

Abstract
Nuclear receptors (NRs) are a superfamily of transcription factors central to regulating many biological processes, including cell growth, death, metabolism, and immune responses. NR-mediated gene expression can be modulated by coactivators and corepressors through direct physical interaction or protein complexes with functional domains in NRs. One class of these domains includes short linear motifs (SLiMs), which facilitate protein-protein interactions, phosphorylation, and ligand binding primarily in the intrinsically disordered regions (IDRs) of proteins. Across all proteins, the number of known SLiMs is limited due to the difficulty in studying IDRs experimentally. Computational tools provide a systematic and data-driven approach for predicting functional motifs that can be used to prioritize experimental efforts. Accordingly, several tools have been developed based on sequence conservation or biophysical features; however, discrepancies in predictions make it difficult to determine the true candidate SLiMs. In this work, we present the ensemble predictor for short linear motifs (EPSLiM), a novel strategy to prioritize the residues that are most likely to be SLiMs in IDRs. EPSLiM applies a generalized linear model to integrate predictions from individual methodologies. We show that EPSLiM outperforms individual predictors, and we apply our method to NRs. The androgen receptor is an example with an N-terminal domain of 559 disordered amino acids that contains several validated SLiMs important for transcriptional activation. We use the androgen receptor to illustrate the predictive performance of EPSLiM and make the results of all human and mouse NRs publically available through the web service http://epslim.bwh.harvard.edu.
The nuclear receptor (NR) superfamily of transcription factors helps control growth and developmental processes by directly regulating expression of target genes (1, 2). NR-mediated transcription can be ligand dependent or ligand independent (3, 4), with coactivators and corepressors providing functional specificity (5–7). NR-mediated signaling is critical for normal cellular function and when dysregulated can contribute to the development of disease, such as cancers and congenital syndromes (8). For instance, the dysregulated interactions between the androgen receptor (AR) and the coregulators TMF, Smad3/Smad4, and Src have been associated with prostate cancer (9, 10). In addition, the interruption of an AR N-terminal to C-terminal (N/C) intramolecular interaction has been found to be a cause of androgen insensitivity syndrome (11). Sequence motifs, including short linear motifs (SLiMs), define the sites of interaction that underlie functional activation of NR coregulators. Characterizing existing motifs and uncovering novel sequence signatures will help define the functional sites of NRs and provide a template for matching interacting molecules. Once defined, the full set of interacting partners and the sites of interaction provide the basis for developing targeted, novel therapies.
NRs have common structural domains including an N-terminal domain (NTD), a DNA-binding domain (DBD), and a ligand-binding domain (LBD) (12). Among these regions, the DBD and LBD are highly conserved in sequence and structure, whereas intrinsically disordered regions (IDRs) show a high degree of variability in sequence composition and length. IDRs have been identified in several locations along NRs, including the hinge region between DBD and LBD, the C-terminal end of most NRs, and comprise a large portion of the NTD for specific NRs, such as AR and the glucocorticoid receptor (13). The functional importance of IDRs in NRs has largely been overlooked because they are relatively difficult to study compared with structured protein regions. Recently, a novel small molecule, EPI-001, which targets an IDR in ARs, has shown promise as a cancer therapeutic agent. The molecule acts by binding to the AR NTD and blocks transactivation by inhibiting the AR interactions with coregulators and androgen response elements on target genes (14, 15). The unstructured native state of the IDRs provides the flexibility to assume new conformations when an interacting partner or coregulator is present (16). Moreover, IDRs located in the activating function regions of NRs change conformation due to site-specific interaction with coregulators, N/C intramolecular interactions, or modifications by kinases (17–19).
SLiMs (also known as eukaryotes linear motif or molecular recognition features) are short stretches of protein sequences generally located in IDRs; these sites are targets for posttranslational modifications, cleavage, and ligand-binding modules in protein-protein interactions (20). So far, only a small number of SLiMs have been validated through experiments because of their unique biophysical properties (e.g., high hydrophobicity and larger interaction interfaces) and their location in unstructured, disordered regions (21). Computational methods to predict SLiMs provide a rational basis for which costly and time-intensive follow-up experimental work can be focused. Moreover, providing the most accurate predictions will reduce the likelihood of following false predictions. Several computational tools have been developed to predict potential SLiMs. The Eukaryote Linear Motif database provides data for experimentally validated SLiMs and tools for searching existing SLiM patterns (22). ANCHOR and MoRFpred use physicochemical features of proteins to predict binding sites in IDRs (23, 24). SLiMPrints evaluates the sequence-level evolutionary conservation for motif prediction (25), whereas SLiMPred uses a neural network-based method to consider multiple structural context features, including relevant solvent accessibility and predicted secondary structures (26). These algorithms perform well, but there are discrepancies between the predictions. In addition, their performance for predicting SLiMs in NRs has not been comprehensively characterized.
Ensemble methods are a class of algorithms that integrate multiple individual classifiers to produce a final set of predictions (27). They are based on the phenomenon that the collective wisdom of multiple independent predictions will produce more robust results than any individual approach (28). Ensemble methods have been widely used in solving system biology problems such as protein classification (29), protein structure prediction (30), gene motif prediction (31), gene network inference (32), and microarray analysis (33). Here, we present a novel ensemble predictor for SLiMs (EPSLiM) that first estimates the robustness of biophysical properties under single substitution mutations in protein disordered regions and then integrates predictions from the aforementioned SLiM predictors (Figure 1). The predictions on NRs produced by EPSLiM can facilitate future studies in analyzing the relations between NR coregulators and NR functions. By comparing the prediction performance of all SLiM predictors, we find that EPSLiM is the top performing method. We illustrate the accuracy of EPSLiM by exploring predictions in AR, with several previously validated motifs and also novel predictions. We have applied EPSLiM to all human and mouse NRs and make these results publically accessible through a searchable web interface, http://epslim.bwh.harvard.edu.
Figure 1.

Schematic view of the EPSLiM model. EPSLiM integrates the outputs of 4 individual predictors (CM-ANCHOR, MoRFpred, SLiMPred, and SLiMPrints) to predict SLiMs in intrinsically disordered regions of proteins. First, the same set of protein sequences are input into each of the predictors. These outputs are then integrated into one set of predictions using a generalized linear model. Within the CM-ANCHOR method, simulated mutations are introduced into the input protein sequences to determine which residues have the most impact on protein binding motifs. This effect is measured by comparing the mutated sequence scores from the motif predictor, ANCHOR, with the wild-type protein sequence. After sampling each residue in the protein 100 times, the average score is calculated. To ensure that the residue is in a disordered region, IUPred scores are incorporated to calculate the final CM-ANCHOR score for each residue.
Materials and Methods
Data
Experimentally validated SLiMs were downloaded from the Eukaryotic Linear Motif (ELM) database (22), containing 2245 SLiMs in 1438 proteins. To characterize the data set for predicting binding motifs in NRs, 396 of the 1438 proteins (comprising 216 411 non-ELM residues and 623 ELMs with 3583 ELM residues) were selected for further analysis because they had disordered regions with length and proportion similar to those of NRs and contained at least 1 true-positive ligand binding SLiM (filtering criteria: disordered length of 50–400, total length of 260–1000, and disordered percentage of <60%, with a LIG ELM instance). To compare predictors constructed using organism-specific data, we selected a subset that includes only human proteins (214 proteins).
EPSLiM
Individual predictors
The EPSLiM predictor is an ensemble predictor, in which predictions of individual algorithms were made, then the individual outputs were integrated using a generalized linear model to produce the final set of predictions. The individual methods tested for building the EPSLiM model are ANCHOR (23), MoRFpred (24), SLiMPrints (25), SLiMPred (26), and a modified ANCHOR method that incorporates simulated mutations that we present in the following section. Predictions from each method were made using publically accessible executable programs or web services. Based on data from the ELM database, the experimentally validated SLiMs in the compiled dataset were annotated as 1 (true positive) or 0 (others).
Computational mutated ANCHOR (CM-ANCHOR)
Among the aforementioned individual predictors, SLiMPrints and SLiMPred are fully or partially based on sequence conservation (25, 26). MoRFpred also considers sequence similarity, although it is a minor factor considered in the model (24). ANCHOR is the only method that makes predictions solely on the biophysical properties of the sequence itself (23). To investigate the effect of mutations on potential binding sites based on biophysical properties, we systematically mutated each base in a protein's IDR and measured the resulting binding potential using ANCHOR. The result of this analysis, which we termed CM-ANCHOR, is a measure of how each residue influences potential SLiM sites in the protein.
For each residue in a protein, 100 random mutations were introduced using the substitution frequency table as defined by the probabilities of amino acid replacements in the BLOSUM62 matrix (34). Next, the ANCHOR score for the protein was recalculated for the mutant sequences. The average ANCHOR score differences between the 100 mutant proteins and the wild-type protein were compiled into a matrix. From the calculated ANCHOR score differences, we observed that the influence of a mutation did not extend past 100 bp from the mutation site (Supplemental Figure 1 http://press.endocrine.org/doi/suppl/10.1210/me.2013–1006/suppl_file/me-14–1006.pdf); thus, raw CM-ANCHOR scores were calculated within a window ± 100 bp.
In the matrix of ANCHOR score differences for each protein and displayed in Supplemental Figure 2, the average score for each residue position (column) represents the influence a mutation at a defined residue has on neighboring residues. We further calculated the average of ANCHOR score differences for each residue as
| (1) |
where ep,i is the average ANCHOR score differences for residue i in protein p induced by mutations within a range of 100 bp, αp,i,j is the ANCHOR score difference for residue affected by mutation on a residue at a distance of j residues, and N is the count of residues within the window in which the mutation simulation was performed (±100 bp). Ep is the vector containing the average ANCHOR score differences of all residues of protein p.
The average ANCHOR score differences were then normalized by global scaling from 0 to 1:
| (2) |
where ε is a vector of average ANCHOR score differences for all proteins, p. min(ε) and max(ε) are the minimum and maximum ANCHOR score differences measured for all proteins. The raw and normalized average ANCHOR score differences at residue i of protein p are represented as ep,i and e′p,i.
ANCHOR scores of ordered proteins are much lower than those of disordered regions, which leads to smaller ANCHOR changes during the mutation simulation mentioned above. Because only the disordered regions of proteins are targeted, we further incorporated the residue scores for the disordered region predictor, IUPred (35), to distinguish ordered regions from the insensitive residues in disordered regions as follows:
| (3) |
where dp,i is the prediction from IUPred. The values, e″p,i were scaled from 0 to 1 (equation 2), and define the CM-ANCHOR score.
Integrative model evaluation
Together with the CM-ANCHOR score, the 4 published predictors of SLiMs (ANCHOR, MoRFpred, SLiMPred, and SLiMPrints) were integrated to build the EPSLiM predictor. A series of models were tested to identify the best strategy to integrate the results of individual prediction methods, including generalized linear regression, elastic net regularization, random forests, and support vector machines. These methods were implemented as follows.
Generalized linear model (Gaussian).
Generalized linear regression with a Gaussian link function (identity) was applied with the generalized linear model function, glm( ), in R statistical programming software (36), representing the following model:
| (4) |
Generalized linear model (binomial).
The response in our dataset is binary (true or false SLiMs); thus, we tested a generalized linear regression using the R function glm( ) with a binomial logit link function:
| (5) |
The outputs were scaled from 0 to 1 (equation 2). This generalized linear model was selected as the integration method based on a comparative analysis of integration strategies (as shown in the next section). The prediction threshold was determined through cross-validation as described below.
Elastic net regularization.
To avoid overfitting, both generalized linear models were analyzed again with the glmnet (Lasso and elastic-net regularized generalized linear models) R package (37). Lambda.1se (the largest value of λ such that error is within 1 SE of the minimum) was used as the as the penalty parameter. The prediction outputs were normalized using equation 2.
Random forest.
To account for potential nonlinear relationships in the data, a random forest model was implemented using the randomForest R package (38). Considering the unbalanced training dataset, the parameter class weight was based on the ratio between motif residues and nonmotif residues in the training dataset. Default settings were used for the other parameters. The prediction outputs, which represent the probability of being classified as positive, were calculated with the predict( ) function.
Support vector machine.
A support vector machine model with a radial basis kernel was tested using the e1071 R package (39). To keep the two classes (SLiM and non-SLiM) balanced, parameter class weights were set based on the ratio between motif and nonmotif sites. The prediction outputs were calculated with the predict( ) function.
Cross-validation
Three-fold cross-validation was used to evaluate the prediction accuracy of all models. The full dataset of 396 proteins (216 411 non-ELM residues and 3583 ELM residues) was randomly and evenly split into 3 groups. The protein sequences have different lengths; thus, to ensure that the residue content was balanced, we required that the number of residues in each group differed by no more than ±3%. One of the groups was held out for testing, and the remaining two groups were combined to train predictors. Performance was evaluated by calculating the true-positive rate and false-positive rate as reflected by the receiver operating characteristic (ROC) curve. A true positive was defined as a known ELM residue that was predicted to be within a SLiM site. A false positive was defined as a residue predicted to be within a SLiM site but was not known to be an ELM residue. To determine the threshold for a positive residue, the true-positive rate (TPR = true positives/positives) was plotted against the corresponding false-positive rate (FPR = false positives/negatives). The area under the ROC curve (AUROC) was calculated and defines a predictor's overall performance (all calculations were made using the ROCR R package [40]). This procedure was repeated for each of the 3 groups (folds) in the 3-fold cross-validation and for a total of 10 iterations. Plots reflect the average performance as measured over the 10 iterations.
Results
Variable, model, and threshold selection
There is an array of approaches that can be taken for constructing an ensemble predictor; thus, we first compared the performance of 4 popular models, including generalized linear model (with Gaussian or binomial link function), elastic net regression (with Gaussian or binomial link function), random forest, and support vector machine. Based on 10 interactions of 3-fold cross-validation (see Materials and Methods), the generalized linear model with the binomial link function showed the highest performance with a measured AUROC = 0.8364 (Figure 2) and was thus selected to be the integrative model for the EPSLiM predictor.
Figure 2.
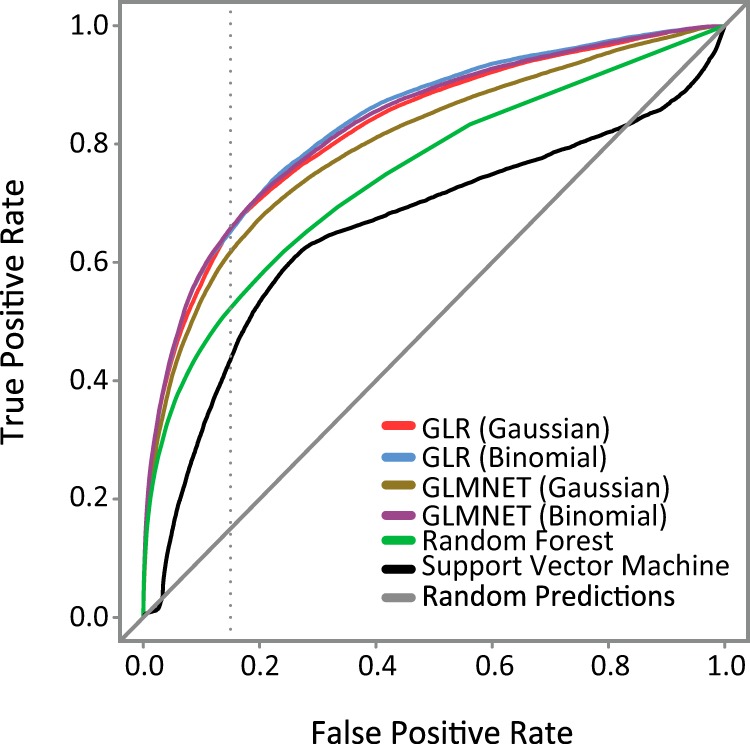
Performance evaluation for different integrative models. Individual integrative models were evaluated using ROC curves. The top performing model, generalized linear regression (GLR) with a binomial link function, was designated as the EPSLiM model. The dotted gray line was placed at the 15% FPR. Corresponding TPRs and the AUROCs are reported in Table 1. A 15% FPR corresponds to the threshold of 0.02 for the EPSLiM model.
We also considered the effect that different combinations of individual methods would have on the performance of an ensemble predictor. We found that the ensemble predictor with the best performance consisted of CM-ANCHOR, MoRFpred, SLiMPred, and SLiMPrints; adding ANCHOR to the group did not improve the performance of the model (Supplemental Table 1) because of the redundant information provided by ANCHOR and CM-ANCHOR. We investigated the predictive performance of ensemble predictors with these methods selectively excluded and found that lower performance was shown with exclusion of CM-ANCHOR (AUROC = 0.8080) than with exclusion of ANCHOR (AUROC = 0.8364) (Supplemental Table 1). Thus, we chose to include CM-ANCHOR and exclude ANCHOR from the EPSLiM model. The final EPSLiM predictor coefficients were fit using the full set of training data (see Materials and Methods).
To identify predicted SLiMs, a threshold of 0.02 was selected using the ROC curves and reflects a balance of TPR and FPR. On ROC curves, the optimal threshold corresponds to the point nearest to the upper left corner (FPR = 0%; TPR = 100%). We tested a series of other thresholds by shifting the FPR from 5% to 50% and recording the corresponding TPR (upplemental Table 2). From the observed results, points between FPR = 15% and 30% had relatively close distances to the upper left corner. Because of the limited amount of experimentally validated SLiMs, the accuracy of true negatives in the training dataset is lower than that of true positives. Although this finding indicates that the FPR is possibly underestimated, we chose a conservative FPR = 15%. Accordingly, residues with an EPSLiM score of ≥0.02 were considered positive predictions, corresponding to an TPR = 65% and FPR = 15%.
Table 1.
Integrative Model Performance
| TPR, % | AUROC | |
|---|---|---|
| Generalized linear regression (Gaussian) | 65 | 0.8261 |
| Generalized linear regression (binomial) | 65 | 0.8364 |
| Elastic-net (Gaussian) | 62 | 0.8107 |
| Elastic-Net (binomial) | 65 | 0.8331 |
| RandomForest | 52 | 0.7491 |
| Support vector machine | 44 | 0.6602 |
AUROCs for different integrative models are shown. The TRP is reported at a 15% FPR. The top performing model, generalized linear regression with a binomial link function, was designated as the EPSLiM model. A 15% FPR and 65% TPR for the EPSLiM predictor corresponds to a threshold of 0.02.
Performance comparison across individual methods
The EPSLiM model integrates the predictions from 4 individual methods (Figure 1). To demonstrate the optimal performance of the EPSLiM model, we compared the predictions from all individual methods with that of EPSLiM using the same cross-validation method for evaluating the full EPSLiM predictor. The results show that 3 of the other 4 individual predictors had an AUROC of ≤0.7, whereas EPSLiM was the only predictor that had an AUROC of >0.8. The best performing individual predictor, SLiMPred, achieved a performance of AUROC = 0.7802. In addition, at an FPR = 15%, whereas EPSLiM produces a TPR = 65%, the individual predictors achieve TPRs between 36% and 54% (Figure 3). Note that SLiMPrints produces binary predictions, which can only be presented as a single point on the ROC plots. At this single point, the FPR = 4.5% and the TPR = 28%. The corresponding TPR = 43% for EPSLiM is the best performance of all the predictors. The performance of ANCHOR, which was excluded from the final model as an individual predictor, was found to be AUROC = 0.6764 (Supplemental Figure 3), and a TPR = 33% at FPR = 15% (Supplemental Table 2). Given the full set of evaluations, we conclude that EPSLiM produces the most accurate results for predicting SLiMs in intrinsically disordered protein regions.
Figure 3.
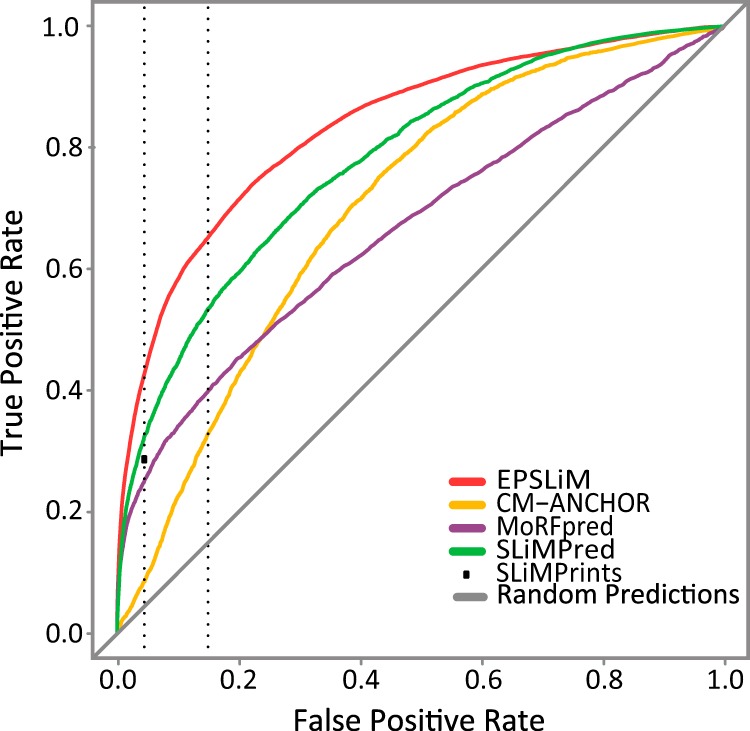
Performance evaluation for SLiM predictors. Individual SLiM predictors were evaluated by measuring the AUROCs. The EPSLiM model had the best performance (AUROC = 0.8364), whereas the remaining predictors showed a much lower performance (AUROC = <0.7). The SLiMPrints method generates binary predictions and thus is represented as a single point corresponding to a 4.5% FPR. Represented as a dotted gray line, a 4.5% FPR corresponds to a 28% TPR. The second dotted gray line represents the 15% FPR, which corresponds to a 65% TPR at a threshold of 0.02 for EPSLiM. Corresponding TPRs for all methods are available in Supplemental Table 2.
Predicting SLiMs for NRs
To discover SLiMs in the disordered regions of NRs, we applied EPSLiM to NRs collected for human and mouse using the threshold defined in the previous section. There are 46 sequences for each organism, all of which contain IDRs of different lengths. In the ELM database, human retinoic acid receptor α (RARA), human nuclear hormone receptor NUR/77 (NR4A1), and mouse steroidogenic factor 1 (NR5A1) have 6 SLiM instances recorded in total. Although there are only a few SLiMs validated by experiment in nuclear hormone receptors, EPSLiM is able to identify 4 of the motifs.
Comparing a human-specific to a universal EPSLiM model
The EPSLiM model coefficients are determined by the training dataset. To test the influence of different training datasets, we compared the performance of human-specific and nonorganism-specific training datasets. From the ROC plots, the human-specific EPSLiM model achieved an AUROC = 0.8310, compared with AUROC = 0.8364 using the nonorganism-specific training data. Using the established threshold = 0.02, the human-specific predictor showed the same TPR = 65% and a similar but slightly higher FPR = 17% compared with that for the nonorganism-specific model (Supplemental Figure 4). When the prediction results on human and mouse NRs (92 proteins), with a threshold = 0.02, were compared, the nonorganism-specific model was able to predict 3036 residues as SLiM sites and the human-specific model predicted 2959 residues as SLiM sites. More than 90% of the two prediction sets were the same (Figure 4). Because of the slightly better performance, the EPSLiM model used to predict SLiMs on NRs was trained using the nonorganism-specific dataset.
Figure 4.
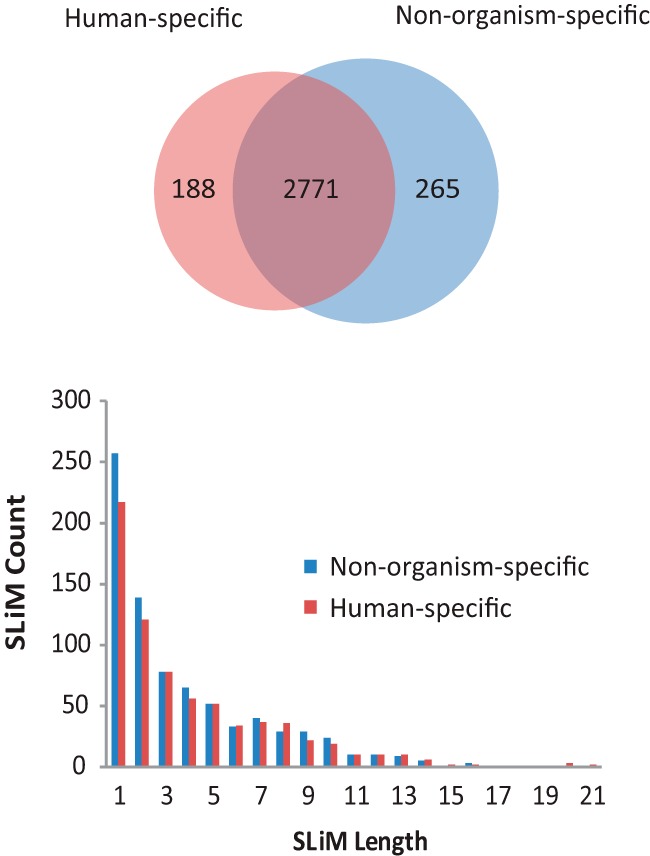
Evaluating the impact of training datasets. SLiMs predicted from EPSLiM using 2 different training datasets were compared. Predictions on 92 nuclear hormone receptors were made with a human-specific or nonorganism-specific dataset. With the threshold of 0.02 (Figures 2 and 3), there were 2771 overlapping predictions representing 93% of the total predictions with the human-specific dataset and 91% of the total predictions with the nonorganism-specific dataset. The histogram displays the number of predicted SLiM regions for different disordered region lengths. Both sets of predictions are highly similar, supporting the use of the nonorganism-specific dataset within the EPSLiM model for predictions in human and nonhumans.
Web server of EPSLiM
To make the EPSLiM model publically accessible, we developed a web service that can be found at http://epslim.bwh.harvard.edu. Users can input NR protein names and accession identification numbers and then access the individual SLiM prediction results (Figure 5). For each protein, general information and line plots of prediction results are displayed with the sequence disorder status annotated above. For easy reference and interpretation, a heat map of binarized predictions is also generated. The web service provides a link to download the fasta file of the protein and the EPSLiM results.
Figure 5.

The EPSLiM web interface displaying results for AR. The EPSLiM web service allows users to search for human and mouse nuclear receptor predictions. The web interface will display basic information on the protein (name and function). Next, the results of the SLiM predictors are displayed as a line plot and a discretized heat plot. The results from the IUPred disordered region predictor are also displayed. For AR, the NTD is largely disordered; multiple SLiMs were predicted by each method.
SLiM predictions and validation in AR
In total, EPSLiM predicted 37 SLiMs of varying length in AR (Figure 6A), of which 16 overlap with already identified ELM patterns (Supplemental Table 3). Here, we explore 3 cases that highlight the prediction accuracy of EPSLiM and also discuss several novel predictions. We use the AR NTD as an example because it is an intrinsically disordered region and is responsible for interacting with many AR coactivators and corepressors (41, 42). In addition, an N/C intramolecular interaction between the AR NTD and AR LBD is essential for AR dimerization and for binding to the target gene (11).
Figure 6.
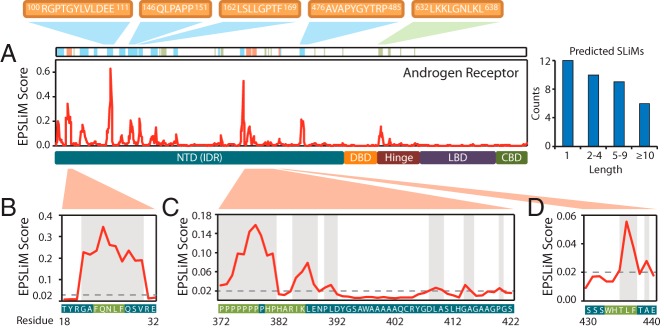
EPSLiM predictions on disordered regions of AR. A, In AR, EPSLiM predictions are observed in the NTD and hinge region. Highlighted in red are predicted SLiMs with supporting experimental validation. Highlighted in blue are regions with high EPSLiM scores that match patterns found in the ELM database. Highlighted in green are regions with high EPSLiM scores but no additional supporting information. B, An EPSLiM peak was detected at AR 21–30, which overlaps with AR 23–27 (FXXLF), a validated SLiM that is important in the intramolecular interaction between the AR N-terminal and C-terminal domains. The gray dashed line is the EPSLiM threshold; the protein sequence and position are located below the EPSLiM line plot with the validated SLiM highlighted in green. C, Six regions in AR 378–422 were predicted by EPSLiM, the binding area for the AR-Src interaction. Highlighted in green, the first two predictions in this region overlap with 2 key binding motifs for Src (AR 372–378 and AR 380–386). D, AR 423–427 (WXXLF) is another validated SLiM that is responsible for the AR N/C intramolecular interaction.
The first verification of the EPSLiM predictions is for the proline-rich region of AR NTD between the residues 372 and 422 (Figure 6C) (43). In humans, an SH3 domain mediates the interaction between AR and Src. Induced by steroid hormone or epidermal growth factor, the AR-Src interaction activates Src and affects the S phase of the cell cycle (44). A deletion at either AR 372–385 or AR 372–378 will abolish the AR-Src interaction, and a deletion at AR 380–386 results in a weak activation of Src (10). From the EPSLiM result, 6 regions (AR 372–381, 385–388, 390–391, 409–411, 415–416, and 420) were predicted to be binding SLiMs. For the first 2 regions, we can observe a clear peak with EPSLiM scores of 0.032 to 0.158, far greater than the 0.02 threshold; the neighboring residues score drop far below the threshold (Figure 6C). Accuracy for each EPSLiM prediction was calculated as accuracy = (true positives + true negatives)/(positives + negatives). The accuracy of this peak ranges from 90% to 98% based on the cross-validation evaluation. In addition, AR 372–381 matches an SH3 domain defined in the ELM database (LIG_SH3_3; … [PV]… P) (22).
The second and third examples of verified EPSLiM predictions involve two separate 5-amino acid short peptides that are specific to AR and mediate the interactions with coregulatory proteins and also the N/C intramolecular interaction of AR. The motif, FXXLF (AR 23–27), interacts with the AR LBD and forms a head-tail structure that regulates gene expression. In addition, by interaction with this motif, cyclin D1 can inhibit this head-tail structure to repress AR function (45). A clear spike in the EPSLiM scores can be seen at the FXXLF motif with scores of >0.185 (Figure 6B), which corresponds to an accuracy of 98%. Positive EPSLiM scores were also discovered for the neighboring residues, which fall into the N/C intramolecular interaction region, defined as AR 1−36; a total of 23 of the 36 residues were predicted by EPSLiM. The motif, WXXLF (AR 433–437), a major transactivation motif in AR activation function 5 (AF5) region, is also involved in the N/C intramolecular interaction and mediates ligand-independent AR activity (46, 47). The EPSLiM score of the last 3 residues in the WXXLF motif were predicted with scores varying from 0.022 to 0.056, corresponding to accuracy between 86% and 94% (Figure 6D).
Aside from the already experimentally validated sites, EPSLiM predicted previously uncharacterized but high-scoring regions. One of these interesting regions is AR 100–111 (RGPTGYLVLDEE), which shows the highest EPSLiM score in AR (Figure 6A). Of the 11 residues, 9 have EPSLiM scores >0.1, which corresponds to an accuracy of >97%. With use of the ELM database (22), the FHA phosphopeptide ligand binding site (LIG_FHA_1:..(T)..[ILV].), the SH2 ligand binding site (LIG_SH2_STAT5: Y[VLTFIC]..), and the SUMO protein binding site (LIG_SUMO_SBM_1: [ILV](.[ILV]|[ILV]|[ILV].) [ILV][STDE]{1, 10}) were matched to this region. Besides predictions in the AR NTD, a few residues were also predicted as SLiMs in the hinge region (Figure 6A). The EPSLiM scores of AR 631–638 (LKKLGNLKL) and AR 650–653 (SPTE) vary from 0.024 to 0.156, which are the only two peaks outside of the AR NTD. Interestingly, it has been reported that this region is responsible for recognizing and interacting with AR response elements (48). The prediction strength and motif patterns together unveiled possible sequence features that are attractive for future experimental investigation.
Discussion
Intrinsically disordered regions are present in most NRs and often in stretches of many contiguous residues (>30 residues), such as those in AR, the glucocorticoid receptor, the estrogen receptor, and the progesterone receptor (13). SLiMs in IDRs of NRs play an important role in protein-protein interactions related to NR transcriptional regulation and have been targeted to develop anticancer therapeutic agents (14, 15, 49). The difficulties in targeting specific regions containing SLiMs has motivated the development of computational tools to predict SLiMs from protein sequences, which can then be targeted for further experimental investigation.
In this study, we present the ensemble predictor for SLiMs (EPSLiM) to predict potential binding motifs in intrinsically disordered regions of proteins. The advantage of ensemble prediction is that the best predictions are sampled from multiple independent methods, thus producing the most robust set of predictions (28). We trained EPSLiM with an ELM annotated dataset of proteins that match the overall protein length and IDR length of NRs. We also introduced CM-ANCHOR, which aims to characterize the effect of single residue mutations on the binding potential of SLiMs. Along with CM-ANCHOR, the EPSLiM model integrates SLiMPred, MoRFPred, and SLiMPrints. The fit of coefficient in the generalized linear model implemented in EPSLiM is useful in understanding the weight of each individual predictor as it relates to the full model. In this instance of EPSLiM, we found that MoRFpred was assigned the largest weight, SLiMPred had the second highest weight, and SLiMPrints and CM-ANCHOR had similarly lower weights.
The predictions produced by EPSLiM can facilitate the identification of novel SLiMs, which can help guide experimental discovery of coregulator binding sites in NRs. We demonstrated 3 instances of experimentally supported predictions that were validated in the AR NTD, but the tool provided many additional predictions with high EPSLiM scores that contain interesting predicted binding sites. In particular, AR 100–111 contains a high proportion of residues with prediction accuracy greater than 97%. In addition, AR 631–638 and AR 650–653 show similarly high prediction accuracy. These sites represent potentially novel SLiMs for future research efforts.
The limitation of EPSLiM is the small number of experimentally determined SLiMs. Because of the difficulty in experimentally validating SLiMs, the number of true-positive and true-negative motifs is limited, leading to an unbalanced ratio of positives and negatives in the training and test datasets. Thus, it should also be noted that the results we present are overly conservative, because there are probably true-positive motifs not included in the ELM database; these motifs will be counted as false-positive predictions in the current performance evaluation.
Computational biology prediction tools provide a fast and data-driven set of results that can help guide experiments. The implementation of EPSLiM facilitates the study of functional intrinsically disordered regions and particularly the relation between intrinsically disordered regions and coregulators of NRs. To verify the predictive performance of EPSLiM, future work will include the necessary step of experimentally validating predictions. As the number of validated SLiMs grows, the prediction accuracy of EPSLiM will also increase, thus providing a more powerful community tool for future research.
Additional material
Supplementary data supplied by authors.
Acknowledgments
Current address for J.C.C.: Department of Pharmacology, School of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO 80045.
This work was supported by the Evans Foundation (merit award to R.J.), Brigham and Women's Hospital startup funds (S.B. and R.J.), the National Institute on Aging (Grant 5 R01 AG037193-11), and the National Science Foundation (Grant CCF-1219007).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AR
- androgen receptor
- AUROC
- area under the receiver operating characteristic curve
- CM-ANCHOR
- computational mutated ANCHOR
- DBD
- DNA-binding domain
- ELM
- Eukaryotic Linear Motif
- EPSLiM
- ensemble predictor for short linear motifs
- IDR
- intrinsically disordered region
- LBD
- ligand-binding domain
- N/C
- N-terminal to C-terminal
- NTD
- N-terminal domain
- NR
- nuclear receptor
- ROC
- receiver operating characteristic
- SLiM
- short linear motif.
References
- 1. Beato M, Herrlich P, Schütz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. [DOI] [PubMed] [Google Scholar]
- 2. Owen GI, Zelent A. Origins and evolutionary diversification of the nuclear receptor superfamily. Cell Mol Life Sci. 2000;57:809–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rachez C, Lemon BD, Suldan Z, et al. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature. 1999;398:824–828. [DOI] [PubMed] [Google Scholar]
- 4. Hörlein AJ, Näär AM, Heinzel T, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. [DOI] [PubMed] [Google Scholar]
- 5. Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab. 2002;13:55–60. [DOI] [PubMed] [Google Scholar]
- 6. Lonard DM, O'Malley B W. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700. [DOI] [PubMed] [Google Scholar]
- 7. Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Nuclear receptor coactivators and corepressors. Mol Endocrinol. 1996;10:1167–1177. [DOI] [PubMed] [Google Scholar]
- 8. Lonard DM, Lanz RB, O'Malley BW. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28:575–587. [DOI] [PubMed] [Google Scholar]
- 9. Rahman M, Miyamoto H, Chang C. Androgen receptor coregulators in prostate cancer: mechanisms and clinical implications. Clin Cancer Res. 2004;10:2208–2219. [DOI] [PubMed] [Google Scholar]
- 10. Migliaccio A, Varricchio L, De Falco A, et al. Inhibition of the SH3 domain-mediated binding of Src to the androgen receptor and its effect on tumor growth. Oncogene. 2007;26:6619–6629. [DOI] [PubMed] [Google Scholar]
- 11. Langley E, Kemppainen JA, Wilson EM. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J Biol Chem. 1998;273:92–101. [DOI] [PubMed] [Google Scholar]
- 12. Kumar R, Thompson EB. The structure of the nuclear hormone receptors. Steroids. 1999;64:310–319. [DOI] [PubMed] [Google Scholar]
- 13. Krasowski MD, Reschly EJ, Ekins S. Intrinsic disorder in nuclear hormone receptors. J Proteome Res. 2008;7:4359–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andersen RJ, Mawji NR, Wang J. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. [DOI] [PubMed] [Google Scholar]
- 15. Sadar MD. Small molecule inhibitors targeting the “Achilles' heel” of androgen receptor activity. Cancer Res. 2011;71:1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bain DL, Heneghan AF, Connaghan-Jones KD, Miura MT. Nuclear receptor structure: implications for function. Annu Rev Physiol. 2007;69:201–220. [DOI] [PubMed] [Google Scholar]
- 17. Garza AM, Khan SH, Kumar R. Site-specific phosphorylation induces functionally active conformation in the intrinsically disordered N-terminal activation function (AF1) domain of the glucocorticoid receptor. Mol Cell Biol. 2010;30:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He B, Minges JT, Lee LW, Wilson EM. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J Biol Chem. 2002;277:10226–10235. [DOI] [PubMed] [Google Scholar]
- 19. Yeh S, Hu YC, Rahman M, et al. Increase of androgen-induced cell death and androgen receptor transactivation by BRCA1 in prostate cancer cells. Proc Natl Acad Sci USA. 2000;97:11256–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Diella F, Haslam N, Chica C, et al. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–6603. [DOI] [PubMed] [Google Scholar]
- 21. Mészáros B, Tompa P, Simon I, Dosztányi Z. Molecular principles of the interactions of disordered proteins. J Mol Biol. 2007;372:549–561. [DOI] [PubMed] [Google Scholar]
- 22. Dinkel H, Michael S, Weatheritt RJ, et al. ELM—the database of eukaryotic linear motifs. Nucleic acids Res. 2012;40(Database issue):D242–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mészáros B, Simon I, Dosztanyi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol. 2009;5(5):e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Disfani FM, Hsu WL, Mizianty MJ, et al. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics. 2012;28:i75–i83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davey NE, Cowan JL, Shields DC, Gibson TJ, Coldwell MJ, Edwards RJ. SLiMPrints: conservation-based discovery of functional motif fingerprints in intrinsically disordered protein regions. Nucleic Acids Res. 2012;40:10628–10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mooney C, Pollastri G, Shields DC, Haslam NJ. Prediction of short linear protein binding regions. J Mol Biol. 2012;415:193–204. [DOI] [PubMed] [Google Scholar]
- 27. Dietterich TG. Ensemble methods in machine learning. Mult Classifier Syst. 2000;1857:1–15. [Google Scholar]
- 28. Surowiecki J. The Wisdom of Crowds : Why the Many Are Smarter Than the Few and How Collective Wisdom Shapes Business, Economies, Societies, and Nations. New York, NY: Doubleday; 2005. [Google Scholar]
- 29. Martelli PL, Fariselli P, Casadio R. An ENSEMBLE machine learning approach for the prediction of all-alpha membrane proteins. Bioinformatics. 2003;19(suppl 1):i205–i211. [DOI] [PubMed] [Google Scholar]
- 30. Shen HB, Chou KC. Ensemble classifier for protein fold pattern recognition. Bioinformatics. 2006;22:1717–1722. [DOI] [PubMed] [Google Scholar]
- 31. Wijaya E, Yiu SM, Son NT, Kanagasabai R, Sung WK. MotifVoter: a novel ensemble method for fine-grained integration of generic motif finders. Bioinformatics. 2008;24:2288–2295. [DOI] [PubMed] [Google Scholar]
- 32. Marbach D, Costello JC, Küffner R, et al. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9(8):796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qiu P, Wang ZJ, Liu KJ. Ensemble dependence model for classification and prediction of cancer and normal gene expression data. Bioinformatics. 2005;21:3114–3121. [DOI] [PubMed] [Google Scholar]
- 34. Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dosztányi Z, Csizmók V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347:827–839. [DOI] [PubMed] [Google Scholar]
- 36. R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 37. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 38. Liaw A, Matthew Wiener M. 2002 Classification and Regression by randomForest. R News. 2002;2(3):18–22. [Google Scholar]
- 39. Meyer D, Hornik K, Weingessel A, Leisch F. e1071: misc functions of the department of statistics (e1071). Vienna, Austria: Technische Universität Wien; 2012. [Google Scholar]
- 40. Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. [DOI] [PubMed] [Google Scholar]
- 41. Hsiao PW, Chang C. Isolation and characterization of ARA160 as the first androgen receptor N-terminal-associated coactivator in human prostate cells. J Biol Chem. 1999;274:22373–22379. [DOI] [PubMed] [Google Scholar]
- 42. McEwan IJ, Lavery D, Fischer K, Watt K. Natural disordered sequences in the amino terminal domain of nuclear receptors: lessons from the androgen and glucocorticoid receptors. Nucl Recept Signal. 2007;5:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Migliaccio A, Castoria G, Di Domenico M, et al. Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19:5406–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Migliaccio A, Di Domenico M, Castoria G, et al. Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res. 2005;65:10585–10593. [DOI] [PubMed] [Google Scholar]
- 45. Burd CJ, Petre CE, Moghadam H, Wilson EM, Knudsen KE. Cyclin D1 binding to the androgen receptor (AR) NH2-terminal domain inhibits activation function 2 association and reveals dual roles for AR corepression. Mol Endocrinol. 2005;19:607–620. [DOI] [PubMed] [Google Scholar]
- 46. He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000;275:22986–22994. [DOI] [PubMed] [Google Scholar]
- 47. Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion independent prostate cancer cells. Cancer Res. 2007;67:10067–10077. [DOI] [PubMed] [Google Scholar]
- 48. McEwan IJ. Intrinsic disorder in the androgen receptor: identification, characterisation and drugability. Mol Biosyst. 2012;8:82–90. [DOI] [PubMed] [Google Scholar]
- 49. Myung JK, Banuelos CA, Fernandez JG, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.