na+ channel function by virtue of whole-cell patch-clamp recordings in CHL1610 cells stably transfected with rat Nav1.2, a voltage-gated Na+ channel highly expressed in the brain. Compound 4 caused a concentration-dependent, voltage-dependent, frequency-dependent block of these channels, with virtually no inhibition at −107 mV with an IC50 value of 36 μM at −67 mV (N = 3). In this assay phenytoin, lamotrigine, and topiramate evinced similar Na+ channel blockade with with IC50 values of 22, 35, and 97 μM, respectively; however, gabapentin had no effect on Na+ channel activity (up to 300 μM). Thus, it appears that inhibition of voltage-gated Na+ channels may contribute to the anticonvulsant activity of 4.
Compound 4 was studied for N-type calcium ion channel activity in the neuronal (N-type) subgroup of voltage-activated calcium channels.23 We employed two different in vitro assays involving the same heterologous cell line expressing the α, β, and α2-δ subunits, which together comprise the functional ion channel. Sulfamide 4 inhibited calcium influx in response to depolarization (fluorescence-based assay) with an IC50 of 34 μM. In a whole-cell, patch-clamp experiment with low-frequency stimulation (0.07 Hz), intended to measure N-type channel activity directly, 4 caused a concentration-dependent increase in inhibition, with an IC50 of 70 μM. Comparing high-frequency (5-Hz) and low-frequency stimulation at 100 μM, 4 showed a marked increase in inhibition, 53 ± 6%, at 5 Hz vs 0.07 Hz, 37 ± 7% (N = 5 cells at each frequency). From these results, 4 is viewed as having moderate potency in blocking use-dependent, N-type calcium ion channels. Inhibition of voltage-activated calcium channels by other anticonvulsant drugs, such as lamotrigine, felbamate, levetiracetam, and topiramate, is thought to contribute to their pharmacological activity in treating epilepsy, neuropathic pain, and migraine.23
Since human mutations in the gene that encodes the voltage-gated potassium ion channel KCNQ2 (Kv7.2) lead to a type of epilepsy, 4 was examined at 300 μM for its effect on whole-cell, patch-clamp recordings of cells stably transfected with rat KCNQ2 under an M-current protocol.24-26 At a membrane potential of −50 mV (N = 5 cells), there was a statistically significant 2.4-fold increase in KCNQ2 current and 2-fold increase in decay kinetics. At an elevated concentration of 750 μM, 4 showed a 4-fold increase in current. From these results, 4 is a KCNQ2 channel opener, particularly at −50 mV.
To investigate the effects on excitatory and inhibitory amino acid receptor currents, whole-cell, patch-clamp studies in cultured primary mouse cortical neurons at −70 mV were performed in collaboration with the NINDS. Sulfamide 4 at 100 μM (N = 5) had statistically significant effects (p < 0.05) with 38% inhibition of the current evoked by 1 μM glycine and 10 μM N-methyl-D-aspartic acid, 15% inhibition of the current evoked by 50 μM kainic acid, and 24% increase in the current evoked by 5 μM γ-aminobutyric acid.26
Compound 4 was profiled in 50 different receptor-binding assays that were conducted at a fixed concentration of 10 μM by the contract laboratory Cerep.16a,27 Thus, 4 was found to have virtually no affinity for GABA and central benzodiazepine receptors, nor for chloride ion channels. The Cerep panel indicated that 4 (at 10 μM) interacts weakly with the cholecystokinin-A receptor (21% inhibition), histamine H1 receptor (23% inhibition), and the Na+-channel site 2 (21% inhibition), and moderately with the kappa-opioid receptor (45% inhibition); however, a follow-up dose-response study with these receptors did not confirm the inhibition results. Dose-response studies indicated weak affinity for the serotonin 5HT2c receptor (29 μM) and the dopamine transporter (16 μM).
ADME Properties of 4
The absorption, distribution, metabolism, and elimination (ADME) properties of 4 were examined.28 Metabolic stability in vitro was assessed by using rat and human liver microsomes (LM). After a 60-min incubation period in the microsomal preparations, the percentages remaining were 92% in rat LM and 100% in human LM, which suggests very high stability to hepatic transformation. The half-life of 4 in a standard human LM preparation was >100 min. A minor hydroxylation metabolite was found in human LM incubations, but the oxidation site on the benzene ring could not be established by MS–MS analysis. Later in vivo studies determined that the minor aromatic hydroxylation mainly occurs at the 7-position.
Pharmacokinetic results in rats and dogs lent support to 4 as a development candidate. In adult male rats, 4 was administered as an i.v. dose of 2 mg/kg and an oral dose of 10 mg/kg. The oral mean Cmax, tmax, F, t1/2, and AUC (total exposure) values in plasma were 9090 ng/mL (33 μM), 53 min, 95%, 8.2 h, and 53,200 ng-h/mL. Linear, dose-related increases in exposure were observed at 10, 30, and 300 mg/kg. After i.v. administration at 2 mg/kg, the volume of distribution at steady state (Vdss) was 390 mL/kg and the clearance (CL) was 96 mL/h-kg. At 45 mg/kg, p.o., the plasma to brain ratio in rats was 1:1 at 1–6 h post-dosing; at 6 h the concentrations in plasma and brain were 61 and 55 μM, respectively. In female beagle dogs, 4 was administered as an i.v. dose of 2 mg/kg and an oral dose of 10 mg/kg. The oral mean Cmax, tmax, F, t1/2, and AUC values in plasma were 11,500 ng/mL (41 μM), 55 min, 83%, 20 h, and 212,000 ng-h/mL. After i.v. administration at 2 mg/kg, the Vdss and CL values were 630 mL/kg and 30 mL/h-kg, respectively. Overall, 4 posseses high oral bioavailability in rats and dogs, with an extended duration in plasma.
CONCLUSION
Compound 4 exhibited excellent broad-spectrum anticonvulsant activity in rodents against audiogenic, electrically-induced, and chemically-induced seizures, with very weak inhibition of human CA-II (IC50 = 35 μM). This low level of CA-II inhibition probably excludes this pharmacological mechanism as a meaningful source of the anticonvulsant action of 4, which is consistent with our perspective that CA inhibition is not an important factor in the anticonvulsant activity of topiramate.10a,b Our mechanistic studies suggest that several mechanisms contribute to the observed pharmacological profile of 4, but that no single mechanism is likely to be a major contributor. Since 4 inhibited Na+, kainate, and KCNQ2 channels to varying degrees, while moderately potentiating GABA current and inhibiting N-methyl-D-aspartic acid current, its action at several targets appears to be responsible for the observed neurostabilizing effects. Given that 4 limited seizure spread and elevated seizure threshold in preclinical animal models, it has the potential to be effective in refractory, or pharmacoresistant, epilepsy. Also, the anticonvulsant pharmacology suggests applicability for treating multiple forms of epilepsy, such as generalized tonic-clonic, complex partial, and myoclonic seizures, at dose levels that confer a favorable safety margin (therapeutic index ≥ 20). Compound 4 was nominated for clinical development and subsequently advanced into human clinical studies.
EXPERIMENTAL SECTION
General Chemical Procedures
Details for general methods are provided in our previous papers.6,10 Methodology for preparing various sulfamide products is also discussed in our previous papers.6,10 Synthetic examples are given for illustrative purposes. Flash-column chromatography was performed with silica gel. The structures of all new compounds were consistent with their 1H NMR and EI-MS mass spectra. The purity of target compounds was confirmed by analytical reverse-phase HPLC and elemental microanalyses results were within 0.4% of theorectical values (see Supporting Information). Elemental microanalysis was performed by either Robertson Microlit Laboratories, Inc., Madison, NJ, or Quantitative Technologies, Inc. (QTI), Whitehouse, NJ.
(2S)-(–)-N-((6-Chloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (4) and Regioisomer (2S)-(–)-N-((7-Chloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (10b). Example of Method A
4-Chlorocatechol (6.94 g, 0.048 mol) and potassium carbonate (6.64 g, 0.048 mol) were stirred in DMF (200 mL) and (R)-glycidyl tosylate (9.12 g, 0.040 mol) was added. The mixture was stirred at 60 °C for 22 h, cooled to room temperature, and diluted with ice water (600 mL). The mixture was extracted with ethyl ether (3 times) and the combined organic solution was washed with 10% aqueous potassium carbonate (3 times), twice with brine, dried (MgSO4), and concentrated in vacuo. The resulting oil (7.60 g, 0.038 mol) of 6-chloro- and 7-chloro- (S)-(2,3-dihydrobenzo[1,4]dioxin-2-yl)methanols (Ia/Ib) was dissolved in pyridine (50 mL) and the solution was cooled to 0 °C. p-Toluenesulfonyl chloride (7.24 g, 0.038 mol) was added and the reaction mixture was stirred at room temperature for 20 h. The reaction was diluted with 1 N HCl (0.75 L) and extracted with ethyl ether (3 × 250 mL). The combined organic solution was washed twice with 1 N HCl, once with water, twice with brine, dried (MgSO4), and concentrated in vacuo. The resulting oil (12.1 g) was purified by flash-column chromatography (heptane/ethyl acetate, 2:1) to yield the tosylate derivatives as a clear, colorless oil (9.80 g, 69%). The oil (9.8 g, 27.7 mmol) was combined with potassium phthalimide (8.14 g, 44 mmol) in DMF (100 mL) and heated at reflux for 1 h. The mixture was cooled to room temperature and poured slowly, portionwise, into vigorously stirring ice water (600 mL), followed by stirring for 30 min. The resulting white solid was filtered, washed several times with water, and dried in air to yield a white powdery phthalimide (IIa/IIb, 7.8 g, 86%). This material was reacted with hydrazine (1.50 g, 47 mmol) in EtOH (115 mL) and the mixture was heated at reflux for 2 h, cooled to room temperature, acidified with 1 N HCl (to pH 1) with stirring, and stirred for 30 min. The white solid was filtered and rinsed with fresh EtOH (solid discarded). The filtrate was evaporated in vacuo to a solid, which was partitioned between ethyl ether and dilute aqueous NaOH. The organic solution was washed once with brine, dried (Na2SO4), and evaporated in vacuo to a yield a light yellow oil (3.94 g, 84%) comprised of (S)-(6-chloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methylamine (IIIa) and (S)-(7-chloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methylamine (IIIb), obtained as a ca. 3:1 ratio of 6-Cl:7-Cl isomers by reverse-phase HPLC. This amine mixture (3.94 g) was dissolved in 2-propanol (100 mL) and 1 N HCl in ethyl ether was added until pH 1 was attained. The hydrochloride salt that precipitated was filtered (2.65 g) and recrystallized from methanol/2-propanol to yield white crystals (2.02 g), which were partitioned between dichloromethane and dilute aqueous NaOH. The organic solution was washed once with brine, dried (Na2SO4), and evaporated in vacuo to yield purified (S)-(6-chloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methylamine as a white solid (IIIa, 1.59 g), [α]D −67.8 (c 1.51, CHCl3). This solid (1.55 g, 7.75 mmol) and sulfamide (1.50 g, 15.5 mmol) in 1,4-dioxane (50 mL) were heated at reflux for 2 h, cooled to room temperature, and evaporated in vacuo to yield a solid. Purification by flash-column chromatography (CH2Cl2/MeOH, 20:1) yielded 4 as a white solid (1.34 g, 62%), which was recrystallized from ethyl acetate/hexanes to afford white crystals mp 98–99 °C; [α]D −59.9 (c 1.11, MeOH); MS (ES) m/z 277 (M-H); 1H NMR (CDCl3) δ 3.45 (m, 2H), 4.05 (dd, J = 7.1, 11.5 Hz, 1H), 4.29 (dd, J = 2.4, 11.5 Hz, 1H), 4.40 (m, 1H), 4.55 (s, 2H), 4.76 (m, 1H), 6.81 (m, 2H), 6.90 (d, J = 2.2 Hz, 1H). Anal. Calcd. (C9H11ClN2O4S): C, 38.78; H, 3.98; N, 10.05. Found: C, 38.80; H, 3.67; N, 9.99. Determination of a single-crystal X-ray structure for one polymorph (“form VI”) confirmed the absolute configuration for 4.28
The filtrates from crystallization of amine hydrochloride salt, in the above preparation, were evaporated in vacuo to yield a solid (ca. 1:1 ratio of 6-Cl:7-Cl isomers), which was partitioned between CH2Cl2 (200 mL) and 0.5 N NaOH (50 mL). The organic solution was washed once with brine, dried (Na2SO4), and concentrated in vacuo to an oil, which was purified via reverse-phase HPLC (10–50% acetonitrile containing 0.16% CF3CO2H) to yield enriched 7-chloro amine (IIIb). This material (0.94 g, 4.7 mmol) and sulfamide (0.90 g, 9.4 mmol) in 1,4-dioxane (25 mL) were refluxed for 2.5 h, cooled to room temperature, and evaporated in vacuo to an oil, which was purified by flash-column chromatography (CH2Cl2/MeOH, 10:1) to yield 10b as a white solid (0.85 g, 65%). A sample was recrystallized from ethyl acetate/hexane to give white solid, mp 116-118 °C; [α]D −40.8 (c 1.17, MeOH); MS (ES) m/z 277 (M-H); 1H NMR (CDCl3/CD3OD) δ 3.38 (m, 2H), 4.04 (dd, J = 7.0, 11.6 Hz, 1H), 4.30 (dd, J = 2.3, 11.6 Hz, 1H), 4.37 (m, 1H), 6.81 (m, 2H), 6.88 (d, J = 0.7 Hz, 1H). Anal. Calcd (C9H11ClN2O4S): C, 38.78; H, 3.98; N, 10.05; S, 11.51. Found: C, 38.79; H, 3.59; N, 9.97; S, 11.41.
(2R)-(–)-N-((6-Chloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (ent-4)
The (R)-enantiomer of 4 (ent-4) was prepared in the same manner as for the (S)-enantiomer (4), but with the use of (S)-glycidyl tosylate as a reactant. The same chemical transformations were employed to obtain the final crude product, which was recrystallized from ethyl acetate/hexanes to afford white crystals of ent-4, mp 99–100 °C; [α]D 59.7 (c 1.33, MeOH); MS (ES) m/z 277 (M–H); 1H NMR (CDCl3) δ 3.45 (m, 2H), 4.05 (dd, J = 7.1, 11.6 Hz, 1H), 4.29 (dd, J = 2.4, 11.6 Hz, 1H), 4.40 (m, 1H), 4.56 (s, 2H), 4.76 (m, 1H), 6.81 (m, 2H), 6.90 (d, J = 2.2 Hz, 1H). Anal. Calcd (C9H11ClN2O4S): C, 38.78; H, 3.98; N, 10.05; S, 11.51. Found: C, 38.77; H, 3.64; N, 9.87; S, 11.62.
N-((2,3-Dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (5)
Commercially available rac-2,3-dihydro-1,4-benzodioxin-2-ylmethylamine (4.4 g, 26 mmol) and sulfamide (5.1 g, 53 mmol) were combined in 1,4-dioxane (100 mL) and refluxed for 2 h. The reaction was cooled to room temperature and a small amount of solid was filtered and discarded. The filtrate was evaporated in vacuo and the residue was purified by using flash-column chromatography (CH2Cl2:MeOH, 10:1) to yield a white solid, which was recrystallized from CH2Cl2 to yield title compound 5 as a white solid, mp: 97.5–98.5 °C; MS (ES) m/z 243 (M–H); 1H NMR (DMSO-d6) δ 3.10 (m, 1H), 3.20 (m, 1H), 3.97 (dd, J = 6.9, 11.4 Hz, 1H), 4.28 (m, 2H), 6.68 (bd s, 3H, NH), 6.85 (m, 4H). Anal. Calcd (C9H12N2O4S): C, 44.25; H, 4.95; N, 11.47; S, 13.13. Found: C, 44.28; H, 4.66; N, 11.21; S, 13.15.
(2S)-(–)-N-((2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (S-5)
According to Method A, catechol (13.2 g, 0.12 mol) and potassium carbonate (16.6 g, 0.12 mol) were stirred in DMF (250 mL) and (R)-glycidyl tosylate (22.8 g, 0.10 mol) was added, followed by stirring at 60 °C for 24 h. The reaction was worked up to yield a white solid, which was purified by flash-column chromatography (CH2Cl2:MeOH, 50:1) to yield (2S)-(2,3-dihydrobenzo[1,4]dioxin-2-yl)methanol. This alcohol was processed according to Method A to give crude (2S)-(2,3-dihydrobenzo[1,4]dioxin-2-yl)methylamine as a light yellow oil. The oil was purified by flash-column chromatography (CH2Cl2:MeOH, 10:1) to yield a purified oil. A portion of this oil (4.82 g, 29 mmol) in 2-propanol (250 mL) was treated with 1 N HCl (30 mL), heated on steam bath until homogeneous, and let cool to room temperature. After 3 h, the mixture was ice cooled for 2 h to give a flaky solid, which was recrystallized from 2-propanol to yield a white solid HCl salt, [α]D −69.6 (c 1.06, EtOH). This white solid was partitioned between CH2Cl2 and dilute NaOH; the organic solution was dried (NaSO4) and concentrated in vacuo to yield oily (2S)-(2,3-dihydrobenzo[1,4]dioxin-2-yl)methylamine, [α]D −57.8 (c 1.40, CHCl3). The oil (2.1 g, 12.7 mmol) and sulfamide (2.44 g, 25.4 mmol) were refluxed in 1,4-dioxane (75 mL) for 2 h and the crude product was purified by flash-column chromatography (CH2Cl2:MeOH, 10:1) to yield a white solid, which was recrystallized from CH2Cl2 to yield the title compound as a white crystalline solid, mp 102–103 °C; [α]D −45.1 (c 1.05, MeOH); 1H NMR (DMSO-d6) δ 3.10 (dd, J = 6.9, 13.7 Hz, 1H), 3.20 (dd, J = 5.5, 13.7 Hz, 1H), 3.97 (dd, J = 6.9, 11.4 Hz, 1H), 4.3 (m, 2H), 6.81 (bd s, 3H, NH), 6.86 (m, 4H). Anal. Calcd (C9H12N2O4S): C, 44.25; H, 4.95; N, 11.47; S, 13.13. Found: C, 44.20; H, 4.69; N, 11.40; S, 13.22.
N-((2,3-Dihydrobenzo[1,4]dioxin-2-yl)methyl)-N-methylsulfamide (6)
Racemic (2,3-dihydro-1,4-benzodioxin-2-yl)methylamine (825 mg, 5 mmol) in ethyl formate (15 mL) was refluxed for 30 min and evaporated in vacuo to yield N-((2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)formamide as an oil. This oil in diethyl ether (25 mL) was treated with 1 M LiAlH4 in THF (9.0 mL, 9.0 mmol) at 0 °C and stirred for 5 h at room temperature. The reaction was cooled in an ice bath and quenched with water (0.50 mL), followed by 3 N NaOH (0.50 mL) and water (0.50 mL). The mixture was stirred at room temperature for 1 h and the solid was filtered off. The filtrate was evaporated in vacuo to yield a residue, which was partitioned between 1 N HCl and diethyl ether. The aqueous phase was basified with 1 N NaOH and extracted with diethyl ether. The organic phase was dried (MgSO4) and evaporated in vacuo to yield N-((2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)-N-methylamine as an oil (0.40 g). MS (ES) m/z 180 (MH+); 1H NMR (CDCl3) δ 2.50 (s, 3H), 2.85 (m, 2H), 4.02 (dd, J = 7.9, 11.6 Hz, 1H), 4.30 (m, 2H), 6.85 (m, 4H). A solution of this amine (380 mg, 2.1 mmol) and sulfamide (820 mg, 8.5 mmol) in 1,4-dioxane (15 mL) was refluxed for 1.5 h and evaporated in vacuo to yield a crude residue. The residue was purified via flash-column chromatography (ethyl acetate/heptane, 1:1) and the resultant solid was recrystallized from ethyl acetate/hexane to yield 6 as a white solid (330 mg), mp 97–98 °C. MS (ES) m/z 257 (M–H); 1H NMR (CDCl3) δ 2.99 (s, 3H), 3.40 (dd, J = 5.9, 14.9 Hz, 1H), 3.51 (dd, J = 6.7, 14.9 Hz, 1H), 4.05 (dd, J = 6.5, 11.5 Hz, 1H), 4.29 (dd, J = 2.3, 11.5 Hz, 1H), 4.46 (m, 1H), 4.52 (bd s, 2H), 6.86 (m, 4H). Anal. Calcd (C10H14N2O4S): C, 46.50; H, 5.46; N, 10.85; S, 12.41. Found: C, 46.48; H, 5.65; N, 10.90; S, 12.07.
N-((2,3-Dihydrobenzo[1,4]dioxin-2-yl)methyl)-N’,N’-dimethylsulfamide (7)
Racemic 2,3-dihydro-1,4-benzdioxin-2-ylmethylamine (8.25 g, 5.0 mmol) and triethylamine (1.52 g, 15 mmol) were combined in DMF (10 mL) and cooled in an ice bath as dimethylsulfamoyl chloride (1.44 g, 10 mmol) was added. The reaction mixture was stirred for 3 h with continued cooling, after which the reaction mixture was partitioned between ethyl acetate and water, and the organic solution was washed with brine, dried (MgSO4) and evaporated in vacuo to yield an oil. The oil was purified using flash-column chromatography (EtOAc:heptane, 1:1) to yield a white solid, which was recrystallized (EtOAc/hexanes) to yield title compound 7 as a white floccular solid, mp 76–78 °C; MS (ES) 273 (MH+); 1H NMR (CDCl3) δ 2.82 (s, 6H), 3.36 (m, 2H), 4.04 (dd, J = 7.0, 11.4, 1H), 4.27 (dd, J = 2.3, 11.4 Hz, 1H), 4.35 (m, 1H), 4.59 (bd m, 1H, NH), 6.87 (m, 4H). Anal. Calcd (C11H16N2O4S): C, 48.52; H, 5.92; N, 10.29; S, 11.78. Found: C, 48.63; H, 5.62; N, 10.20; S, 11.90.
N-(Chroman-2-ylmethyl)sulfamide (8). Example of Method B
Chroman-2-carboxylic acid (4.5 g, 25 mmol) and 1-hydroxybenzotriazole (3.86 g, 25 mmol) were combined in CH2Cl2 (40 mL) and DMF (10 mL). 1-Ethyl-3-dimethylaminopropylcarbodiimide (EDC; 4.84 g, 25 mmol) was added at room temperature and the mixture was stirred for 30 min; ammonium hydroxide (28%, 2.26 mL, 33.4 mmol) was added and the mixture was stirred for 16 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and water (50 mL), and the pH was adjusted to ~3 (pH paper) by addition of 1 N HCl. The organic phase was separated and the aqueous phase was extracted twice with CH2Cl2. The combined organic solution was dried (Na2SO4) and evaporated in vacuo to yield an oil, which was purified with flash-column chromatography (ethyl acetate) to yield the carboxamide as a white solid (4.35 g, 97%). 1H NMR (CDCl3) δ 2.10 (m, 1H), 2.40 (m, 1H), 2.75–2.95 (m, 2H), 4.54 (dd, J = 3.0, 9.4 Hz, 1H), 5.50 (bd s, 1H), 6.55 (bd s, 1H), 6.85–6.95 (m, 2H), 7.10–7.20 (m, 2H). The amide (5.35 g, 30 mmol; from several preparations) in THF (90 mL) was treated with 1 M LiAlH4 in THF (36 mL, 36 mmol) with stirring and the mixture was stirred at room temperature for 20 h. The reaction was cooled in an ice bath, quenched (1.4 mL of water, 1.4 mL of 3 N NaOH, 2.8 mL of water), and stirred for 2 h at room temperature. The solids were filtered; the filtrate was dried (K2CO3) and evaporated in vacuo to an oil, which was purified via flash-column chromatography (ethyl acetate/MeOH/ammonium hydroxide, 90:9:1) to yield chroman-2-ylmethylamine (3.50 g, 71%) as an oil, MS (ES) m/z 164 (MH+). A mixture of this amine (1.63 g, 10 mmol) and sulfamide (1.92 g, 20 mmol) in 1,4-dioxane (50 mL) was refluxed for 2 h, cooled, and evaporated in vacuo to an oil, which was purified via flash-column chromatography (CH2Cl2/MeOH, 10:1) to give a white solid. Recrystallization from ethyl acetate/hexane furnished 8 as a white solid (1.52 g, 63%), mp 100–101 °C; MS (ES) m/z 241 (M–H); 1H NMR (CDCl3) δ 1.85 (m, 1H), 2.05 (m, 1H), 2.85 (m, 2H), 3.35 (m, 1H), 3.50 (m, 1H), 4.22 (m, 1H), 4.59 (s, 2H), 4.91 (s, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.87 (dd, J = 7.3, 7.5 Hz, 1H), 7.10 (m, 2H). Anal Calcd. (C10H14N2O3S): C, 49.57; H, 5.82; N, 11.56; S, 13.23. Found: C, 49.57; H, 5.80; N, 11.75; S, 13.33.
N-(2-(2,3-Dihydrobenzo[1,4]dioxin-2-yl)ethyl)sulfamide (9)
Potassium cyanide (2.05 g, 31.5 mmol) was added to 2-bromomethyl(2,3-dihydrobenzo[1,4]dioxole) (6.87 g, 30 mmol) in DMSO (90 mL) and the mixture was stirred at room temperature for 20 h. The reaction mixture was diluted with water (250 mL) and extracted twice with diethyl ether. The organic solution was washed with water, washed twice with brine, dried (Na2SO4), and evaporated in vacuo to yield 2-cyanomethyl-(2,3-dihydrobenzo[1,4]dioxole) as a white solid (4.90 g, 93%). 1H NMR (CDCl3) δ 2.78 (d, J = 6.1 Hz, 2H), 4.08 (dd, J = 6.2, 11.6 Hz, 1H), 4.31 (dd, J = 2.3, 11.5 Hz, 1H), 4.50 (m, 1H), 6.89 (m, 4H). This material was dissolved in THF (50 mL), treated with 1 M BH3-THF (80 mL, 80 mmol), and the mixture refluxed for 5 h, then stirred at room temperature for 16 h. With ice-bath cooling, 2 N HCl was added until pH 1 was achieved. The reaction mixture was stirred for 1 h at room temperature and evaporated in vacuo to yield an oil. The oil was partitioned between 3 N NaOH and diethyl ether, and the diethyl ether solution was washed with brine, dried (Na2SO4), and evaporated in vacuo to yield crude 2-(2,3-dihydrobenzo[1,4]dioxin-2-yl)ethylamine (2.80 g, 58%), MS (ES) m/z (MH+) 180. This amine (2.80 g, 15.6 mmol) in 1,4-dioxane (100 mL) was combined with sulfamide (3.0 g, 31 mmol) and heated to reflux for 2 h. The solution was cooled and evaporated in vacuo to give an orange solid, which was purified by flash-column chromatography (CH2Cl2/MeOH, 10:1) to yield a white solid. The solid was recrystallized from CH2Cl2 to yield 9 as a white solid (2.12 g), mp 101–103 °C; MS (ES) m/z (M–H) 257; 1H NMR (CDCl3) δ 1.94 (dd, J = 6.5, 12.9, 2H), 3.43 (dd, J = 6.4, 12.9 Hz, 2H), 3.94 (dd, J = 7.4, 11.3 Hz, 1H), 4.30 (m, 2H), 4.52 (s, 2H), 4.70 (m, 1H), 6.86 (m, 4H). Anal. Calcd (C10H14N2O4S): C, 46.50; H, 5.46; N, 10.85; S, 12.41. Found: C, 46.48; H, 5.60; N, 10.81; S, 12.41.
(2S)-(–)-N-((6-Fluoro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (10e)
A DMF (200 mL) solution of 4-fluoro-2-hydroxyacetophenone (8.94 g, 58 mmol) and potassium carbonate (8.84 g, 64 mmol) was treated with (R)-glycidyl m-nitrophenylsulfonate (15.0 g, 58 mmol) and the reaction stirred at 40–45 °C for 7 h. The reaction was cooled to room temperature and poured into water (500 mL) and extracted with t-butyl methyl ether (3 × 350 mL). The combined organic solution was washed twice with brine, dried (MgSO4), and evaporated in vacuo to white solid (11.4 g, 94%). This solid and m-chloroperbenzoic acid (MCPBA, 14.6 g, 65 mmol) in CH2Cl2 (160 mL) was refluxed for 4 h, then let stand at room temperature for 16 h. An additional amount of MCPBA was added (8 g) and the mixture was refluxed for 4 h and then stirred at room temperature for 16 h. The white solid m-chlorobenzoic acid was filtered and rinsed several times with CH2Cl2. The filtrate was added to 10% aqueous sodium bisulfite (250 mL), stirred for 10 min, and added cautiously to saturated aqueous sodium bicarbonate. The separated organic solution was washed twice with saturated NaHCO3, once with brine, dried (MgSO4), and evaporated in vacuo to an oil (11.7g). To a stirred solution of this material (11.4 g, 50.4 mmol) in MeOH (100 mL) at room temperature was added dropwise 25% NaOMe in MeOH (14.4 mL, 63 mmol) over 15 min, followed by stirring for 2 h. The reaction was evaporated in vacuo to a solid, which was partitioned between CH2Cl2 and water (200 mL each). The organic phase was separated and washed with water, twice with brine, dried (MgSO4), and evaporated in vacuo to afford the title alcohol as a white solid (7.50 g, 81%). MS (ES) m/z 185 (MH+); 1H NMR (CDCl3) δ 2.01 (t, J = 6.2 Hz, 1H), 3.75–3.95 (m, 2H), 4.10 (m, 1H), 4.21 (m, 1H), 4.29 (dd, J = 2.1, 11 Hz, 1H), 6.5–6.7 (m, 2H), 6.81 (dd, J = 5.4, 8.9 Hz, 1H). This alcohol was converted into (2S)-(–)-N-((6-fluoro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (10e) by using the relevant four synthetic steps described in Method A, involving sequential treatment with p-TsCl, potassium phthalimide, hydrazine, and sulfamide (see above).
(2S)-(–)-N-((6,7-Dichloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (10h)
According to Method A, 4,5-dichlorocatechol (8.6 g, 48 mmol), potassium carbonate (6.64 g, 48 mmol), and (R)-glycidyl tosylate (9.12 g, 40 mmol) were reacted in DMF (200 mL) to afford (2S)-2-(6,7-dichloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methanol, as a viscous oil. This alcohol was converted into (2S)-(–)-N-((6-fluoro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (10e) by using the relevant four synthetic steps described in Method A, involving sequential treatment with p-TsCl, potassium phthalimide, hydrazine, and sulfamide (see above). Crude 10h was purified by flash-column chromatography (CH2Cl2:MeOH, 20:1) to yield a white solid, which was recrystallized from ethyl acetate/hexanes to yield a purified white crystalline solid, mp 119–121 °C; [α]D −53.4 (c 1.17, MeOH); MS (ES) [M–H] 311; 1H NMR (DMSO-d6) δ 3.15 (m, 2H), 4.05 (dd, J = 6.5, 11.5 Hz, 1H), 4.35 (m, 2H), 6.68 (bd s, 2H), 6.91 (bd s, 1H), 7.20 (s, 1H), 7.22 (s, 1H). Anal. Calcd (C9H10Cl2N2O4S): C, 34.52; H, 3.22; N, 8.95; Cl, 22.64; S, 10.24. Found: C, 34.52; H, 3.22; N, 8.95; Cl, 22.64; S, 10.24.
(2S)-(–)-N-((7-Methyl-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl)sulfamide (10i)
According to Method A, 4-methylcatechol was converted to the target compound, a white solid that was recrystallized from ethyl acetate/hexane to give a purified white solid, MS (ES) [M–H] 257; 1H NMR (CDCl3) δ 2.25 (s, 3H), 3.45 (m, 2H), 4.03 (dd, J = 6.9, 11.4 Hz, 1H), 4.28 (m, 1H), 4.40 (m, 1H), 4.57 (bd s, 1H), 4.80 (m, 1H), 6.66 (m, 2H), 6.76 (m, 1H), 7.20 (s, 1H), 7.22 (s, 1H). Anal. Calcd (C10H14N2O4S): C, 46.50; H, 5.46; N, 10.85; S, 12.41. Found: C, 46.65; H, 5.60; N, 10.84; S, 12.61.
N-((Benzo[1,3]dioxol-2-yl)methyl)sulfamide (11a)
Catechol (10.26 g, 93.2 mmol), sodium methoxide (25% by weight in MeOH, 40.3 g, 186 mmol), and methyl dichloroacetate (13.3 g, 93.2 mmol) were combined in dry MeOH (100 mL), and the solution was heated at reflux overnight. The reaction was cooled to room temperature, acidified with concentrated HCl, and reduced in volume under vacuum to about 50 mL. Water was added and the mixture was extracted with ethyl ether (3 × 100 mL). The combined organic solution was dried (MgSO4) and concentrated. The brown semisolid was flash-column chromatographed (ethyl acetate/hexane, 2:98) to yield methyl benzo[1,3]dioxole-2-carboxylate as a colorless oil, MS (ESI) m/z 195 (MH+); 1H NMR (300 MHz, CDCl3) δ 1.33 (t, J =7 Hz, 3H), 4.34 (q, J = 7 Hz, 2H), 6.29 (s, 1H), 6.89 (broad, 4H). This material (7.21 g, 40.0 mmol) was treated with ammonium hydroxide (29% in water, 10 mL) and enough acetonitrile to make the mixture homogeneous (~5 mL). The solution was stirred for 2 h at room temperature and diluted with distilled water. Benzo[1,3]dioxole-2-carboxamide precipitated as a white solid, which was collected by filtration and used without further purification, MS (ESI) m/z 160 (MH+); 1H NMR (300 MHz, DMSO-d6) δ 6.30 (s, 1H), 6.86 (m, 2H), 6.94 (m, 2H), 7.72 (s, broad, 1H), 7.99 (s, broad, 1H). This amide (5.44 g, 32.9 mmol) was dissolved in THF (100 mL) and LiAlH4 (1 M in THF, 39.5 mL, 39.5 mmol) was added slowly at room temperature with stirring. The reaction was stirred at 23 °C for 24 h. Distilled water was added carefully to destroy the excess LiAlH4. Aqueous NaOH (3 N, 100 mL) was added and the solution was extracted with ethyl acetate (3 × 100 mL). The combined organic solution was washed with water, dried (MgSO4), and concentrated to yield (benzo[1,3]dioxol-2-yl)methylamine as a colorless oil, MS (ESI) m/z 152 (MH+); 1H NMR (300 MHz, CDCl3) δ 3.13 (d, J = 4 Hz, 2H), 6.09 (t, J = 4 Hz, 1H), 6.87 (m, 4H). This amine (2.94 g, 19.4 mmol) and sulfamide (3.74 g, 38.9 mmol) were combined in dry 1,4-dioxane (50 mL) and the mixture was heated at reflux overnight. The reaction was concentrated and the residue was flash-column chromatographed (2–10% acetone in CH2Cl2) to yield 11a as a white solid, MS (ESI) m/z 230 (MH+); 1H NMR (300 MHz, CDCl3) δ 3.64 (d, J = 4 Hz, 2H), 4.62 (broad, 1H), 4.79 (broad, 1H), 6.25 (t, J = 4 Hz, 1H), 6.87 (m, 4H).
N-((3,4-Dihydro-2H-benzo[b][1,4]dioxepin-3-yl)methyl)sulfamide (12)
Catechol (IV, 5.09 g, 46.2 mmol) and potassium carbonate were combined in acetonitrile and refluxed for 1 h. 2-Chloromethyl-3-chloro-1-propene (5.78 g, 46.2 mmol) was added and the reaction was refluxed for 24 h. The solution was cooled to room temperature and filtered. The filtrate was evaporated; the residue was diluted with water and extracted with ethyl ether (3x). The combined organic solution was dried (MgSO4) and concentrated in vacuo. Flash-column chromatography (ethyl ether/hexane, 2:98) gave 3-methylene-3,4-dihydro-2H-benzo[b][1,4]dioxepine (V) as a colorless oil, MS (ESI) m/z 163 (MH+); 1H NMR (300 MHz, CDCl3) δ 4.76 (s, 4H), 5.07 (s, 2H), 6.94 (m, 4H). This material (5.00 g, 30.8 mmol) was dissolved in dry THF (100 mL) and borane-THF (1.0 M in THF, 10.3 mL) was added at 0 °C. After stirring at 23 °C for 5 h, hydroxylamine-O-sulfonic acid (6.97 g, 61.6 mmol) was added to aminate the organoborane intermediate29 and the reaction was heated at reflux overnight. The reaction was cooled to room temperature and aqueous NaOH (3 N, 100 mL) was added. The solution was extracted with ethyl acetate (3 × 100 mL), and the combined organic solution was dried (MgSO4) and concentrated under vacuum. Purification by chromatography (2–8% MeOH in CH2Cl2) yielded ((3,4-dihydro-2H-benzo[b][1,4]dioxepin-3-yl)methyl)amine (VI) as a colorless oil, MS (ESI) m/z 180 (MH+); 1H NMR (300 MHz, DMSO-d6) δ 2.30 (m, 1H), 2.72 (d, J = 4 Hz, 1H), 3.16 (d, J = 4 Hz, 1H), 3.33 (broad, 2H), 4.07 (m, 2H), 4.21 (m, 2H), 6.92 (m, 4H). This amine (2.90 g, 16.2 mmol) and sulfamide (3.11 g, 32.4 mmol) were combined in dry 1,4-dioxane (60 mL) and heated at reflux overnight. Chloroform was added and the precipitate was removed by filtration. The filtrate was concentrated under vacuum and purified by flash-column chromatography (2–8% acetone in CH2Cl2) to yield 12 as an off-white solid, MS (ESI) m/z 259 (MH+); 1H NMR (300 MHz, DMSO-d6) δ 2.39 (m, 1H), 3.00 (m, 2H), 4.04 (m, 2H), 4.19 (m, 2H), 6.59 (broad, 2H), 6.71 (broad, 1H), 6.92 (m, 4H).
Maximal Electroshock Seizure (MES) Test in Mice and Rats
The MES test procedure described by Swinyard et al. was employed.14 In experiments with mice, a 60-Hz current of 50-mA intensity was applied through corneal electrodes for a 0.2-s duration; with rats, seizure activity was induced by delivery of a 150-mA current (0.2 s) to the cornea. Both procedures caused immediate hind-limb tonic extension. Absence of tonic extension suggests that the test compound is able to prevent the spread of seizure discharge in neural tissue. Efficacy in this model is very predictive of clinical utility against tonic and/or clonic generalized seizures.
Male CD-1 mice (Charles River Laboratories; 22–28 g) were fasted overnight prior to administering test compounds. Compounds were administered orally (p.o.) as a suspension in 0.5% aqueous methylcellulose at various time points (0.5–8 h) prior to seizure testing. Ten mice per dose and eleven doses were used to establish ED50 values; i.e., the calculated dose required to block the hind-limb tonic-extensor component of the maximal electroshock seizure in 50% of the mice tested. The ED50 values, determined at the time of peak activity, were calculated by nonlinear regression using the Sigmoidal Emax model (Pharsight WinNonlin Program).
Additional MES testing was performed as described above (by NeuroAdjuvants) except with CF-1 mice (Charles River Laboratories; 18–25 g) and the ED50 values were determined at 2 h following oral (p.o.) administration.
In studies conducted at the NINDS, male Sprague–Dawley rats (Simonsen Laboratories; 100–150 g) were food deprived just prior to testing. Eight rats per dose and four doses (test compound in 0.5% aqueous methylcellulose) were used to establish ED50 values, which were determined at the time of peak activity after oral administration.
Chemically-Induced Seizures in Mice.14b,19,30
These studies were conducted at the NINDS. Three convulsant compounds, bicuculline (Bic), picrotoxin (Pic), and pentylenetetrazol (PTZ), were used to induce seizures in male CF-1 albino mice (Charles River Laboratories; 18–25 g). Fifteen minutes post-dosing with either vehicle (0.5% aqueous methylcellulose) or test compound, Bic (2.7 mg/kg), Pic (3.15 mg/kg), or PTZ (85 mg/kg) were administered subcutaneously (s.c.) at doses calculated to induce forelimb clonic seizures for 3 s in 97% of the mice (CD97). Animals not displaying a clonic seizure within the prescribed time frame, 30 min (Bic or PTZ) or 45 min (Pic), were considered protected. Eight mice per dose and at least four doses were used to establish ED50 values. Anticonvulsant activity against these three convulsants indicates an ability to protect against threshold seizures, i.e., to raise the seizure threshold.
The intravenous pentylenetetrazol (iv PTZ) seizure threshold test was also performed.31 In this case, the test compound does not have to completely prevent seizure occurrence, but just delay its appearance, indicating a the ability to modify seizure threshold. The iv PTZ test can determine if a compound can increase (anticonvulsant) or decrease (proconvulsant) seizure threshold. Two doses of the compound were employed, the first dose corresponding to the ED50 for protection against MES seizures and the second dose corresponding to the median toxic dose (TD50; dose that produces rotorod impairment in 50% of mice tested). Nine mice per dose were injected i.p. with vehicle (0.5% aqueous methylcellulose) or test compound. Fifteen minutes post-dosing, a convulsant solution of PTZ (0.5% PTZ in 0.9% saline containing 10 USP units/mL of heparin sodium) was infused into the tail vein at a constant rate of 0.34 mL/min. The time in seconds from the start of the infusion to the appearance of the first twitch and the onset of clonus was recorded. The times to each end point were converted to mg/kg of PTZ for each mouse in the vehicle and test compound groups. An increase in the dose of PTZ to produce a first twitch or clonic seizure suggests that the test compound can increase seizure threshold (anticonvulsant).
Audiogenic Seizures in Mice32
These studies were conducted at the NINDS. The ability of the test compound to prevent sound-induced seizures was evaluated in adult male and female Fring’s AGS mice (20–25 g; in-house breeding colony at the University of Utah, Salt Lake City). Individual mice were placed into a Plexiglass cylinder (diam, 15 cm; ht, 18 cm) fitted with an audio-transducer and exposed to a sound stimulus of 110 db (11 kHz) delivered for 20 s. Sound-induced seizures are characterized by wild running followed by loss of righting reflex with forelimb and hindlimb tonic extension. Drug-treated mice that did not display hindlimb tonic extension were considered protected. There were eight mice per dose and a minimum of four doses, which established an ED50 value at the time to peak effect. Test compound or vehicle (0.5% aqueous methylcellulose) was administered i.p. 1 h prior to sound induction.
Kindling Studies in Rats19
Adult male Sprague–Dawley rats (300–400 g) were surgically implanted with bipolar electrodes placed in the hippocampus. Rats were kindled by repetitive electrical stimulation (50 Hz, 10-s train of 1-ms biphasic 200-μA pulses every 30 min for 6 h every other day for a total of 60 stimulations) resulting in stage 5 bilateral motor seizures. One week later, the rats received 2–3 supra-threshold stimulations every 30 min before treatment with test compound to ensure stability of the behavioral seizure stage and after-discharge duration. Fifteen minutes after the last stimulation, a single dose of vehicle or test compound was administered i.p.; after 15 min, each rat was stimulated every 30 min for 3–4 h. After each stimulation, individual seizure scores and after-discharge durations were recorded. The group scores (mean ± SEM) were calculated for each parameter, with eight rats per dose and at least four doses being used to establish the ED50 values. Efficacy was measured in terms of the ability of a compound to modify the seizure score (severity of spread) and after-discharge duration (excitability) of the generalized seizures.
Voltage-Gated Na+ Channels33
Sodium channel activity was studied by using a whole-cell patch-clamp technique in CHL1610 cells that stably express rat Nav1.2. A 45-s preconditioning pulse (at −107 and −67 mV) was followed by 3 s of brief (5 ms) depolarizations to −7 mV at 10 Hz. The membrane potential during the time between each depolarization was the same as the preconditioning voltage. The interval between each preconditioning pulse was 15 s, during which time the cell was held at −107 mV. The extracellular solution perfusing the cell contained NaCl (132 mM), KCl (5.4 mM), CaCl2 (1.8 mM), MgCl2 (0.8 mM), HEPES (10 mM), and glucose (10 mM) at pH 7.4. The pipette solution contained CsCl (45 mM), CsF (100 mM), EGTA (5.0 mM), HEPES (10 mM), and glucose (5.0 mM) at pH 7.4. Data were acquired both in the absence and presence (after 2–3 min application) of the test compound. All experiments were performed at 22 °C. The peak current amplitude during the 30th 5-ms depolarization to −7 mV was used to determine percent inhibition by the compounds tested. To obtain the 50% inhibitory concentration (IC50) value, the concentration–response data were fitted to a logistic function of the form: R = 100 − 100/(1 + C/IC50)p, where R is the percentage inhibition, p is the slope coefficient, and C is the concentration of the test compound.
Voltage-Gated N-type Ca2+ Channels33c,34
Compounds were prepared as 1 M solutions in DMSO and diluted to the indicated concentrations in either “patch-clamp control buffer,” containing 121 mM triethylamine-HCl, 10 mM BaCl2, 1.0 mM MgCl2, 10 mM HEPES, and 10 mM glucose (pH 7.4) or, for calcium-imaging studies, Hank’s Balanced Salt Solution (Invitrogen), to which was added 2.5 mM CaCl2, 20 mM HEPES, and 0.1% bovine serum albumin. MVIIA was prepared as a 200 μM stock solution in patch-clamp control buffer containing 0.3% bovine serium albumin. The N-type voltage-gated calcium channel stable cell line was generated with HEK cells by expressing rat Cav2.2 (α1B) subunit in pcDNA3.1 (Genbank# AAO53230) and α2δ and β3 in pBudCE4.1 vectors under selection by 400 μg/mL of G418 and 200 μg/mL of Zeocin, respectively. Cells were clonally isolated, expanded, and screened by Western blot analyses, and then further tested for expression of characteristic N-type calcium currents by whole-cell patch-clamp. Rat Cav2.2-containing channels were tested by using a calcium indicator dye detection system (FDSS, Hamamatsu) and the whole-cell patch-clamp technique (EPC-10 amplifier with Pulse software, HEKA). For the FDSS assay, peak responses to 50 mM KCl stimulation were recorded. Responses with different concentrations of test compounds were normalized to the peak control response to 50 mM KCl (set to 100%) and the peak response in the presence of 200 nM MVIIA (set to 0%). The data from four wells for each concentration were averaged, and the averaged points were subjected to a nonlinear regression curve fit (GraphPad Prism) to determine the IC50 values. For whole-cell patch-clamp, current (in pA) was measured in a 20-ms window surrounding the peak current evoked during the step to +20 mV for each pulse. The amount of current at the end of a given drug concentration was normalized to the control level determined at the start of the experiment. The percent inhibition of a given concentration of test compound was calculated as: 100 − [100 × (pA in compound)/(pA in control)]. For determining the IC50, the percent inhibition at each concentration (1, 10, 30, 100 and 300 μM in 3, 6, 10, 8, and 5 cells, respectively) was averaged across all cells tested at that concentration and the average data for all concentrations tested were fit to the same logistic function as above. Where applicable, data were analyzed by a Student’s t-test and are expressed as the mean ± SEM.
Neurotoxicity in Mice35
To determine sedative and/or ataxic side effects for the test compounds, the standard rotorod toxicity test was performed. The time of peak effect for minimal motor impariment on the rotorod was determined following administration of 100 mg/kg (i.p.) of compound. Mice were observed by a trained technician on a rotating rotorod (6 rpm) for 1 min to check for motor impairment, ataxia, or other signs of behavioral toxicity. Motor impairment was defined as the inability of a mouse to maintain equilibrium for 60 s in three consecutive trials on the rotorod.
Carbonic Anhydrase Inhibition
The inhibition of CA-II was performed by the pH shift method, which involves the hydration of CO2. We described the procedure for this assay in earlier papers.10a-d An analogous assay method was performed by Cerep, an independent contract laboratory.16,17
Supplementary Material
Acknowledgments
We thank Jung Lee, Leonard Hecker, Tasha Hutchinson, and Yan Wang for technical assistance. We are grateful to the National Institute of Neurological Disorders and Stroke (NINDS) for conducting studies involving anticonvulsant pharmacological models, with special thanks to James Stables.
ABBREVIATIONS USED
- ADME
adsorption, distribution, metabolism, and excretion
- AUC
area under the curve
- Bic
bicuculine
- CA
carbonic anhydrase
- CNS
central nervous system
- GABA
γ-aminobutyric acid
- LM
liver microsomes
- MES
maximal electroshock seizure
- NMDA
N-methyl-D-aspartic acid
- Pic
picrotoxin
- PTZ
pentylenetetrazol
Footnotes
Supporting Information
Elemental microanalyses and accurate mass data for the test compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.World Health Organization. [2 June 2013];Media Center, Epilepsy Fact Sheet. http://www.who.int/mediacentre/factsheets/fs999/en/index.html.
- 2.(a) White HS, Smith MD, Wilcox KS. Mechanisms of action of antiepileptic drugs. Int Rev Neurobiol. 2007;81:85–110. doi: 10.1016/S0074-7742(06)81006-8. [DOI] [PubMed] [Google Scholar]; (b) Calabresi P, Galletti F, Rossi C, Sarchielli P, Cupini LM. Antiepileptic drugs in migraine: from clinical aspects to cellular mechanisms. Trends Pharmacol Sci. 2007;28:188–195. doi: 10.1016/j.tips.2007.02.005. [DOI] [PubMed] [Google Scholar]; (c) Landmark CJ. Antiepileptic drugs in non-epilepsy disorders: relations between mechanisms of action and clinical efficacy. CNS Drugs. 2008;22:27–47. doi: 10.2165/00023210-200822010-00003. [DOI] [PubMed] [Google Scholar]; (d) Ettinger AB, Argoff CE. Use of antiepileptic drugs for nonepileptic conditions: psychiatric disorders and chronic pain. Neurotherapeutics. 2007;4:75–83. doi: 10.1016/j.nurt.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zaremba PD, Bialek M, Blaszczyk B, Cioczek P, Czuczwar SJ. Non-epilepsy uses of antiepileptic drugs. Pharmacol Rep. 2006;58:1–12. [PubMed] [Google Scholar]; (f) Maryanoff BE. Pharmaceutical “gold” from neurostabilizing agents: topiramate and successor molecules. J Med Chem. 2009;52:3431–3440. doi: 10.1021/jm900141j. [DOI] [PubMed] [Google Scholar]
- 3.(a) Maryanoff BE, Nortey SO, Gardocki JF, Shank RP, Dodgson SP. Anticonvulsant O-alkyl sulfamates. 2,3:4,5-Bis-O-(1-methylethylidene)-β-D-fructopyranose sulfamate and related compounds. J Med Chem. 1987;30:880–887. doi: 10.1021/jm00388a023. [DOI] [PubMed] [Google Scholar]; (b) Shank RP, Gardocki JF, Vaught JL, Davis CB, Schupsky JJ, Raffa RB, Dodgson SJ, Nortey SO, Maryanoff BE. Topiramate: preclinical evaluation of a structurally novel anticonvulsant. Epilepsia. 1994;35:450–460. doi: 10.1111/j.1528-1157.1994.tb02459.x. [DOI] [PubMed] [Google Scholar]; (c) Maryanoff BE, Margul BL. Topiramate. Drugs Future. 1989;14:342–344. [Google Scholar]; (d) Maryanoff BE. Sugar sulfamates for seizure control: discovery and development of topiramate, a structurally unique antiepileptic drug. Curr Top Med Chem. 2009;9:1048–1061. doi: 10.2174/156802609789630938. [DOI] [PubMed] [Google Scholar]
- 4.TOPAMAX® (topiramate) attained peak, annual global sales of $2.7 billion in 2008, as indicated in the 2008 Johnson & Johnson Annual Report, p 23 (http://www.jnj.com/wps/wcm/connect/a9a1fd804d5592ab9b16db1bc89e29c7/2008_Annual_Report.pdf?MOD=AJPERES).
- 5.Shank RP, Maryanoff BE. Molecular pharmacodynamics, clinical therapeutics, and pharmacokinetics of topiramate. CNS Neurosci Ther. 2008;14:120–142. doi: 10.1111/j.1527-3458.2008.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maryanoff BE, Costanzo MJ, Nortey SO, Greco MN, Shank RP, Schupsky JJ, Ortegon ME, Vaught JL. Structure-activity studies on anticonvulsant sugar sulfamates related to topiramate. Enhanced potency with cyclic sulfate derivatives. J Med Chem. 1998;41:1315–1343. doi: 10.1021/jm970790w. [DOI] [PubMed] [Google Scholar]
- 7.Parker MH, Smith-Swintosky VL, McComsey DF, Huang Y, Brenneman D, Klein B, Malatynska E, Plata-Salaman C, White HS, Milewski ME, Herb M, Finley MFA, Liu Y, Lubin ML, Qin N, Iannucci R, Leclercq L, Cuyckens F, Reitz AB, Maryanoff BE. Novel broad-spectrum anticonvulsants containing a sulfamide group: advancement of N-((benzo[b]thien-3-yl)methyl)sulfamide (JNJ-26990990) into human clinical studies. J Med Chem. 2009;52:7528–7536. doi: 10.1021/jm801432r. [DOI] [PubMed] [Google Scholar]
- 8.Some sulfamide-based anticonvulsants with different structures have been reported: Gavernet L, Cabrera MJD, Bruno-Blanch LE, Estiu GL. 3D-QSAR design of novel antiepileptic sulfamides. Bioorg Med Chem. 2007;15:1556–1567. doi: 10.1016/j.bmc.2006.06.010.. Gavernet L, Barrios IA, Cravero MS, Bruno-Blanch LE. Design, synthesis, and anticonvulsant activity of some sulfamides. Bioorg Med Chem. 2007;15:5604–5614. doi: 10.1016/j.bmc.2007.05.024.. Gavernet L, Elvira JE, Samaja GA, Pastore V, Sella Cravero M, Enrique A, Estiu G, Bruno-Blanch LE. Synthesis and anticonvulsant activity of amino acid-derived sulfamides. J Med Chem. 2008;52:1592–1601. doi: 10.1021/jm800764p..
- 9.For a review on biologically active sulfamides, see: Winum J-V, Scozzafava A, Montero J-L, Supuran CT. The sulfamide motif in the design of enzyme inhibitors. Expert Opin Ther Pat. 2006;16:27–47. doi: 10.1517/13543776.16.1.27.
- 10.(a) Maryanoff BE, McComsey DF, Costanzo MJ, Hochman C, Smith-Swintosky V, Shank RP. Comparison of sulfamate and sulfamide groups for the inhibition of carbonic anhydrase-II by using topiramate as a structural platform. J Med Chem. 2005;48:1941–1947. doi: 10.1021/jm040124c. [DOI] [PubMed] [Google Scholar]; (b) Klinger AL, McComsey DF, Smith-Swintosky V, Shank RP, Maryanoff BE. Inhibition of carbonic anhydrase-II by sulfamate and sulfamide groups: an investigation involving direct thermodynamic binding measurements. J Med Chem. 2006;49:3496–3500. doi: 10.1021/jm058279n. [DOI] [PubMed] [Google Scholar]; (c) Shank RP, McComsey DF, Smith-Swintosky VL, Maryanoff BE. Examination of two independent kinetic assays for determining the inhibition of carbonic anhydrases I and II: structure-activity comparison of sulfamates and sulfamides. Chem Biol Drug Design. 2006;68:113–119. doi: 10.1111/j.1747-0285.2006.00423.x. [DOI] [PubMed] [Google Scholar]; (d) Shank RP, Smith-Swintosky VL, Maryanoff BE. Carbonic anhydrase inhibition. Insight into the characteristics of zonisamide, topiramate, and the sulfamide cognate of topiramate. J Enz Inh Med Chem. 2008;23:271–276. doi: 10.1080/14756360701507001. [DOI] [PubMed] [Google Scholar]; (e) Maryanoff BE, McComsey DF, Lee J, Smith-Swintosky VL, Wang Y, Minor LK, Todd MJ. Carbonic anhydrase-II inhibition. What are the true enzyme inhibitory properties of the sulfamide cognate of topiramate? J Med Chem. 2008;51:2518–2521. doi: 10.1021/jm7015649. [DOI] [PubMed] [Google Scholar]
- 11.Nelson WL, Powell ML, Dyer DC. Absolute configuration of glycerol derivatives. 7. Enantiomers of 2-[[[2-(2,6-dimethoxyphenoxy)ethyl]amino]methyl]-1,4-benzodioxane (WB-4101), a potent competitive α-adrenergic antagonist. J Med Chem. 1979;22:1125–1127. doi: 10.1021/jm00195a024. [DOI] [PubMed] [Google Scholar]
- 12.Delgado A, Leclerc G, Lobato MC, Mauleon D. Short and enantioselective synthesis of (R)- and (S)-2-hydroxymethyl-1,4-benzodioxane. Tetrehedron Lett. 1988;29:3671–3674. [Google Scholar]
- 13.Birch AM, Bradley PA, Gill JC, Kerrigan F, Needham PL. N-Substituted (2,3-dihydro-1,4-benzodioxin-2-yl)methylamine derivatives as D2 antagonists/5-HT1A partial agonists with potential as atypical antipsychotic agents. J Med Chem. 1999;42:3342–3355. doi: 10.1021/jm9910122. [DOI] [PubMed] [Google Scholar]
- 14.(a) Swinyard EA, Brown WC, Goodman LS. Comparative assays of antiepileptic drugs in mice and rats. J Pharmacol Exp Ther. 1952;106:319–330. [PubMed] [Google Scholar]; (b) Swinyard EA. Laboratory evaluation of antiepileptic drugs: review of laboratory methods. Epilepsia. 1969;10:107–119. doi: 10.1111/j.1528-1157.1969.tb03838.x. [DOI] [PubMed] [Google Scholar]
- 15.Irwin S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia. 1968;13:222–257. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- 16.(a) Cerep, an independent contract laboratory, can be contacted at www.cerep.com. (b) The IC50 value for acetazolamide, a reference standard, was <10 nM.
- 17.Shingles R, Moroney JV. Measurement of carbonic anhydrase activity using a sensitive fluorometric assay. Anal Biochem. 1997;252:190–197. doi: 10.1006/abio.1997.2305. [DOI] [PubMed] [Google Scholar]
- 18.Details on these pharmacological attributes will be published separately.
- 19.White HS, Woodhead JH, Wilcox KS, Stables JP, Kupferberg HJ, Wolf HH. General principles: discovery and preclinical development of antiepileptic drugs. In: Levy RH, Mattson RH, Meldrum BS, Perruca E, editors. Antiepileptic Drugs. 5. Lippincott, Williams and Wilkins; Philadelphia, PA: 2002. pp. 36–48. [Google Scholar]
- 20.(a) Lothman EW, Salerno RA, Perlin JB, Kaiser DL. Screening and characterization of antiepileptic drugs with rapidly recurring hippocampal seizures in rats. Epilepsy Res. 1988;2:367–379. doi: 10.1016/0920-1211(88)90048-4. [DOI] [PubMed] [Google Scholar]; (b) Lothman EW, Perlin JB, Salerno RA. Response properties of rapidly recurring hippocampal seizures in rats. Epilepsy Res. 1988;2:356–366. doi: 10.1016/0920-1211(88)90047-2. [DOI] [PubMed] [Google Scholar]
- 21.(a) Soderpalm B. Anticonvulsants: aspects of their mechanisms of action. Eur J Pain. 2002;6:3–9. doi: 10.1053/eujp.2001.0315. [DOI] [PubMed] [Google Scholar]; (b) Liu G, Yarov-Yarovoy V, Nobbs M, Clare JJ, Scheuer T, Catterall WA. Differential interactions of lamotrigine and related drugs with transmembrane segment IVS6 of voltage-gated sodium channels. Neuropharmacology. 2003;44:413–422. doi: 10.1016/s0028-3908(02)00400-8. [DOI] [PubMed] [Google Scholar]; (c) White HS, Smith MD, Wilcox KS. Mechanisms of action of antiepileptic drugs. Intl Rev Neurobiol. 2007;81:85–110. doi: 10.1016/S0074-7742(06)81006-8. [DOI] [PubMed] [Google Scholar]; (d) Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res. 2006;69:273–294. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lombardo AJ, Kuzniecky R, Powers RE, Brown GB. Altered brain sodium channel transcript in human epilepsy. Mol Brain Res. 1996;35:84–90. doi: 10.1016/0169-328x(95)00194-w. [DOI] [PubMed] [Google Scholar]
- 23.(a) Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. 2004;10:685–692. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]; (b) Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- 24.Wang H-S, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 25.Gribkoff VK. The therapeutic potential of neuronal KCNQ channel modulators. Expert Opin Ther Targets. 2003;7:737–748. doi: 10.1517/14728222.7.6.737. [DOI] [PubMed] [Google Scholar]
- 26.More details for these biological studies with 4 will be published separately. For information on the experimental methods used with GABA, NMDA, and kainate, see: Hertz E, Yu ACH, Hertz L, Juurlink BHJ, Schousboe A. Preparation of primary cultures of mouse cortical neurons. In: Shahar A, De Vellis J, Vernadakis A, Haber B, editors. A Dissection and Tissue Culture Manual of the Nervous System. Alan R. Liss, Inc.; New York: 1989. pp. 183–186.. Skeen GA, White HS, Twyman RE. The dihydropyridine nitrendipine reduces N-methyl-D-aspartate (NMDA)-evoked currents of rodent cortical neurons through a direct interaction with the NMDA receptor-associated ion channel. J Pharmacol Exp Ther. 1994;271:30–38.. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp technique for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archiv : Eur J Physiol. 1981;391:85–100. doi: 10.1007/BF00656997..
- Compound 4 was tested at 10 μM with the following receptors and ion channels by Cerep: adenosine A1, A2A, A3; α-1, α-2, β-1 adrenergic; angiotensin-1; benzodiazepine (central); bradykinin-2; cholecystokinin-A; CCR1 chemokine; dopamine D1, D2S; endothelin-A; GABA; galanin-2; histamine H1, H2; interlukin-8B; melanocortin-4; melatonin-1; muscarinic M1, M2, M3; neurokinin NK2, NK3; neuropeptide Y1, Y2; neurotensin-1; delta-2, kappa, mu opioid; nociceptin (ORL1); serotonin 1A, 1B, 2A, 3, 5A, 6, 7; somatostatin; vasoactive intestinal peptide-1; vasopressin V1a; Ca channel L-type, SK-type; KV channel; Na channel (site 2); Cl channel; norepinephrine and dopamine transporters.
- Experimental details will be published separately.
- 29.Brown HC, Kim KW, Srebnik M, Singaram B. Organoboranes for synthesis. 7. An improved general synthesis of primary amines from alkenes via hydroboration–organoborane chemistry. Tetrahedron. 1987;43:4071–4078. [Google Scholar]
- 30.White HS, Wolf HH, Woodhead JH, Kupferberg HJ. The National Institutes of Health Anticonvulsant Drug Development Program: screening for efficacy. Adv Neurol. 1998;76:29–39. [PubMed] [Google Scholar]
- 31.Orloff MJ, Williams HL, Pfeiffer CC. Timed intravenous infusion of metrazol and strychnine for testing anticonvulsant drugs. Proc Soc Exp Biol Med. 1949;70:254–257. doi: 10.3181/00379727-70-16891. [DOI] [PubMed] [Google Scholar]
- 32.(a) White HS, Patel S, Meldrum BS. Anticonvulsant profile of MDL 27,266: an orally active, broad-spectrum anticonvulsant agent. Epilepsy Res. 1992;12:217–226. doi: 10.1016/0920-1211(92)90076-6. [DOI] [PubMed] [Google Scholar]; (b) Lothman EW, Salerno RA, Perlin JB, Kaiser DL. Screening and characterization of antiepileptic drugs with rapidly recurring hippocampal seizures in rats. Epilepsia. 1988;2:367–379. doi: 10.1016/0920-1211(88)90048-4. [DOI] [PubMed] [Google Scholar]
- 33.(a) Köhling R. Voltage-gated sodium channels in epilepsy. Epilepsia. 2002;43:1278–1295. doi: 10.1046/j.1528-1157.2002.40501.x. [DOI] [PubMed] [Google Scholar]; (b) Liu Y, Qin N, Reitz T, Wang Y, Flores CM. Inhibition of the rat brain sodium channel Nav1.2 after prolonged exposure to gabapentin. Epilepsy Res. 2006;70:263–268. doi: 10.1016/j.eplepsyres.2006.03.007. [DOI] [PubMed] [Google Scholar]; (c) Benjamin ER, Pruthi F, Olanrewaju S, Ilyin VI, Crumley G, Kutlina E, Valenzano KJ, Woodward RM. State-dependent compound inhibition of Nav1.2 sodium channels using the FLIPR Vm dye: on-target and off-target effects of diverse pharmacological agents. J Biomol Screening. 2006;11:29–39. doi: 10.1177/1087057105280918. [DOI] [PubMed] [Google Scholar]
- 34.(a) Lukyanetz EA, Shkryl VM, Kostyuk PG. Selective blockade of N-type calcium channels by levetiracetam. Epilepsia. 2002;43:9–18. doi: 10.1046/j.1528-1157.2002.24501.x. [DOI] [PubMed] [Google Scholar]; (b) Kuzmiski JB, Barr W, Zamponi GW, MacVicar BA. Topiramate inhibits the initiation of plateau potentials in CA1 neurons by depressing R-type calcium channels. Epilepsia. 2005;46:481–489. doi: 10.1111/j.0013-9580.2005.35304.x. [DOI] [PubMed] [Google Scholar]
- 35.Dunham MS, Miya TA. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc Sci Ed. 1957;46:208–209. doi: 10.1002/jps.3030460322.. (b) Studies were conducted by NeuroAdjuvants.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.