

Abstract
Two-photon excitation microscopy is an alternative to confocal microscopy that provides advantages for three-dimensional and deep tissue imaging. This unit will describe the basic physical principles behind two-photon excitation and discuss the advantages and limitations of its use in laser-scanning microscopy. The principal advantages of two-photon microscopy are reduced phototoxicity, increased imaging depth, and the ability to initiate highly localized photochemistry in thick samples. Practical considerations for the application of two-photon microscopy will then be discussed, including recent technological advances. This unit will conclude with some recent applications of two-photon microscopy that highlight the key advantages over confocal microscopy and the types of experiments which would benefit most from its application.
Keywords: fluorescence, microscopy, two-photon excitation, confocal microscopy
INTRODUCTION
The effective sensitivity of fluorescence microscopy in thick samples is limited by out-of-focus background signal. This introduces a diffuse background fluorescence that does not encode any spatial information, and which therefore acts to reduce the image contrast. In a confocal microscope, a confocal pinhole is used to reject the out-of-focus background and produce an unblurred image which corresponds to a thin (<1 μm) “optical section.” This greatly reduces the limitation of imaging extended samples and enables high-resolution 3-D imaging. Two-photon excitation microscopy (a type of nonlinear microscopy, also known as multi-photon microscopy; Denk et al., 1990) is an alternative to confocal microscopy that provides clear advantages for three-dimensional imaging. In particular, two-photon excitation microscopy excels at high-resolution imaging in intact thick tissues such as brain slices, embryos, whole organs, and live animals (intra-vital imaging). This unit will describe the basic physical principles of two-photon excitation and discuss the advantages and limitations of its use in laser-scanning microscopy. Practical considerations for this technique will be highlighted in order to illustrate the utility of this technique and demonstrate some of its physical limitations. Finally, several applications of two-photon excitation microscopy will be described, which illustrate the key advantages provided by this technique, which make possible experiments that could not otherwise have been performed.
Before utilizing any optical-sectioning approach, it is important to consider whether the appropriate technique has been selected that is best suited to answer the biological question at hand. For fluorescence microscopy in relatively thick samples, two-photon excitation often provides the most attractive solution, although complementary three-dimensional fluorescence microscopy methods each have particular benefits that can make them better suited for certain experiments.
Confocal microscopy uses a pinhole to reject out-of-focus background fluorescence. Thus, this technique allows optical-sectioning for three-dimensional imaging in thicker samples than conventional wide-field fluorescence microscopy. However, while the excitation light generates fluorescence throughout the specimen, signal is collected only from the focal plane. The absorption of the excitation light will causes photobleaching and photodamage throughout the specimen, which can cause significant problems, especially in live samples. The penetration depth in confocal microscopy is also limited by sample scattering of both the excitation and emission photons, as well as absorption of excitation energy.
Two-photon excitation provides optical sectioning for three-dimensional imaging, but in contrast to confocal microscopy there is no absorption and fluorescence (and thus no photobleaching and phototoxicity) above and below the plane of focus. Consequently, it can be less perturbing to live samples due to the reduced phototoxicity incurred throughout the sample. In addition, the ability to image at depth in the sample is degraded less by sample scattering of excitation and emission photons. Thus, two-photon excitation microscopy is used, in preference to confocal microscopy, for experiments that require large image depths in live tissue or in small animals. However, because the photophysics involved with two-photon excitation is different from conventional fluorescence excitation, deleterious effects may occasionally be observed with two-photon excitation for certain fluorophores, and this in turn can limit the applicability of this method for optical sectioning in thin samples.
Selective-plane illumination microscopy [SPIM, also known as light sheet microscopy (Huisken et al., 2004)] is a recently developed technique that can also provide optical sectioning in thicker tissue samples. In ‘epi-illumination,’ generally used in conventional or confocal microscopes, fluorescence is collected back through the same path as excitation illumination, via the microscope objective. This provides certain limits to the spatial resolution in the axial (z) direction relative to lateral (x-y) directions. In SPIM, a light sheet illuminates a plane within the sample from one side, perpendicular to the imaging axis. Fluorescence is collected and imaged as normal. Thus higher axial resolution can be achieved (limited to the light sheet thickness), and excitation scattering poses less of a problem in imaging. Photobleaching and photodamage will also be reduced compared to confocal microscopy, as fluorescence is only generated at the focal plane—thus, photobleaching only occurs at the focal plane as in two-photon microscopy. SPIM also lends itself well to tomographic imaging, as the sample can be rotated to multiple imaging positions. However, SPIM requires a specialized microscope system, and the system-sample geometry will limit the range of samples that can be imaged effectively. The fundamental depth limit will also be less than with two-photon microscopy, and well localized excitation will not be possible. Thus, while SPIM can also provide several advantages over confocal microscopy, these are limited to certain samples, embryo imaging being one notable example.
TWO-PHOTON EXCITATION
Principles of Two-Photon Excitation
The concept of two-photon excitation was first proposed theoretically by Maria Göppert-Mayer in her doctoral dissertation (Göppert-Mayer, 1931). The units of two-photon absorption cross-section (GM) are named in her honor (1 GM = 10−50cm4sec-photon−1). Two-photon excitation was then observed experimentally after the invention of the laser (Kaiser and Garrett, 1961). Thus, much of the theoretical and experimental background is very well understood. Before describing two-photon excitation, let us first briefly consider conventional ‘one-photon excitation.’ A fluorophore existing in the ground state (S0) can absorb a single photon that excites the fluorophore to a higher energy state (S1, an excited state). After a short period of time in the excited state, the fluorophore relaxes back to its ground state by emitting a photon of light. To efficiently excite the fluorophore, the excitation photon should have a wavelength (λ1p) that corresponds to an energy which matches the energy of the excited state of the fluorophore (ES1).
Two-photon excitation arises from the simultaneous absorption of two photons in a single event. Each photon has half the energy as in the corresponding single-photon absorption event. The energy of a photon is inversely proportional to its wavelength; therefore, in two-photon excitation, the photons should have a wavelength (λ2p) of approximately twice that of the photons required to achieve an equivalent transition under one-photon excitation. For example if a fluorophore efficiently absorbs light at 400 nm under conventional excitation, it could be excited by two simultaneous photons at approximately 800 nm (Fig. 4.11.1). Thus, rather than fluorescence being excited by UV-visible illumination, it can be excited by infra-red illumination. The resulting excited state from which emission occurs is the same singlet state as that excited during conventional one-photon absorption. Thus, the fluorescence emission after two-photon excitation is exactly the same as that generated in normal one-photon excitation (Denk et al., 1995). Two-photon excitation depends on the simultaneous absorption of two photons, so the resulting fluorescence emission intensity depends on the square of the excitation intensity. This quadratic dependence gives rise to the enhanced excitation provided by a pulsed laser source, as well as the intrinsic optical section capability of two-photon excitation microscopy (see Two-Photon Excitation in Laser-Scanning Microscopy. below).
Figure 4.11.1.
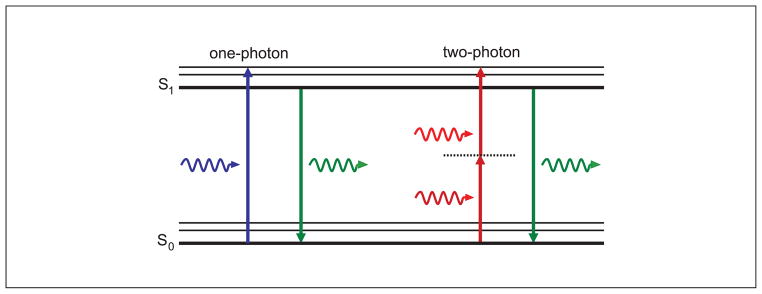
Jabłoński (energy-level) diagram of conventional one-photon excitation (left) and nonlinear two-photon excitation (right) of fluorescence. In each case, the absorption of photon(s) generates an excited state from which the molecule can relax by emitting a fluorescent photon. Thus, the path to the excited state follows a different path under either one- or two-photon absorption, leading to different absorption spectra. However, fluorescence is emitted from the same excited state producing identical emission spectra.
The requirement for two-photon excitation is that the photons are absorbed at exactly the same time, within ~10−18 sec. For there to be a significant probability that two photons will be incident on the fluorophore at exactly the same time, a much higher photon flux is required (~106 times more) compared to one-photon excitation. Therefore, a much higher laser power is required compared to that used under one-photon excitation. Instead of simply using a high-power CW laser source (which would be impractical for many reasons), the photon density is increased by the tight focusing of an ultra-short pulsed laser source. In a pulsed laser source, all of the photons are concentrated into discrete pulses such that the peak power of the pulse is enhanced relative to the time-averaged power—there is a much greater probability of two photons being incident at a fluorophore at the same time. The shorter the laser pulse, the greater the concentration of photons in time and thus the higher the peak power relative to the average power. In this way, by using an ultra-short pulsed laser source, there can be a significant probability of two-photon absorption occurring while still maintaining low incident power (Denk et al., 1995). The ‘two-photon enhancement’ is the ratio of the peak power to the average power, which is equivalent to the pulse duration relative to the pulse frequency. Many solid-state mode-locked pulsed laser sources can generate pulses of ~100 fsec duration at ~100 MHz repletion rate, such that the peak power is enhanced by a factor of 105 to 106.
In addition to the concentration of photon flux in time, the focusing of the illumination using a high numerical aperture (NA) objective creates a spatial concentration of photon flux, thus further increasing the likelihood that two photons can be incident on a fluorophore at exactly the same time.
A related nonlinear process called three-photon excitation can also be used in biological experiments (Maiti et al., 1997). Three-photon excitation works in a similar way to two-photon excitation, where three photons must interact with a fluorophore at exactly the same time. To achieve a sufficiently high photon flux density for this to occur, only another 10 times more power is required compared to two-photon excitation (not a further ~106 increase in power). Therefore, three-photon absorption can be readily achieved. An excitation wavelength of ~3 times the conventional excitation wavelength will be required: for example, to excite a fluorophore normally excited at ~300 nm, a 900-nm source can be used. Thus, three-photon excitation can be used to excite fluorophores that normally require deep-ultraviolet wavelengths, which is normally problematic with a conventional microscope. Even higher-order absorptions have been experimentally demonstrated, such as four-photon absorption (Chu et al., 2005), although it is not clear what advantages these may provide in biological imaging.
Two-Photon Excitation in Laser-Scanning Microscopy
The advantages provided by two-photon excitation in laser-scanning microscopy arise from the physical principle that two-photon absorption depends on the square of the excitation intensity. Following focusing of the pulsed infra-red source by a high-NA objective, the illumination converges to a diffraction-limited spot at the focal plane. As the focal plane is approached, the photon density becomes greater, and so the probability of two photons interacting with a fluorophore at the same time increases. Due to the quadratic dependence of two-photon absorption on excitation intensity, the probability of two-photon absorption at the center of the focus is substantially greater than outside of the focus, and thus significant two-photon absorption occurs only at the center of the focus. This is illustrated and compared to conventional one-photon absorption in Figure 4.11.2.
Figure 4.11.2.

Description of how two-photon absorption occurs only at the focal plane in a microscope. (A) Upon focusing of mode-locked pulsed infrared illumination, the density of photons becomes increasingly greater until at the focal plane. At the focal plane, due to the quadratic dependence of two-photon absorption on intensity, the photon density is increased sufficiently for two-photon absorption to occur. Outside of the focal plane, negligible two-photon absorption occurs, and thus no fluorescence is generated. Pulsed illumination is required to temporally crowd the photons in time, such that the peak photon density is much greater, to achieve two-photon absorption, whereas the time-averaged power is still low. (B) Single-photon absorption depends linearly on the excitation intensity, and thus occurs throughout the focus, requiring a confocal pinhole to achieve optical sectioning (red dotted line). Graph shows total fluorescence generated at each axial position throughout the focus, illustrating the intrinsic optical sectioning provided by two-photon excited fluorescence. For the color version of this figure, go to http://www.currentprotocols.com/protocol/cb0411.
Under conventional excitation, the intensity increases under focusing, being a maximum at the focal plane. However, the spatially averaged power throughout the focus is constant. For example, if the excitation intensity falls by a factor of 4 outside the focus, the excited fluorescence intensity falls by a factor of 4, but the area covered is increased by a factor of 4. Upon scanning the beam laterally across a sample to build up an image, the average fluorescence generated throughout the axial direction is constant (Fig. 4.11.2). Thus, if there are multiple fluorescent objects at different axial positions, they will all be excited to a similar degree. However, objects outside of the focal plane will be defocused and blurred, which will reduce the contrast and fine details in the image—there is said to be no optical sectioning. In confocal microscopy, the confocal pinhole is used to reject the fluorescence generated outside of the focal plane, providing the optical sectioning.
Under two-photon excitation, while the excitation intensity increases to a similar degree under focusing, the probability of absorption increases by the square of that. For example, if the excitation intensity falls by a factor of 4 outside the focus, the excited fluorescence intensity falls by a factor of 42 = 16, and the area covered increases by a factor of 4. Upon scanning the beam laterally across a sample to build up an image, the average fluorescence generated outside of the focus is still lower (Fig. 4.11.2). Fluorescent objects outside of the focal plane are not excited, and thus optical sectioning is achieved intrinsically due to the nature of the focused two-photon excitation. The temporal concentration of photons using a pulsed laser source achieves significant two-photon absorption at low laser powers, and the spatial concentration of photons achieves high two-photon absorption only at a single localized axial position.
The localization of two-photon excitation to solely the focal plane provides most of the advantages over confocal microscopy. In a confocal microscope, fluorescence is excited throughout the sample, but only signal from the focal plane passes through the confocal pinhole, so background-free data can be collected. In contrast, two-photon excitation only generates fluorescence at the focal plane, so there is no background; thus, no pinhole is required. As the excitation of fluorescence in confocal microscopy occurs throughout the sample, photobleaching and photodamage will also occur throughout the sample. Under two-photon excitation, however, photobleaching will be restricted to the highly localized region in which fluorescence is excited. This dramatic difference can be demonstrated by imaging the photobleaching patterns of each method. Figure 4.11.3 shows the photobleaching pattern in the axial (z) direction, which is generated from repeated scanning of a single image plane in a thick fluorescent object. The confocal microscope laser excites fluorophores and photobleaches throughout the sample, including regions substantially outside of the focal plane. In contrast, two-photon excitation only excites fluorophores and photobleaches at the focal plane. In this sense, two-photon excitation can be considered ‘more efficient’—fluorescence is only generated in those regions of the sample where it is actually detected and imaged. Therefore, in a thick sample, the volume in which photobleaching and phototoxicity occurs is very low relative to the sample volume.
Figure 4.11.3.
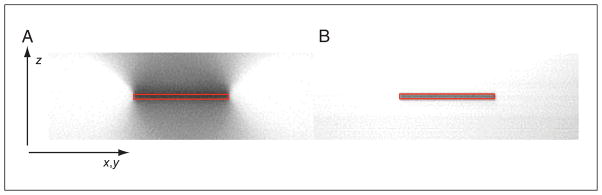
Profile (x-z) of photobleaching caused by one-photon and two-photon excitation. A thick fluorescent object is repeatedly imaged at a single focal position either in a confocal microscope (left) or a two-photon microscope (right) until the focal plane is substantially photobleached. The red box indicates the position of the focal plane from which fluorescence is collected. In the confocal microscope, photobleaching can be seen to extend above and below the focal plane, and additional photobleaching throughout the object. Under two-photon excitation, photobleaching has occurred solely at the focal plane, with no photobleaching outside of the region in which fluorescence was collected. For the color version of this figure, go to http://www.currentprotocols.com/protocol/cb0411.
The reduction of photobleaching throughout the sample due to the highly localized two-photon excitation is one of the main advantages of two-photon excitation. Photo-bleaching and photodamage are some of the main limitations in fluorescence microscopy when imaging live cells and tissue. The damage to cells caused by light interactions is still not fully understood, but limiting it will extend the viability of the biological sample, particularly for long imaging times. In addition, the localization of excitation means that there is no out-of-focus absorption, and thus more excitation light can penetrate through the sample to the plane of focus. Despite these advantages, the spatial resolution is almost the same as that for confocal microscopy, which allows images with high spatial resolution to be acquired.
The localization at which fluorescence is generated also means that two-photon excitation microscopy does not require a pinhole to obtain three-dimensional resolution. This allows flexible detection geometries and more efficient photon detection. As shown in Figure 4.11.4, in conventional descanned detection the emitted fluorescence returns through the same path as the excitation light. This includes the microscope optics and the scanning optics, before the light is spectrally separated and passes through the confocal pinhole to the detector. In confocal microscopy, this geometry is required for the confocal pinhole to selectively transmit only fluorescence generated at the focal region. While this pinhole can be removed or fully opened for two-photon microscopy (A), nondescanned detection paths provide more alternatives: a dichroic mirror, located directly after the objective lens, can reflect the fluorescence through a collector lens to a detector placed in a plane conjugate (equivalent) to the objective back aperture (B); alternatively, the emitted fluorescence is collected directly from the sample using an external detector (C). Using a nondescanned alternative is recommended, since the distance over which the light travels is less, allowing for more efficient detection of scattered fluorescence. There are also fewer intermediate optical elements, allowing for more efficient detection in general. This is particularly important for deep tissue imaging, as described below.
Figure 4.11.4.

Schematic of descanned and nondescanned detection geometries used with a two-photon microscope. Excitation light is raster scanned (x and y scan mirrors) and focused onto the sample by the objective lens. (A) Under descanned detection, the fluorescence emission follows a path returning back along the excitation beam path, first collected by the objective, passing by the scan mirrors, and reflected by a dichroic mirror and focused onto the confocal pinhole, to an ‘internal’ PMT detector. Under two-photon excitation, this pinhole will generally be open. (B) Under non-descanned detection (NDD), the fluorescence emission is collected by the objective and reflected by a dichroic mirror through a transfer lens, which projects the light from the back aperture of the objective onto an ‘internal’ PMT. Under NDD, the position of the dichroic/lens/PMT varies by microscope model, but can be immediately behind the objective in the nose piece. (C) Alternatively, fluorescence emission can be directly detected using an external detector before it reaches the objective.
Mechanism of Deep Sectioning
As already mentioned, one of the key advantages provided by two-photon microscopy is its ability to provide superior imaging deep into thick samples. There are three mechanisms that allow this increased effectiveness in thick samples: (1) the scattering of excitation and fluorescence emission is less detrimental in two-photon microscopy compared to confocal microscopy; (2) excitation light used in two-photon microscopy (infrared) will generally scatter less than the equivalent excitation light used in confocal microscopy (visible, blue-green); and (3) since no two-photon excitation occurs outside the focus, no excitation light will be absorbed, such that more excitation light reaches the focus. These points can all be considered separately.
The first point is perhaps the most important point in describing how two-photon microscopy achieves greater imaging depths compared to confocal microscopy. When the excitation light is focused through heterogeneous biological material, some of the light will scatter. As the imaging depth is increased, a greater proportion of the light will scatter, such that the unscattered fraction (the ballistic photons) decreases in intensity exponentially with depth. In a confocal microscope (Fig. 4.11.5A), excitation light is focused into the sample. Fluorescence generated at the focal plane will pass back through the confocal pinhole and be detected. The signal originating from this will build up a faithful representation of the fluorophore distribution at the focal plane (a). Fluorescence generated above and below the sample will be rejected by the pinhole (b). In the presence of scattering, some fluorescence generated at the focus will scatter outside of the confocal pinhole and not be detected, which will reduce the signal in the image (c). In addition, some excitation light will also scatter outside of the focus and also reduce the amount of fluorescence generated at the focus, again reducing the signal in the image (d). The scattered excitation light will generate fluorescence outside of the focus, which will be rejected by the confocal pinhole (d). However, if this fluorescence also scatters, there will be a small probability that the fluorescence will scatter back into the pinhole and be detected (e). Since this light originates from a random location, it will give rise to a constant background signal. At low imaging depths, this background signal is negligible; however, at high imaging depths, the large amount of scattering that occurs will increase this background signal. Similarly, light absorbed above or below the focal plane will also generate fluorescence that is normally rejected by the confocal pinhole, but may scatter back through the pinhole. Therefore, as the imaging depth is increased and scattering increases, the image signal will decrease and the background signal will increase, severely reducing contrast in the image. This is demonstrated in Figure 4.11.6.
Figure 4.11.5.

Schematic showing the reduced effect of scattering in reducing signal-to-background in a two-photon microscope compared to a confocal microscope. (A) In a confocal microscope, excitation light is focused on the object, and fluorescence generated at the focal plane is collected and passes through the pinhole to be detected (a). Above or below the focal plane, excitation light generates fluorescence, but when collected, this is rejected by the confocal pinhole and not detected (b). In a turbid object, fluorescence originating from the focal plane can scatter (appearing to originate from outside of the focus), and is thus rejected by the confocal pinhole and not detected (c). Excitation light can also scatter and not reach the focus (d). Both c and d will reduce the detected fluorescent signal. The scattered excitation light will also generate out-of-focus fluorescence, which normally will be rejected by the confocal pinhole, as in b. However, there is a small probability that it can scatter and pass through the pinhole and be detected, increasing the background (e). Similarly, there is a small probability that fluorescence originating from outside of the focus can scatter and pass though the pinhole and be detected, also increasing the background. As the amount of scattering increases, the signal will decrease and the background will increase. (B) In a two-photon excitation microscope, fluorescence generated at the focal plane is collected and detected (a). Above or below the focal plane, no fluorescence is generated (b). In a turbid object, fluorescence originating from the focal plane can scatter, but due to the absence of the confocal pinhole it is still collected. The scattered excitation light will also not generate out-of-focus fluorescence (d). Thus as the amount of scattering increases, the signal will reduce to a lesser degree and the background will not increase.
Figure 4.11.6.

A comparison of confocal microscopy and two-photon microscopy with imaging depth, together with descanned and nondescanned detection. A thick, highly scattering pancreatic islet sample was first imaged at ~20 μm under confocal microscopy and two-photon microscopy with descanned and non-descanned detection, with settings maintained such that the peak fluorescence signal is the same in each case. The sample was then imaged at a greater depth of 50 μm, with the same settings retained. Under confocal microscopy, a decrease in image signal and a substantial background reduces the image contrast (a), which is not present under two-photon excitation (b,c). At an even greater imaging depth (~100 μm) under constant settings, no image contrast is visible under confocal microscopy (d). Two-photon descanned detection shows a reduction in signal but maintains image contrast (e), whereas non-descanned detection retains a good signal and image contrast (f).
In two-photon microscopy (Fig. 4.11.5B), excitation light again is focused into the sample, and fluorescence generated at the focus is detected to build up an image (a). As described above, a negligible amount of fluorescence is generated outside the focus, and so no pinhole is required to reject out-of-focus fluorescence (b). In the presence of scattering, some fluorescence generated at the focus will scatter, but this will still be detected (c). While scattering of the excitation light will reduce the amount of fluorescence generated at the focus, none of this excitation light will generate any fluorescence (d). There is practically zero chance that two excitation photons will scatter to exactly the same position at exactly the same time, which would be necessary to generate fluorescence. Therefore, even in the presence of very high scattering at high image depths, no background signal will be generated, thus maintaining a high image contrast. This is also demonstrated in Figure 4.11.6.
The second point is that light at greater wavelengths will generally scatter less in biological samples. Therefore, the infrared illumination used in two-photon microscopy, at approximately double the wavelength of the equivalent blue-green illumination used in confocal microscopy, will scatter much less. Biological tissue is a heterogeneous medium with spatial variations in refractive index that cause the scattering. These variations occur on different spatial scales, and vary themselves between different tissues; therefore, it is not possible to know in advance the precise level of scattering and its dependence on the wavelength of light. However, approximations can be made. In Rayleigh scattering (variations occur on a spatial scale much less than the wavelength), the amount of scattered light is inversely proportional to the fourth power of the light’s wavelength (1/λ4). Using this estimate, 488-nm (one-photon) excitation would be expected to scatter ~7-fold more than 800-nm (two-photon) excitation. In Mie scattering (variations occur on a spatial scale similar to the wavelength), the amount of scattered light scales much less with wavelength (1/λ0.5). Biological scattering is generally intermediate to these approximations, but it is always the case that longer (redder) wavelengths are scattered less than shorter (bluer) wavelengths. Therefore, under two-photon excitation, the effects of scattering on excitation light, described in the first point, are reduced due to the longer wavelength. However, the fluorescence is generally the same under one- or two-photon excitation; therefore, scattering will affect fluorescence emission similarly. Thus, in two-photon microscopy, a greater proportion of excitation light will reach the focus to generate fluorescence compared with confocal microscopy.
The third point is that the lack of two-photon absorption outside of the focus allows more excitation light to reach the focus. In confocal microscopy, fluorescence is generated throughout the sample, so those photons that are absorbed before reaching the focal plane will be unable to generate fluorescence at the focal plane. If there are fluorophores distributed throughout the sample, at greater imaging depths the excitation intensity will decrease, even in the absence of scattering. However, under two-photon excitation, no intermediate absorption will occur, and thus the full excitation power will reach the focus. Thus, under single-photon excitation on a confocal microscope, the level of fluorescence will decrease at increasing image depths. However, under two-photon excitation, the fluorescence will be stable with image depth.
PRACTICAL CONSIDERATIONS FOR TWO-PHOTON EXCITATION MICROSCOPY
Image Resolution
The spatial resolution provided by two-photon excitation microscopy is described in a nearly identical manner to that provided by a perfect confocal microscope—i.e., a nearly fully closed pinhole. The axial (Δz) and lateral (Δρ) spatial resolution of both is dependent on the numerical aperture (NA0) of the microscope objective and the wavelength of light (λ) used.
However two important points need to be made to compare the spatial resolutions provided by each modality:
The wavelength of light for two-photon excitation is approximately twice that for one photon excitation. Thus, the axial and lateral resolution will actually be worse for two-photon microscopy compared to a perfect confocal microscope with an equivalent microscope objective.
For a confocal microscope, the equations above are only valid for a ‘perfect’ confocal microscope, which includes a fully closed pinhole (≪1 AU diameter). This is not generally the case in practical confocal microscopy, and so the axial and lateral resolutions will be slightly poorer than the perfect confocal case (Wilson, 2011).
Thus, the lateral and axial resolution for two-photon excitation is only slightly worse than for confocal microscopy. If an object or feature of interest cannot be resolved in a confocal microscope, particularly when using a fine confocal pinhole, then two-photon microscopy will also be unable to resolve that object. It is important to understand this point when deciding whether a two-photon microscope will be suitable for an imaging experiment. While understood well by experts in the field, this point can often be missed by prospective users in the biomedical research community.
Imaging Thick Samples
As described above, two-photon excitation is more effective than confocal microscopy in thick samples for several reasons: scattered excitation light does not excite any fluorescence, scattering fluorescence emission can still be detected, and infrared wavelengths scatter less than visible wavelengths. To image in deep tissue, long-working-distance objectives must be used. The working distance for a typical non-specialized high-NA objective is ~100 μm, which is far below the penetration depth achievable with two-photon microscopy in biological tissue (see below). In addition, if the immersion medium and objective are not well matched to the refractive index of the tissue, aberrations can occur which will increase with imaging depth and reduce the excitation efficiency (Booth et al., 1998). Therefore, water-dipping objectives are increasingly used for in vivo imaging with two-photon excitation. These have long working distances in excess of 1 mm, with high NA, to achieve efficient focusing and excitation, and rely on water as the immersion medium, which best matches the tissue refractive index compared to oil immersion. Despite this, aberrations will likely still occur due to biological heterogeneity. Some developments aim to minimize this by correcting for aberrations using adaptive optics (Booth et al., 2002).
Another issue is that the imaging depth achievable can often be limited by the labeling efficiency. With increasing depth, it becomes increasingly difficult to introduce fluorescent labels into the cells and tissue of interest. The use of tissue-specific fluorescent protein expression in transgenic animals enhances the usefulness of two-photon excitation in vivo imaging, allowing efficient labeling of tissues at any depth.
Imaging Thin Samples
In general, two-photon excitation microscopy does not provide significant advantages over confocal microscopy or other 3-D imaging techniques for imaging thin samples. This is because the level of photobleaching and phototoxicity between confocal and two-photon microscopy will be comparable in thin samples: the volume of the focal plane to which photobleaching is restricted to in two-photon microscopy is comparable to the volume of the whole thin sample where photobleaching will occur under confocal microscopy. In fact, for some fluorophores, photobleaching at the focal plane is actually increased under two-photon microscopy (Patterson and Piston, 2000; Hopt and Neher, 2001).
However, a two-photon microscope does provide a significant advantage over confocal microscopy for imaging UV-excitable fluorophores, even in relatively thin samples. This is because UV light can be particularly harmful to biological samples compared to infrared light used in two-photon microscopy. As described in the examples section, this can be useful for imaging endogenous fluorophores such as NADH or elastin, as well as UV-excitable dyes such as Laurdan in live cells and tissue.
In addition, confocal microscopes do not generally have UV sources for 3-D imaging, and therefore a two-photon microscope can provide a convenient and flexible alternative. In general, to decide if two-photon microscopy will be beneficial for an imaging experiment, it is always desirable to first perform the experiment using confocal microscopy. Once the limitations of confocal microscopy have been determined for the required measurement, one can then determine whether two-photon microscopy will provide an additional advantage for the experiment.
Fundamental Depth Limit
When imaging in thick samples, there is a fundamental limit to the depth at which images can be effectively acquired. To understand how this arises, it is best to initially think about the fundamental depth limit of confocal microscopy. As described above, at greater imaging depths scattering causes both excitation photons not to reach the focus and fluorescence emission photons not to reach the confocal pinhole. Thus, there is a precipitous loss of signal and reduced signal-to-noise, for which the excitation power can be increased as compensation. However, fluorescence excited outside of the focus can scatter through the confocal pinhole, and thus increase the background and reduce the signal-to-background. A fundamental depth limit will be reached where the probability of scattering is such that the amount of fluorescence from outside the focal region that scatters into the pinhole exceeds the amount of fluorescence from the focal region that does not scatter and reaches the pinhole—see Figure 4.11.6. While this limit may be reached in a thick sample, it is important to remember that fluorescence is being excited throughout the sample, thus causing greater photodamage and phototoxicity in the sample as a whole. This may be a limiting factor in live samples.
Under two-photon microscopy at greater imaging depths, scattering causes excitation photons not to reach the focus; however, these scattered photons will not excite fluorescence. Fluorescence originating from the focus will also scatter; however, the lack of a pinhole, and the closer proximity of a non-descanned detector (see below), will mean that the loss of signal will be less severe. Thus, to compensate for the excitation scattering, the excitation power can be increased without inducing phototoxicity outside of the focal plane. In this way, the power can be increased by a very large amount to achieve greater imaging depths (although for very high powers, some local heating at the sample surface may occur). However, a fundamental depth limit will still be reached where the energy density of ballistic (unscattered) light near the surface of the sample will exceed the attenuated energy density of ballistic light at the focus. This arises because the energy density at the focus increases approximately quadratically as light is focused (and thus two-photon excitation to a power of four); however, the energy density will decrease exponentially with depth due to scattering. Beyond a fundamental depth limit, the effect of scattering will always outweigh the effect of focusing, and thus fluorescence will start to be excited closer to the surface of the sample. This will have a similar effect to the confocal case by producing a fluorescent background and thus decreasing the signal-to-background (Theer et al., 2003).
The fundamental depth at which two-photon microscopy will be possible depends on the level of scattering as well as a number of additional factors, described in detail in Theer and Denk (2006). For a high-contrast object (where fluorescing objects are highly localized and not distributed diffusely), under high-NA (>0.8) illumination, the fundamental depth limit at which image contrast can still be resolved is approximately 6 to 7 mean free path scattering distances. In relatively weakly scattering biological material such as neuronal tissue (scattering distance ~200 μm), this translates into a fundamental depth in excess of 1.2 mm for 900-nm illumination (Theer et al., 2003). For denser tissues with shorter scattering distances, this fundamental depth will be less. For example, in epithelial tissue, imaging depths of ~370 μm have been demonstrated where the scattering distance was shown to be approximately 90 μm. In this case, a lower-contrast object was imaged where fluorescence was less well localized, and thus the imaging depth was limited to ~4 mean free paths (Durr et al., 2011). This can be improved further using longer-wavelength excitation light, which scatters less in biological tissue; therefore, the use of red fluorophores should allow greater imaging depths.
Absorption Spectrum
Two-photon absorption spectra are generally very different from their corresponding one-photon absorption spectra, in contrast to the identical emission spectrum. The difference in the absorption spectrum stems from the very different excitation route that the fluorophore takes, as described above. As a very rough approximation, the absorption maximum for two-photon excitation is approximately twice that under one-photon excitation. This applies well to fluorescent proteins such as GFP, YFP, and CFP, along with some other conventional fluorophores. However, for many of the orange to far-red fluorescent proteins, there exists a much stronger blue-shifted absorption band that corresponds to exciting a higher energy state (Drobizhev et al., 2011). For example, TagRFP (Abs peak = 555 nm under 1 photon absorption) has a weak two-photon absorption band centered around 1100 nm, but has a much stronger two-photon absorption band centered around 780 nm (equivalent to 390 nm under one-photon excitation).
In general, it is very challenging to theoretically determine the two-photon excitation spectrum based on the fluorophore structure; therefore, the spectrum needs to be determined empirically. This is complex to accurately determine compared to the spectrum for one-photon absorption spectra. Fortunately, over the past several years, many commonly used fluorophores and fluorescent proteins have had their two-photon excitation spectra determined by specialized labs. Therefore, the best course of action is to look up these spectra for the optimal wavelengths (see, for example, Xu and Webb 1996; Albota et al., 1998; Bestvater et al., 2002; Wokosin et al., 2004; Spiess et al., 2005; Drobizhev et al., 2009; Drobizhev et al., 2011). The two-photon absorption spectra are often broader than their one-photon equivalents, and, since the latest laser sources for two-photon excitation are automatically tunable, a wavelength scan may often suffice for finding a good absorption band.
Localized Photochemistry
Two-photon excitation also allows the highly localized initiation of photochemical reactions. Generally, these chemistries involve ultraviolet light–induced reactions, and as described above, two-photon excitation can beneficially substitute for UV light. Since two-photon absorption only occurs in the focal region, the UV-activation can be highly localized to a region of less than 1 μm3. Examples of photochemical reactions include ‘uncaging’ of a fluorophore—that is, the photochemical conversion of a nonfluorescent molecule into a fluorescent molecule. In a similar manner, biological signaling molecules such as ATP, glutamate, and Ca2+ can be ‘uncaged’ from an inactive molecule into an active molecule with UV light. The efficiency of uncaging a molecule is highly variable, and this is continually being optimized for caged compounds. An important parameter for two-photon uncaging is the uncaging time. Photoreactions can often be slow (in the millisecond to second range) such that the compound can diffuse several micrometers within the sample before activating. This can reduce the spatial localization of the activation, thus reducing some of the advantage two-photon activation provides. Uncaging efficiency can also vary between UV-excitation and two-photon excitation; therefore, this needs to be determined for a caged compound before its application. Nevertheless many efficient compounds have been developed which are applied in many studies, as described in the next section.
On occasion, photoactivation of a caged compound over a large area or volume is required. In these cases, two-photon excitation may not be the most appropriate means of photoactivation, as the beam would have to be scanned over the activation region one focal volume at a time, which may be overly time consuming. In this case, UV illumination may be preferable, and an aperture can also be used to direct the exposure to a required area in the sample.
Laser Sources
Most of the instrumentation required for a two-photon microscope is based on that for a confocal microscope. However, the laser excitation source is one of the main differences. A good two-photon excitation laser source should have an infrared wavelength corresponding to the desired fluorophore excitation band (see above). The source must also be able to be ultra-short pulsed, typically 100-fsec in duration, so as to generate the high peak power necessary for two-photon absorption to occur. A high power >100 mW is also desirable to account for losses in the optical path.
While a laser power of up to only a few mW is required in live samples, losses can occur due to various optical elements, particularly the microscope objective, which generally has a low transmission at infrared wavelengths (although this is being improved by microscope manufacturers). In addition, to fully utilize the NA of the microscope objective, the back aperture is ‘overfilled.’ The Gaussian laser beam incident on the back of the microscope objective is expanded to a much greater area than the objective lens area, so that the region of the back aperture is evenly illuminated. This means the light which is focused can form a smaller focus to give a higher energy density and more efficient two-photon excitation.
The first demonstration of two-photon excitation microscopy used a mode-locked dye laser source (Denk et al., 1990). However, today, the most commonly used source available is the titanium-sapphire (Ti:sapphire) solid-state laser. With broadband optics, which are now nearly standard in the systems from the major manufacturers, wavelengths between 700 nm and 1000 nm are possible. Pulses down to 100 fsec and peak powers of 1 to 2 W are also readily achieved using solid-state pump sources. Automated tuning across the full wavelength spectrum is now possible with systems from the major manufacturers [for example, Chameleon from Coherent Inc. (http://www.coherent.com/); or Mai-Tai from New-port (http://www.newport.com/)], allowing versatility and ease-of-use. These automated laser systems do not require specialized cooling or power supply equipment, and they do not require user alignment, which is critical for those labs without specialized knowledge of laser systems. However, these laser systems are considerably more expensive than most of those used for confocal microscopy, and this is probably one of the main barriers to the wider use of two-photon microscopy.
New commercial Ti:sapphire laser systems are also available with additional features to optimize excitation for deep-tissue imaging. Dispersion pre-compensation allows ultra-short pulses to be maintained despite the optical path, microscope objective, and intermediate tissue, which would normally cause dispersion and pulse broadening and thus less efficient excitation (e.g., the Chameleon Vision series). This allows increased fluorescence signal from the sample and therefore increased imaging depths. For example, dispersion compensation allowed pulse widths down to 20 fsec to be maintained, allowing a 11-fold increase in fluorescence signal (Xi et al., 2009). Regenerative amplifier systems are also available (e.g., as supplied by Coherent Inc. or Newport) to further increase the peak pulse power relative to the average power, which is necessary when imaging at >1 mm depths (Beaurepaire et al., 2001). Sources including optical-parametric oscillators and amplifiers (OPO, OPA) provide tunable infra-red illumination over a broader range of wavelengths, allowing simultaneous multi-color imaging and imaging at >1000 nm (Mahou et al., 2012). While some red-shifted fluorophores can be excited at wavelengths provided by a Ti:sapphire laser (see absorption spectra, above), when this is not possible, OPO or OPA laser sources covering excitation wavelengths in excess of 1000 nm can be used. However, these laser systems are more costly and challenging to maintain.
Laser Power
The power required to excite fluorophores in a sample has an optimal limit. Fluorescence intensity increases with increasing power, with this having a linear dependence under one-photon excitation and a quadratic dependence under two-photon excitation. At a certain power level, the fluorophore becomes saturated. Saturation occurs at powers that cause a significant proportion of the fluorescent molecules to exist in their excited rather than ground state. Thus, a fluorophore is already in the excited state when another photon is incident. This occurs at ~1 mW at the sample for one-photon excitation and ~50 mW at the sample for two-photon excitation (Denk et al., 1990). Therefore, more photons will not excite more molecules, but importantly any additional excitation energy around the saturation level will contribute to increased photobleaching and phototoxicity.
The saturation power can be determined by measuring the fluorescence signal as a function of the excitation power. It is important that the excitation power at the sample be measured (e.g., using a power meter just above the objective) and the ‘laser setting’ itself not be relied upon. As the power is increased, the fluorescence signal should increase quadratically: plotted on a log-log scale the gradient should be 2. Above a certain power level, the increase will start to level off with the log-log plot having a gradient <2. At this point the excitation is starting to saturate.
Even below this level of power, photo-bleaching and photodamage can still occur, which may be sufficient to perturb the biological specimen. To assess photobleaching, a time-course measurement can be made in a stationary sample, with the duration of measurement being comparable to that planned for the experiment. Decreases in fluorescence over the measurement time will reflect photobleaching (for example, see Rocheleau et al., 2004). To assess perturbation of biological function, it should be noted that trivial cell-viability tests (such as esterase activity or dye exclusion) do not always accurately reflect cellular photodamage. Often, a more functional test is more rigorous and informative. For example, the viability of hamster embryos was confirmed by their continued development (Squirrell et al., 1999), and the viability of pancreatic islets was confirmed by their maintenance of normal glucose-stimulated NAD(P)H response (Bennett et al., 1996).
Detectors
A two-photon microscope can utilize the same detection system as a confocal microscope—namely, the internal PMTs following descanned detection. However, since the fluorescence only originates at a point on the focal plane, no pinhole is required. Therefore, the pinhole can be set to a maximum opening. For thinner samples, this will generally be sufficient; however, when imaging in deeper samples, scattering of the fluorescence emission can lead to a degradation in collected signal upon descanned detection. This is because the scattered light can exit the sample at a high off-axis angle and thus miss the detector, and not be detected, after traversing the relatively long optical path back through the scanning optics.
Descanning is not required in two-photon microscopy; therefore, fluorescence emission can be sent straight to an optical detector placed as close to the objective as possible for non-descanned detection (see Figure 4.11.4). Since a reduced optical path is traversed, more of the scattered fluorescence emission light will be collected at the detector, giving a more robust detection for deep tissue imaging (see Figure 4.11.6). The lack of a pinhole means that it is critical that effective light shielding be used, since room light (and even that from a computer monitor) can be collected that will increase the background signal. Although the NDD is generally placed at one of the other ports on the microscope, some of the newest confocal microscopes place a PMT immediately after the objective to further minimize the detection path length.
Conventional bi-alkali PMTs generally have a poor quantum efficiency of typically 10% to 20%; for every 10 photons incident, only 1 or 2 will register as being detected. Newer PMTs such as GaAsP or GaAsBiP are available that have increased quantum efficiency of ~40%, thus increasing the sensitivity several fold (e.g., on the Zeiss LSM780 Quasar detector). Photon-counting PMTs are also starting to be deployed in confocal and two-photon microscopy. Rather than integrating the current output produced by a burst of incident photons, each individual current spike produced by a single photon is counted. For low signal levels, such as imaging NADH autofluorescence, this reduces the noise and allows more sensitive detection (Benninger et al., 2008).
Wide-area detectors (see Fig. 4.11.4) placed immediately behind the microscope objective (i.e., non-descanned epi-detection) or in front of the objective (i.e., in transmission mode) have been shown to further increase the collection efficiency of fluorescence that is highly scattered as a result of ultra-deep imaging. Imaging depths up to 3 mm in turbid samples such as brain tissue have been reported using such detectors (Crosignani et al., 2011).
Dichroic Filters
There are some important differences in the dichroic filters used to separate excitation light from fluorescence emission in two-photon microscopy compared to confocal microscopy. The general concept is the same in that the fluorescence emission is at a different wavelength to the excitation light; hence, the fluorescence signal can be separated with a dichroic mirror. In a confocal microscope, the fluorescence is at a longer wavelength, and thus a long-pass (LP) dichoic mirror will generally be utilized. However, in two-photon microscopy, a short-pass (SP) dichroic mirror will be used (if the layout is such that a SP is used in the confocal case, a LP will be used in a two-photon case). Generally, an SP filter centered at <700 nm will be available, allowing the full range of excitation wavelengths available from a Ti:sapphire laser (700 nm to 1000 nm), which can excite most available fluorophores (see above). For far-red fluorophores, a SP dichoic filter centered at <700 nm may block fluorescence emission, therefore a longer wavelength SP dichroic can be available, for example centered at 740 nm or higher.
In addition, the band-pass filters used to select a specific wavelength band for detection must include additional infrared blocking. While the fluorescence signal detected in a confocal and two-photon excitation microscope may be similar, the excitation powers used in two-photon excitation are orders of magnitude higher. Even accounting for the reduced detection quantum efficiencies at infrared wavelengths, very high-efficiency infrared blocking must be used to prevent excitation bleed-through swamping any detected fluorescence and damaging the detector. The main filter manufacturers [e.g., Chroma (http://www.chroma.com/) and Semrock (http://www.semrock.com/)] now sell ‘two-photon’ versions of their filters, which include an additional coating for high-efficiency infrared blocking. However, if a user requires a conventional filter, a separate infra-red blocker such as blue glass filter (BG-39) can also be used.
FLIM
Fluorescence lifetime imaging (FLIM) is a specialized fluorescence imaging technique, in which the lifetime of the excited state of a fluorophore is measured: this is approximately the time between a photon being absorbed by the fluorophore and a fluorescent photon being reemitted. The excited-state lifetime is dependent on a number of factors including the structure of a fluorophore and environmental properties of the fluorophore. As such, FLIM has a number of applications, principally to obtain functional information regarding a sample as opposed to structural information (Levitt et al., 2009). In general, it provides a robust and quantitative measure of changes in fluorescence. One main application of FLIM is for quantifying protein-protein interactions in live cells and tissue via FRET, where it is the gold standard approach (Verveer et al., 2000). Additional applications include resolving different fluorescent species that have overlapping absorption and emission spectra, which is particularly important in imaging tissue autofluorescence (Stringari et al., 2011) and metabolic imaging of NAD(P)H and FAD (Skala et al., 2007).
At the heart of FLIM is a laser source that provides either rapidly modulated or pulsed illumination where the pulse time is much less than the fluorescence lifetime (typical fluorescence lifetimes are of the order 100 psec to 10 nsec duration). Such a laser source can be expensive; however, a two-photon excitation source satisfies such a criterion. Therefore, only a fast-response PMT detector (sometimes included with a confocal/two-photon microscope) and suitable detection electronics are additionally required [e.g., as supplied by PicoQuant (http://www.picoquant-usa.com/), Becker & Hickl (http://www.becker-hickl.com/), ISS (http://www.iss.com/)]. Therefore, FLIM is a relatively inexpensive add-on to a two-photon microscope, which can provide several additional capabilities.
Utilization in Other 3-D Techniques
Two-photon excitation microscopy is most commonly deployed, as described above, in a laser scanning (confocal) microscope, and is applied as in the examples below to provide deep tissue imaging, reduced phototoxicity, and highly localized photochemistry. However, two-photon microscopy can also be combined with other optical section approaches to yield even further improvements. Surface plane illumination microscopy (SPIM, also known as light sheet microscopy) is an alternative optical-sectioning approach used for imaging thick samples, particularly embryo development (see above). While single-photon excitation is most commonly used, recent developments have utilized two-photon excitation for further improvements: namely, providing isotropic spatial resolution (x-y resolution and z resolution are the same, unlike in confocal or two-photon microscopy) at high imaging depths, as well as maintaining low photodamage (Planchon et al., 2011). The use of two-photon excitation in this approach can be considered to be specialized, and therefore one would be advised to use these only if uniformity of spatial resolution at high imaging depths is specifically required.
EXAMPLES OF TWO-PHOTON EXCITATION MICROSCOPY
In this section, we will highlight several applications of two-photon microscopy to demonstrate experimental situations in which two-photon microscopy is more advantageous compared to confocal microscopy. Many studies have utilized two-photon microscopy over the past two decades, as reviewed in Benninger et al. (2008). The examples highlighted below all make use of the three main advantages of two-photon microscopy described above: increased imaging depths; reduced photodamage, particularly with UV excitation; and highly localized excitation to initiate localized photochemistry. Specific details regarding the experiments can be found in the cited references.
Deep Tissue Imaging
As mentioned already, two-photon microscopy allows deeper tissue imaging due to the reduced effect of scattering and absorption on image degradation, as well as the reduced scattering at infrared wavelengths. Therefore, imaging depths exceeding 1 mm are achievable in biological tissue. From these properties, two-photon microscopy can be applied to imaging whole-organ preparations and imaging tissue in small-animal models such as mice or zebrafish. These small animal models allow the use of genetically encoded fluorescent proteins or biosensors, which can be localized throughout the tissue of interest. This contrasts with exogenous dyes, which are limited by the depth at which the dye can be delivered. Fluorescent proteins can be used to label the localization of specific proteins, whereas biosensors can be used to monitor an environmental property such as ion concentration.
Imaging neuronal tissue is one of the most common applications of two-photon microscopy for deep-tissue imaging, and applications include imaging combinations of electrical activity, neuronal architecture, and blood flow. The spatial resolution of two-photon microscopy is sufficient to resolve synaptic structures such as dendritic spines, and imaging depths through several layers of the cortex are achievable, accessing many physiological functions of the brain in small animals. Sur and coworkers measured in vivo how different cellular populations (astrocytes and neurons) responded to visual stimuli, utilizing microinjected calcium dye (Schummers et al., 2008). By obtaining subcellular resolution at depths in excess of 100 μm, they could quantify the tuned response to different orientations and spatial frequencies of visual stimulation, and found astrocytes responded in a similar manner to neurons. To monitor electrical activity, membrane potential dyes can be used, or calcium activity can be monitored either through microinjected calcium dye (as above), electroporation of calcium dye, AM loading of calcium dyes, or more recently, genetically encodable calcium sensors such as a FRET-based sensor (Mower et al., 2011). An alternative genetically encodable calcium sensor is a circular-permutated GCamP3, and Kerr and coworkers utilized this to image electrical activity at up to 800 μm depth (Mittmann et al., 2011), allowing the quantification of activity at layer 5 of the adult somatosensory cortex. Image quality was improved at these depths by utilizing a regenerative amplifier laser source to achieve sufficient peak powers. Furthermore, the genetically encoded calcium sensor labeled only deep neurons, and provides a high-contrast object to avoid the problem of homogeneous labeling, which can limit the maximum image depth, as discussed above (Theer and Denk, 2006). In another example, by using GCamp3, Svoboda and coworkers tracked the electrical activity of large populations of neurons over weeks as mice learned sensory motor tasks (Huber et al., 2012). This was assisted by skull windows to enable a consistent location to be repeatedly imaged, allowing the same neurons to be studied over the duration of the experiment. As mice learned to link sensing and object identification with reward, the authors could track how the activity of specific neurons and networks corresponded to certain behavioral features and how these changed during the learning process.
Another common area where two-photon microscopy is applied for deep tissue imaging is immune cell dynamics. Real-time imaging of immune cell migration, locomotion, and trafficking, and the direct interaction of immune cells with other cells, can be achieved in isolated lymph nodes, as well as in vivo (Miller et al., 2003; Friedman et al., 2010). Immune cells can be labeled with fluorescent proteins using a transgenic mouse model, or they can be isolated and labeled with conventional fluorophores and re-delivered to a desired mouse model. Utilizing resonant scanning, acquisition speeds of up to video rate allowed neutrophil motility rates of ~100 μm/sec to be resolved, with velocities in blood vessels up to ~1 mm/sec possible (Looney et al., 2011). Two-photon microscopy has been applied to image the spatiotemporal characteristics of the immune system during allograft rejection (Celli et al., 2011). By using GFP-expressing transgenic mice, the authors could track the infiltration of specific immune cell types and their motilities over days during rejection. They found that infiltration by monocytes and dendritic cells was a hallmark of the transplant rejection, and that they contribute to the activation of T cells, which ultimately leads to rejection.
In studying the kidney, two-photon microscopy has been applied to study fluid transport in vivo in rodents. This includes imaging blood flow via red blood cell velocity, such as in a mouse model of kidney transplantation (Imamura et al., 2010) where p53 siRNA improved blood flow following renal injury. Filtration rate and proximal tubular uptake rates of fluorophores have also been measured using two-photon microscopy in vivo to assess renal function. Renal function can be physiologically quantified via the glomerular filtration rate, by quantifying the redistribution rate of two different-sized fluorophores from blood vessels to proximal tubules in vivo (Wang et al., 2010). This method also avoids the use of radioactive tracers.
Other recent applications of two-photon microscopy include cancer biology applications, where it has been applied to examine the movement of xenografts over time (Sanz-Moreno et al., 2011). Depletion of certain signaling pathways reduced tumor cell mobility. In developmental biology, two-photon microscopy can be used to generate an intrinsic autofluorescence signal that allowed cell motility to be tracked in zebrafish embryos, and thus the lineage tree to be determined up to the 512-cell stage (i.e., over 9 cell divisions), with minimal phototoxicity (Olivier et al., 2010).
The ability to image at depths of ~1 mm provides access to many organ regions that would not be possible to image with confocal microscopy. However, even in small animals, the range of sites than can be imaged with two-photon microscopy using a conventional microscope arrangement is still limited. The development of miniature two-photon microscope systems and endoscopic or in vivo light delivery has broadened the range of sites than can be accessed. With such miniaturization, a two-photon microscope system can be mounted on freely moving mice, allowing longitudinal imaging studies (Flusberg et al., 2005; Piyawattanametha et al., 2009). Parameters studied in this manner include in vivo imaging of neuronal activity, blood flow, and architecture. These micro-endoscopes can be delivered to several millimeters depth in the rodent brain, with imaging depths from this point of 600 to 700 Kμm still possible (Barretto et al., 2011), allowing the imaging of neuronal architecture and blood flow over several weeks to ~1 year. Noninvasive in vivo imaging of muscle fiber structure and autofluorescence has also benefited from this development, allowing in vivo imaging in mice and even in humans (Llewellyn et al., 2008).
Reduced Phototoxicity
Two-photon excitation of fluorescence only occurs at the focal plane, such that photo-bleaching and photodamage is much lower than for confocal microscopy where fluorescence and thus photodamage occurs throughout the sample. Thus, two-photon microscopy is better suited to maintain viability and minimize perturbations to live thick tissue samples compared to confocal microscopy. This was demonstrated soon after the initial application of two-photon microscopy in imaging mammalian embryo development (Squirrell et al., 1999). Mitochondrial staining could be continually imaged for ~24 hr using two-photon microscopy without significant photodamage. In fact, the embryos continued to develop in a healthy way, and after implantation could produce healthy pups. However, under confocal imaging, even in unstained embryos, most embryos failed to develop further, and no viable pups could be generated. In a similar manner, two-photon microscopy has been combined with light-sheet microscopy (SPIM) to provide faster imaging of fruit fly or zebrafish embryo development, without compromising normal biology or development (Truong et al., 2011).
UV illumination is particularly photodamaging to cells and tissue; therefore, two-photon microscopy provides an even greater advantage compared to conventional illumination at UV wavelengths. As discussed above, even in relatively thin samples such as cells, two-photon microscopy provides reduced phototoxicity. This has been applied most prominently to image NAD(P)H autofluorescence for assessment of cellular metabolism. NADH and NADPH [often described together as NAD(P)H] absorb optimally in the rage of ~360 nm (equivalent to 710 nm), and autofluorescent NADH increases relative to nonfluorescent NAD+ upon glycolysis and citric-acid-cycle metabolism. Two-photon microscopy has been applied to measure NADH in a number of tissues to investigate cellular metabolism. For example, in the pancreatic islets, two-photon microscopy provides sufficient three-dimensional spatial resolution to resolve the NAD(P)H in the cytoplasm and NADH in the mitochondria, to separately assess glycolysis and citric-acid-cycle metabolism respectively (Rocheleau et al., 2002, 2004). This is illustrated in Figure 4.11.7. These parameters can then be monitored over time, requiring repeated illumination, which would be much more photodamaging using UV illumination. Time-dependent elevations in NAD(P)H, as well as FAD (excited using visible 488-nm conventional excitation), in the presence of different metabolic substrates such a glucose, pyruvate, or methylpyruvate could then be measured to understand the metabolic processes regulating insulin secretion. Two-photon imaging of NAD(P)H has also been applied in pancreatic islets to assess mitochondrial dysfunction following prolonged hyperglycemia induced in a model of diabetes (Benninger et al., 2011).
Figure 4.11.7.
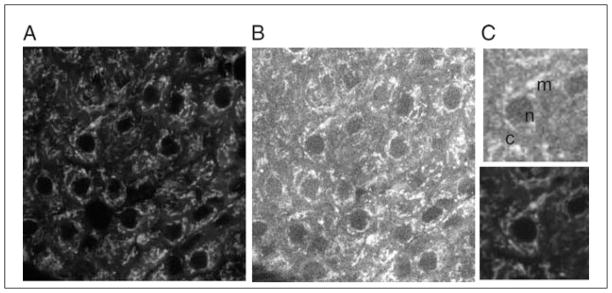
Optical section of Rhodamine123 fluorescence (A) and NAD(P)H autofluorescence (B) from an intact pancreatic islet. A close-up of a single cell for each channel can be seen in (C). NAD(P)H signal arises from the cytoplasm (indicated ‘c’) and mitochondria (indicated ‘m’), the latter being brighter, somewhat punctate, and overlapping with Rhodamine123 fluorescence. Cell nuclei (indicated ‘n’)—where there is little or no NAD(P)H—appear dark.
NAD(P)H has also been imaged for studying cancer, since altered metabolism of glucose generally occurs in cancerous cells. Using two-photon microscopy, changes in NAD(P)H autofluorescence in epithelial cells reflects changes in cellular metabolism (Schafer et al., 2009), which in this case is caused by a loss of matrix adhesion and cell detachment. This helped demonstrate the importance of matrix adhesion and environment in regulating metabolic activity and cell survival. Two-photon microscopy has also been utilized to study tissue biopsy samples at different stages of cancer development by imaging NAD(P)H, as well as FAD autofluorescence, which was also excited using two-photon excitation (Skala et al., 2007). To facilitate comparisons between the highly heterogeneous tissue samples, fluorescence lifetime imaging was also utilized. Since the detected fluorescence intensity could be substantially altered due to absorption and scattering of excitation and emission within the turbid sample, it is difficult to quantify changes in NAD(P)H and FAD from changes in fluorescence intensity alone. Fluorescence lifetime imaging (FLIM, see above) is independent of sample scattering and absorption, and is therefore a more robust measurement, particularly when quantifying changes in NAD(P)H fluorescence. FLIM requires a short-pulsed (<100 psec) laser source, which is already utilized for two-photon microscopy. In this study, as well as in robustly imaging FAD and NAD(P)H, different free and bound populations of these molecules could be resolved due to their differing fluorescent lifetimes but overlapping emission spectra. The authors found a significant increase in the contribution of protein-bound NADH to the fluorescence lifetime signal in precancerous tissue, signifying an altered metabolic state and a potential biomarker.
Imaging Laurdan, a UV-excitable dye sensitive to lipid order, requires two-photon excitation. The resultant fluorescence undergoes a spectral shift according to the lipid order. This allows the spatial mapping of lipid order in cells, for example, to resolve ordered and disorder lipid domains (Gaus et al., 2003). This has been further applied to image lipid order in the cells of live animals such as developing zebrafish. Membrane order could be visualized in multiple tissues throughout the zebrafish, and variations in membrane order within cells could be resolved, such as the ordered membrane on the apical side of gut epithelial cells (Owen et al., 2010).
A class of photoswitchable dyes can also be reversibly converted from a ‘dark’ state to a fluorescent state using UV illumination. However, this has proved to be photodamaging to cells in which the dye was utilized, in contrast to its in vitro application (Mao et al., 2008). Therefore two-photon microscopy was used for the photoconversion, while minimally damaging the cells, to enable high-sensitivity imaging of biomolecular interactions.
Localized Photoactivation
Two-photon microscopy allows high-resolution cellular and subcellular imaging at high imaging depths, and with low phototoxicity. However, the well localized excitation volume also permits precise optically based stimulation experiments. For example the fl excitation focus and ability to substitute for UV absorption allows well localized photochemical reactions to be initiated. As described above, this includes the highly localized activation of ‘caged compounds.’ In fact, this was one of the first applications of two-photon microscopy, where caged neurotransmitter (carbamoylcholine) could be activated to stimulate specific dendrites in a neuron (Denk, 1994). Using two-photon microscopy, photoactivation volumes with lateral and axial FWHM diameters as small as 0.6 and 1.4 μm, respectively, have been demonstrated (Noguchi et al., 2011). This property is well illustrated in Araya et al. (2006), where different dendritic spines in close proximity could be separately or combinatorially activated by focusing two-photon excitation on each spine. The authors determined that the activation of two closely localized spines generated APs that combine linearly at the output. The caged-glutamate CNB-Glu cannot be efficiently activated with two-photon microscopy; therefore, (MNI)-Glu is preferable (Matsuzaki et al., 2001). Dual-color uncaging has also been developed (Kantevari et al., 2010), whereby two separate neurotransmitters (GABA and glutamate in this example) could be ‘uncaged to both block or fire individual action potentials respectively, in a well controlled temporal and spatial manner.
As well as those caged neurotransmitter compounds described above [cholinergic, glutaminergic, and GABAinergic agonists (Matsuzaki et al., 2010)], which have been utilized with two-photon microscopy for well localized activation, other compounds have also been developed. These include caged-IP3 (Kantevari et al., 2006), which has been applied to uncage IP3 within individual astrocytes, enabling the effect of astrocyte Ca2+ signaling on neighboring neural synaptic activity to be studied (Gordon et al., 2009). Two-photon uncaging of caged-calcium has been applied to study calcium induced calcium release. The localized uncaging of calcium in ventricular myocytes caused sufficient calcium to be activated to trigger calcium waves (Lipp et al., 2002). Although other caged compounds, including caged cGMP, ATP, and cAMP, have been developed, many of these exhibit poor two-photon-induced photolysis, and are therefore still awaiting optimization for two-photon microscopy.
So far, we have discussed two-photon localized uncaging of receptor agonists to provide well localized initiation of cellular signaling. Localized photoactivation, however, has broader uses. Photoactivatable fluorophores can also be used as fluorescent tracers, allowing specific three-dimensional volumes to be labeled and the diffusion through space to be monitored. For example, a class of photoactivatable flurophores, LAMP (Dakin et al., 2005), can diffuse through gap junction channels. Upon loading into cells or tissue, the LAMP fluorophore NPE-HCCC2-AM can be activated by two-photon microscopy in a specific cell or populations of cells, and its diffusion into coupled cells monitored (Dakin and Li, 2006). In this way, those cells that are gap-junctionally coupled can be identified, and the level of coupling also quantified from the rate of diffusion. Photoactivatable and photoconvertible fluorescent proteins also allow well localized three-dimensional labeling, which can be applied in vivo. An example of photoactivating PA-GFP in a Drosophila embryo can be seen in Figure 4.11.8. Nussenzweig and coworkers utilized a photoactivatable fluorescent reporter to track specific populations of immune cells to study the mechanism of B cell selection (Victora et al., 2010). PA-GFP was activated in B cells (through use of a PA-GFP transgenic mouse) in specific three-dimensional anatomical regions of lymphatic germinal centers, and their movement between different regions was tracked over many hours. The authors were able to understand the mechanisms by which B cells move between zones that separately control their selection and expansion, and how this is controlled by the interaction of B cells with T helper cells.
Figure 4.11.8.
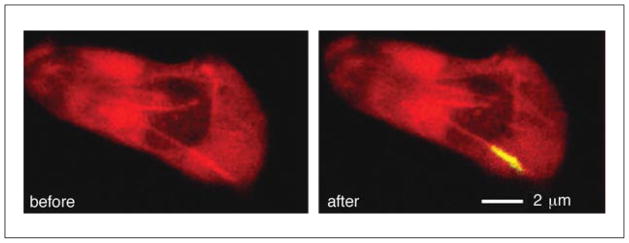
Well localized photoactivation of a photoconvertible fluorescent protein using two-photon excitation. A single neural growth cone in a Drosophila embryo labeled with PA-GFP is highlighted utilizing two-photon excitation. PA-GFP is efficiently converted to bright green fluorescence using two-photon excitation at 800 nm, and is well differentiated from background autofluorescence (red). For the color version of this figure, go to http://www.currentprotocols.com/protocol/cb0411.
Well localized ablation is another method possible with two-photon excitation. The ultra-short pulses associated with two-photon excitation mean that samples undergo well localized photoinduced ablation. For example, specific volumes can be photoablated to disrupt specific cellular populations or regions. This has been used to great effect in studying the blood flow architecture, as specific vessels can be ablated and the resultant blood flow changes measured (Nishimura et al., 2006, 2007; Schaffer et al., 2006). This allows the overall blood flow architecture to be determined by measuring changes in blood flow velocity in vessels downstream of the ablated site. Similarly, specific cell populations in the developing embryo can be disrupted to monitor their role in overall development (Supatto et al., 2005).
An additional interesting application of two-photon excitation is for the targeted optical control of electrical activity in specific cellular populations, which have previously been identified by their electrical activity (O’Connor et al., 2009; Rickgauer and Tank, 2009). ChannelRhodospin is an optically activated ion channel that is widely used in neurosciences to control the electrical activity of genetically specific cellular populations. This is based on the promoter that drives the expression of ChannelRhodopsin (optogenetics). However two-photon microscopy allows further control by allowing specific spatial populations to be activated, such as those that are identified from their activity following a stimulus.
Finally, fluorescence correlation spectroscopy is a single molecule spectroscopy approach in which the diffusion of individual fluorophores in and out of the focal volume is measured (Kim et al., 2007; Fitzpatrick and Lillemeier, 2011). From these measurements, properties of the fluorophore (or the molecule to which the fluorophore is conjugated to) can be determined, such as its diffusion time, size, brightness, and concentration. This can be used to precisely quantify binding, oligormerization, colocalization, and resolving multiple stoichiometries in vitro or in live cells and tissue. In fluorescence correlation spectroscopy (FCS, to measure diffusion time, size and concentration) or single-molecule brightness analysis (to measure molecular brightness or concentration), a smaller focal volume allows the detection of fewer molecules within a specific time window. This allows a better identification of the bursts resulting from individual diffusing fluorophores. The well localized excitation volume provided by two-photon microscopy permits more accurate measurements, particularly in live cells, where the presence of excitation and emission scattering could lead to a greater effective sample probe volume. Two-photon excitation also allows multiple flurophores to be simultaneously excited due to their broader two-photon excitation spectra. This overcomes the problem in single-photon excitation of using multiple excitation wavelengths simultaneously, which requires precise overlapping of the excitation foci and prevention of spectral cross-talk. For example, Williamson and coworkers applied two-photon FCS to resolve intermediate complexes during ribosome formation (Ridgeway et al., 2012). By simultaneously exciting three fluorescent species, the intermediates formed by three separate ribosomal proteins could be measured. The authors discovered several intermediate species that were not previously known to exist thermodynamically during ribosome formation, and this was only possible through two-photon FCS. Muller and coworkers applied two-photon dual-color brightness analysis to determine the stoichiometries and binding curves of two species during ligand-dependent binding of retinoid X receptor to a coactivator transcription factor (Wu et al., 2010). They discovered a nontrivial 3:1 stoichiometry not previously anticipated.
CONCLUSIONS
Two-photon microscopy has great utility for dynamic imaging of live cells and intact tissue and intra-vital imaging in small animals. The advantageous properties of two-photon microscopy originate through the highly localized excitation of the two-photon absorption process and the reduced effect of light scattering in the sample. This enables deep tissue imaging, reduced photodamage, and initiation of well localized photochemistry. As such, many diverse experiments are now possible that would otherwise not be possible with conventional or confocal microscopy approaches. Confocal microscopes can be easily adapted to allow for two-photon microscopy, including the addition of non-descanned detectors and external pulsed infrared laser sources. The widespread availability of automated alignment and laser tuning allows further ease of use for non-specialized labs.
Two-photon microscopy, however, does not provide an improvement in spatial resolution over a well aligned confocal microscope, and, in addition, in thin samples the photodamage may be greater under two-photon excitation compared to conventional fluorescence excitation, particularly for visible flurophores. Therefore, in some circumstances, it is important to note that a confocal microscope or even a conventional wide-field microscope may be more advantageous. Therefore, recognizing when the two-photon microscope is appropriate to use is very important, and the examples described in the previous section illustrate the wide possibilities for improving an experiment using this approach. In the future, we can expect that technological developments (lasers, detectors, in vivo imaging) will further improve the ability to perform two-photon microscopy and enable an even broader range of applications.
LITERATURE CITED
- Albota MA, Xu C, Webb WW. Two-photon fluorescence excitation cross sections of biomolecular probes from 690 to 960 nm. Appl Optics. 1998;37:7352–7356. doi: 10.1364/ao.37.007352. [DOI] [PubMed] [Google Scholar]
- Araya R, Eisenthal KB, Yuste R. Dendritic spines linearize the summation of excitatory potentials. Proc Natl Acad Sci USA. 2006;103:18799–18804. doi: 10.1073/pnas.0609225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto RP, Ko TH, Jung JC, Wang TJ, Capps G, Waters AC, Ziv Y, Attardo A, Recht L, Schnitzer MJ. Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy. Nat Med. 2011;17:223–228. doi: 10.1038/nm.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaurepaire E, Oheim M, Mertz J. Ultra-deep two-photon fluorescence excitation in turbid media. Opt Comm. 2001;188:25–29. [Google Scholar]
- Bennett BD, Jetton TL, Ying G, Magnuson MA, Piston DW. Quantitative sub-cellular imaging of glucose metabolism within intact pancreatic islets. J Biol Chem. 1996;271:3647–3651. doi: 10.1074/jbc.271.7.3647. [DOI] [PubMed] [Google Scholar]
- Benninger RK, Ashby WJ, Ring EA, Piston DW. Single-photon-counting detector for increased sensitivity in two-photon laser scanning microscopy. Opt Lett. 2008;33:2895–2897. doi: 10.1364/ol.33.002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RK, Hao M, Piston DW. Multi-photon excitation imaging of dynamic processes in living cells and tissues. Rev Physiol Biochem Pharmacol. 2008;160:71–92. doi: 10.1007/112_2008_801. [DOI] [PubMed] [Google Scholar]
- Benninger RKP, Remedi MS, Head WS, Ustione A, Piston DW, Nichols CG. Defects in beta cell Ca(2)+ signalling, glucose metabolism and insulin secretion in a murine model of K(ATP) channel-induced neonatal diabetes mellitus. Diabetologia. 2011;54:1087–1097. doi: 10.1007/s00125-010-2039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestvater F, Spiess E, Stobrawa G, Hacker M, Feurer T, Porwol T, Berchner-Pfannschmidt U, Wotzlaw C, Acker H. Two-photon fluorescence absorption and emission spectra of dyes relevant for cell imaging. J Microsc. 2002;208:108–115. doi: 10.1046/j.1365-2818.2002.01074.x. [DOI] [PubMed] [Google Scholar]
- Booth MJ, Neil MAA, Wilson T. Aberration correction for confocal imaging in refractive-index-mismatched media. J Microsc. 1998;192:90–98. [Google Scholar]
- Booth MJ, Neil MA, Juskaitis R, Wilson T. Adaptive aberration correction in a confocal microscope. Proc Natl Acad Sci USA. 2002;99:5788–5792. doi: 10.1073/pnas.082544799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli S, Albert ML, Bousso P. Visualizing the innate and adaptive immune responses underlying allograft rejection by two-photon microscopy. Nat Med. 2011;17:744–749. doi: 10.1038/nm.2376. [DOI] [PubMed] [Google Scholar]
- Chu SW, Chan MC, Tai SP, Keller S, Den-Baars SP, Sun CK. Simultaneous four-photon luminescence, third-harmonic generation, and second-harmonic generation microscopy of GaN. Opt Lett. 2005;30:2463–2465. doi: 10.1364/ol.30.002463. [DOI] [PubMed] [Google Scholar]
- Crosignani V, Dvornikov AS, Gratton E. Enhancement of imaging depth in turbid media using a wide area detector. J Biophotonics. 2011;4:592–599. doi: 10.1002/jbio.201100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakin K, Li WH. Infrared-LAMP: Two-photon uncaging and imaging of gap junctional communication in three dimensions. Nat Methods. 2006;3:959. doi: 10.1038/nmeth1206-959. [DOI] [PubMed] [Google Scholar]
- Dakin K, Zhao Y, Li WH. LAMP, a new imaging assay of gap junctional communication unveils that Ca2+ influx inhibits cell coupling. Nat Methods. 2005;2:55–62. doi: 10.1038/nmeth730. [DOI] [PubMed] [Google Scholar]
- Denk W. 2-Photon scanning photochemical microscopy—mapping ligand-gated ion-channel distributions. Proc Natl Acad Sci USA. 1994;91:6629–6633. doi: 10.1073/pnas.91.14.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-Photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Denk W, Piston DW, Webb WW. Two-photon excitation in laser scanning microscopy. In: Pawley J, editor. Handbook of Biological Confocal Microscopy. Plenum; New York: 1995. pp. 445–458. [Google Scholar]
- Drobizhev M, Tillo S, Makarov NS, Hughes TE, Rebane A. Absolute two-photon absorption spectra and two-photon brightness of orange and red fluorescent proteins. J Phys Chem B. 2009;113:855–859. doi: 10.1021/jp8087379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobizhev M, Makarov NS, Tillo SE, Hughes TE, Rebane A. Two-photon absorption properties of fluorescent proteins. Nat Methods. 2011;8:393–399. doi: 10.1038/nmeth.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr NJ, Weisspfennig CT, Holfeld BA, Ben-Yakar A. Maximum imaging depth of two-photon autofluorescence microscopy in epithelial tissues. J Biomed Opt. 2011;16:026008. doi: 10.1117/1.3548646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick JA, Lillemeier BF. Fluorescence correlation spectroscopy: Linking molecular dynamics to biological function in vitro and in situ. Curr Opin Struct Biol. 2011;21:650–660. doi: 10.1016/j.sbi.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Jung JC, Cocker ED, Anderson EP, Schnitzer MJ. In vivo brain imaging using a portable 3.9 gram two-photon fluorescence microendoscope. Optics Lett. 2005;30:2272–2274. doi: 10.1364/ol.30.002272. [DOI] [PubMed] [Google Scholar]
- Friedman RS, Beemiller P, Sorensen CM, Jacobelli J, Krummel MF. Real-time analysis of T cell receptors in naive cells in vitro and in vivo reveals flexibility in synapse and signaling dynamics. J Exp Med. 2010;207:2733–2749. doi: 10.1084/jem.20091201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaus K, Gratton E, Kable EP, Jones AS, Gelissen I, Kritharides L, Jessup W. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc Natl Acad Sci USA. 2003;100:15554–15559. doi: 10.1073/pnas.2534386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göppert-Mayer M. Über Elementarakte mit zwei Quantensprüngen. Ann Physik. 1931;9:273–294. [Google Scholar]
- Gordon GR, Iremonger KJ, Kantevari S, Ellis-Davies GC, MacVicar BA, Bains JS. Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron. 2009;64:391–403. doi: 10.1016/j.neuron.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopt A, Neher E. Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophysical J. 2001;80:2029–2036. doi: 10.1016/S0006-3495(01)76173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber D, Gutnisky DA, Peron S, O’Connor DH, Wiegert JS, Tian L, Oertner TG, Looger LL, Svoboda K. Multiple dynamic representations in the motor cortex during sensorimotor learning. Nature. 2012;484:473–478. doi: 10.1038/nature11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EH. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Imamura R, Isaka Y, Sandoval RM, Ori A, Adamsky S, Feinstein E, Molitoris BA, Takahara S. Intravital two-photon microscopy assessment of renal protection efficacy of siRNA for p53 in experimental rat kidney transplantation models. Cell Transplant. 2010;19:1659–1670. doi: 10.3727/096368910X516619. [DOI] [PubMed] [Google Scholar]
- Kaiser W, Garrett CGB. Two-photon excitation in CaF2:Eu2+ Phys Rev Lett. 1961;7:229–231. [Google Scholar]
- Kantevari S, Hoang CJ, Ogrodnik J, Egger M, Niggli E, Ellis-Davies GC. Synthesis and two-photon photolysis of 6-(ortho-nitroveratryl)-caged IP3 in living cells. Chembiochem. 2006;7:174–180. doi: 10.1002/cbic.200500345. [DOI] [PubMed] [Google Scholar]
- Kantevari S, Matsuzaki M, Kanemoto Y, Kasai H, Ellis-Davies GC. Two-color, two-photon uncaging of glutamate and GABA. Nat Methods. 2010;7:123–125. doi: 10.1038/nmeth.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SA, Heinze KG, Schwille P. Fluorescence correlation spectroscopy in living cells. Nat Methods. 2007;4:963–973. doi: 10.1038/nmeth1104. [DOI] [PubMed] [Google Scholar]
- Levitt JA, Matthews DR, Ameer-Beg SM, Suhling K. Fluorescence lifetime and polarization-resolved imaging in cell biology. Curr Opin Biotechnol. 2009;20:28–36. doi: 10.1016/j.copbio.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Lipp P, Egger M, Niggli E. Spatial characteristics of sarcoplasmic reticulum Ca2+ release events triggered by L-type Ca2+ current and Na+ current in guinea-pig cardiac myocytes. J Physiol. 2002;542:383–393. doi: 10.1113/jphysiol.2001.013382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn ME, Barretto RP, Delp SL, Schnitzer MJ. Minimally invasive high-speed imaging of sarcomere contractile dynamics in mice and humans. Nature. 2008;454:784–788. doi: 10.1038/nature07104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looney MR, Thornton EE, Sen D, Lamm WJ, Glenny RW, Krummel MF. Stabilized imaging of immune surveillance in the mouse lung. Nat Methods. 2011;8:91–96. doi: 10.1038/nmeth.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahou P, Zimmerley M, Loulier K, Matho KS, Labroille G, Morin X, Supatto W, Livet J, Débarre D, Beaurepaire E. Multicolor two-photon tissue imaging by wavelength mixing. Nat Methods. 2012;9:815–818. doi: 10.1038/nmeth.2098. [DOI] [PubMed] [Google Scholar]
- Maiti S, Shear JB, Williams RM, Zipfel WR, Webb WW. Measuring serotonin distribution in live cells with three-photon excitation. Science. 1997;275:530–532. doi: 10.1126/science.275.5299.530. [DOI] [PubMed] [Google Scholar]
- Mao S, Benninger RK, Yan Y, Petchprayoon C, Jackson D, Easley CJ, Piston DW, Marriott G. Optical lock-in detection of FRET using synthetic and genetically encoded optical switches. Biophys J. 2008;94:4515–4524. doi: 10.1529/biophysj.107.124859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Hayama T, Kasai H, Ellis-Davies GC. Two-photon uncaging of gamma-aminobutyric acid in intact brain tissue. Nat Chem Biol. 2010;6:255–257. doi: 10.1038/nchembio.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Wei SH, Cahalan MD, Parker I. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc Natl Acad Sci USA. 2003;100:2604–2609. doi: 10.1073/pnas.2628040100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann W, Wallace DJ, Czubayko U, Herb JT, Schaefer AT, Looger LL, Denk W, Kerr JN. Two-photon calcium imaging of evoked activity from L5 somatosensory neurons in vivo. Nat Neurosci. 2011;14:1089–1093. doi: 10.1038/nn.2879. [DOI] [PubMed] [Google Scholar]
- Mower AF, Kwok S, Yu H, Majewska AK, Okamoto K, Hayashi Y, Sur M. Experience-dependent regulation of CaMKII activity within single visual cortex synapses in vivo. Proc Natl Acad Sci USA. 2011;108:21241–21246. doi: 10.1073/pnas.1108261109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura N, Schaffer CB, Friedman B, Tsai PS, Lyden PD, Kleinfeld D. Targeted insult to subsurface cortical blood vessels using ultrashort laser pulses: Three models of stroke. Nat Methods. 2006;3:99–108. doi: 10.1038/nmeth844. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci USA. 2007;104:365–370. doi: 10.1073/pnas.0609551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi J, Nagaoka A, Watanabe S, Ellis-Davies GC, Kitamura K, Kano M, Matsuzaki M, Kasai H. In vivo two-photon uncaging of glutamate revealing the structure-function relationships of dendritic spines in the neocortex of adult mice. J Physiol. 2011;589:2447–2457. doi: 10.1113/jphysiol.2011.207100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor DH, Huber D, Svoboda K. Reverse engineering the mouse brain. Nature. 2009;461:923–929. doi: 10.1038/nature08539. [DOI] [PubMed] [Google Scholar]
- Olivier N, Luengo-Oroz MA, Duloquin L, Faure E, Savy T, Veilleux I, Solinas X, Débarre D, Bourgine P, Santos A, Peyriéras N, Beaurepaire E. Cell lineage reconstruction of early zebrafish embryos using label-free nonlinear microscopy. Science. 2010;329:967–971. doi: 10.1126/science.1189428. [DOI] [PubMed] [Google Scholar]
- Owen DM, Magenau A, Majumdar A, Gaus K. Imaging membrane lipid order in whole, living vertebrate organisms. Biophys J. 2010;99:L7–L9. doi: 10.1016/j.bpj.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Piston DW. Photo-bleaching in two-photon excitation microscopy. Biophys J. 2000;78:2159–2162. doi: 10.1016/S0006-3495(00)76762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piyawattanametha W, Cocker ED, Burns LD, Barretto RP, Jung JC, Ra H, Solgaard O, Schnitzer MJ. In vivo brain imaging using a portable 2.9 g two-photon microscope based on a microelectromechanical systems scanning mirror. Opt Lett. 2009;34:2309–2311. doi: 10.1364/ol.34.002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchon TA, Gao L, Milkie DE, Davidson MW, Galbraith JA, Galbraith CG, Betzig E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat Methods. 2011;8:417–423. doi: 10.1038/nmeth.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickgauer JP, Tank DW. Two-photon excitation of channelrhodopsin-2 at saturation. Proc Natl Acad Sci USA. 2009;106:15025–15030. doi: 10.1073/pnas.0907084106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgeway WK, Millar DP, Williamson JR. Quantitation of ten 30S ribosomal assembly intermediates using fluorescence triple correlation spectroscopy. Proc Natl Acad Sci USA. 2012;109:13614–13619. doi: 10.1073/pnas.1204620109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau JV, Head WS, Nicholson WE, Powers AC, Piston DW. Pancreatic islet beta-cells transiently metabolize pyruvate. J Biol Chem. 2002;277:30914–30920. doi: 10.1074/jbc.M202314200. [DOI] [PubMed] [Google Scholar]
- Rocheleau JV, Head WS, Piston DW. Quantitative NAD(P)H/flavoprotein autofluorescence imaging reveals metabolic mechanisms of pancreatic islet pyruvate response. J Biol Chem. 2004;279:31780–31787. doi: 10.1074/jbc.M314005200. [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, Hooper S, Mitter R, Féral CC, Cook M, Larkin J, Marais R, Meneguzzi G, Sahai E, Marshall CJ. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20:229–245. doi: 10.1016/j.ccr.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer CB, Friedman B, Nishimura N, Schroeder LF, Tsai PS, Ebner FF, Lyden PD, Kleinfeld D. Two-photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PloS Biol. 2006;4:e22. doi: 10.1371/journal.pbio.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science. 2008;320:1638–1643. doi: 10.1126/science.1156120. [DOI] [PubMed] [Google Scholar]
- Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, Ramanujam N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci USA. 2007;104:19494–19499. doi: 10.1073/pnas.0708425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess E, Bestvater F, Heckel-Pompey A, Toth K, Hacker M, Stobrawa G, Feurer T, Wotzlaw C, Berchner-Pfannschmidt U, Porwol T, Acker H. Two-photon excitation and emission spectra of the green fluorescent protein variants ECFP, EGFP and EYFP. J Microsc. 2005;217:200–204. doi: 10.1111/j.1365-2818.2005.01437.x. [DOI] [PubMed] [Google Scholar]
- Squirrell JM, Wokosin DL, White JG, Bavister BD. Long-term two-photon fluorescence imaging of mammalian embryos without compromising viability. Nat Biotechnol. 1999;17:763–767. doi: 10.1038/11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringari C, Cinquin A, Cinquin O, Digman MA, Donovan PJ, Gratton E. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc Natl Acad Sci USA. 2011;108:13582–13587. doi: 10.1073/pnas.1108161108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supatto W, Débarre D, Moulia B, Brouzés E, Martin JL, Farge E, Beaurepaire E. In vivo modulation of morphogenetic movements in Drosophila embryos with femtosecond laser pulses. Proc Natl Acad Sci USA. 2005;102:1047–1052. doi: 10.1073/pnas.0405316102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theer P, Denk W. On the fundamental imaging-depth limit in two-photon microscopy. J Opt Soc Am A Opt Image Sci Vis. 2006;23:3139–3149. doi: 10.1364/josaa.23.003139. [DOI] [PubMed] [Google Scholar]
- Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000 micron in living brains by use of a Ti:Al2O3 regenerative amplifier. Optics Lett. 2003;28:1022–1024. doi: 10.1364/ol.28.001022. [DOI] [PubMed] [Google Scholar]
- Truong TV, Supatto W, Koos DS, Choi JM, Fraser SE. Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat Methods. 2011;8:757–760. doi: 10.1038/nmeth.1652. [DOI] [PubMed] [Google Scholar]
- Verveer PJ, Wouters FS, Reynolds AR, Bastiaens PI. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science. 2000;290:1567–1570. doi: 10.1126/science.290.5496.1567. [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Sandoval RM, Campos SB, Molitoris BA. Rapid diagnosis and quantification of acute kidney injury using fluorescent ratio-metric determination of glomerular filtration rate in the rat. Am J Physiol Renal Physiol. 2010;299:F1048–F1055. doi: 10.1152/ajprenal.00691.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T. Resolution and optical sectioning in the confocal microscope. J Microsc. 2011;244:113–121. doi: 10.1111/j.1365-2818.2011.03549.x. [DOI] [PubMed] [Google Scholar]
- Wokosin DL, Loughrey CM, Smith GL. Characterization of a range of fura dyes with two-photon excitation. Biophys J. 2004;86:1726–1738. doi: 10.1016/S0006-3495(04)74241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chen Y, Müller JD. Heterospecies partition analysis reveals binding curve and stoichiometry of protein interactions in living cells. Proc Natl Acad Sci USA. 2010;107:4117–4122. doi: 10.1073/pnas.0905670107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi P, Andegeko Y, Pestov D, Lovozoy VV, Dantus M. Two-photon imaging using adaptive phase compensated ultrashort laser pulses. J Biomed Opt. 2009;14:014002. doi: 10.1117/1.3059629. [DOI] [PubMed] [Google Scholar]
- Xu C, Webb WW. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J Opt Soc Am B Opt Phys. 1996;13:481–491. [Google Scholar]