

Abstract
Alzheimer's disease (AD) is associated with pathological assembly states of amyloid-β protein (Aβ). Aβ-related synaptotoxicity can be blocked by anti-prion protein (PrP) antibodies, potentially allowing therapeutic targeting of this aspect of AD neuropathogenesis. Here, we show that intravascular administration of a high-affinity humanized anti-PrP antibody to rats can prevent the plasticity-disrupting effects induced by exposure to soluble AD brain extract. These results provide an in vivo proof of principle for such a therapeutic strategy.
Keywords: Alzheimer's, drug discovery, immunotherapy, long-term potentiation, prion
Introduction
Alzheimer's disease (AD) presents one of the greatest healthcare and economic challenges globally, but no effective therapy to halt or delay disease progression has been developed. Rather than being a single disease entity, AD may be considered a clinicopathological syndrome with multiple interacting genetic and environmental factors involved in its onset and progression. Aggregation of the amyloid-β protein (Aβ) is believed to be central, with water-soluble nonfibrillar assemblies closely linked to memory impairment (Benilova et al., 2012). Only certain synthetic and cell-derived Aβ preparations are synaptotoxic in vitro at pathophysiologically pertinent low nanomolar concentrations (Benilova et al., 2012). Importantly, soluble extract from AD brain contains potent toxic Aβ assemblies that can rapidly disrupt hippocampal long-term potentiation (LTP; Shankar et al., 2008), a form of synaptic plasticity implicated in memory and learning (Bliss and Collingridge, 1993). Although different assembly forms of Aβ may exert toxicity by different mechanisms (Benilova et al., 2012), it is known that prion protein (PrP) is required for at least some aspects of Aβ toxicity (Laurén et al., 2009; Barry et al., 2011; Freir et al., 2011). Targeting PrP, which has been investigated extensively as a strategy for treating prion disease, is attractive because both constitutive knock-out of PrP expression and ablation of neuronal PrP expression in the adult mouse are without major phenotypes (Nicoll and Collinge, 2009). The murine monoclonal antibodies 6D11 (Laurén et al., 2009), Fab-D13 (Barry et al., 2011), and ICSM35 (Freir et al., 2011), which bind directly to the Aβ-binding site on PrP (Laurén et al., 2009), block Aβ-induced inhibition of LTP, as does ICSM18, which binds to helix-1 of PrP (Freir et al., 2011). 6D11 was shown to reverse behavioral deficits when administered intraperitoneally into an amyloid precursor protein/presenilin-1 mutant mouse model of AD (Chung et al., 2010), but it is uncertain whether this involved a direct action in the nervous system or how the Aβ assemblies in transgenic mouse models relate to those found in human AD brain. It has been suggested that cross-linking of PrP by antibodies that interact close to the Aβ-binding site on PrP triggers neuronal apoptosis in vivo (Solforosi et al., 2004), although we were unable to confirm this (Klohn et al., 2012). Furthermore, cross-linking via this epitope of PrP has been proposed as a cause of PrP-dependent Aβ toxicity (Bate and Williams, 2011) and this region has been suggested to be involved with PrP function and binding interactions (Béland and Roucou, 2012). In contrast, antibodies that bind to only helix-1 of PrP are consistently innocuous, even on direct injection into the hippocampus at 1 mg/ml (Solforosi et al., 2004; Klohn et al., 2012; Ohsawa et al., 2013), which produces a local concentration of up to 6 μm—∼10,000-fold higher than the dissociation constant (Antonyuk et al., 2009). Because clinical therapeutic use would preferentially involve dosing of antibody via intravenous or subcutaneous routes, we sought to determine whether peripheral administration of a humanized anti-PrP antibody could effectively target PrP-Aβ interactions and prevent the disruptive effects of toxic AD brain Aβ assemblies in vivo (Roettger et al., 2013).
Materials and Methods
Antibody development.
Antibody humanization was performed with the complementarity-determining region grafting method (Jones et al., 1986) and human frameworks were chosen on the basis of their conservation of key framework residues (Williams et al., 2010). Candidate humanized antibodies were then expressed as IgG1 molecules and retention of binding to antigen was confirmed by ELISA. The lead candidate was cloned in an IgG4 format selected for reduced effector function before progressing to cell line development using a proprietary glutamine synthetase expression system (Lonza Biologics). The construct encodes a Ser to Pro mutation at the interheavy chain disulfide bond to strengthen the hinge region and reduce the formation of half antibody (Angal et al., 1993). PRN100 was manufactured from the master cell bank at the 200 L scale and purified using a two-step chromatography process to generate highly purified antibody (99.3% by HPLC).
Immunoprecipitation and Western blotting of PrP from normal human brains.
Human brain samples were obtained and used under ethical approval from the National Research Ethics Service (03/133), homogenized to 10% w/v in Dulbecco's Mg2+/Ca2+-free PBS (Invitrogen) with a Dounce homogenizer, and used for immunoprecipitation and Western blotting, as described previously (Khalili-Shirazi et al., 2005), with ICSM18 (D-Gen) as the detection antibody.
Protein expression and purification.
Constructs of human PrP were expressed (Jackson et al., 1999) and purified (Zahn et al., 1997) as described previously. Protein quality was confirmed by SDS-PAGE, MALDI-TOF mass spectrometry, and circular dichroism spectroscopy.
Antibody affinity ELISA.
ELISA plates were coated with recombinant murine PrP91-231 (2.5 μg/ml) in 50 mm sodium carbonate buffer, pH 9.6, washed with PBS (0.05% Tween 20), and blocked with Superblock T20 (Thermo Scientific). PRN100 or ICSM18 diluted in PBS (0.1% bovine γ globulin) were incubated and washed with PBS (0.05% Tween 20) before detecting with species-specific HRP conjugated antibodies (Sigma) in PBST (0.05% Tween 20) coupled to a 3,3′,5,5′-tetramethylbenzidine (BD Biosciences) substrate. Absorbance at 450 nm was read using a Tecan Sunrise plate.
Preparation of human AD brain extract for electrophysiology.
Brain was prepared as described previously (Freir et al., 2011). Brain tissue from a 92-year-old woman with a history of dementia and confirmed AD pathology was obtained and used in accordance with local Institutional Review Board guidelines. Human temporal cortex was homogenized in 1:5 (w/v) ice-cold Tris-buffered saline (TBS), pH 7.4, containing protease inhibitors with 20 passes of a Dounce homogenizer (Fisher Scientific). The homogenized samples were centrifuged at 175,000 × g for 30 min at 4°C and the supernatant (TBS extract) was removed. TBS extract was desalted and Aβ-rich fractions were pooled and the Aβ content estimated using a sensitive immunoprecipitation/Western blot protocol. Known amounts of synthetic Aβ1-42 were electrophoresed on the same gel to allow quantification of Aβ in the TBS extract. After transfer, membranes were immunoblotted with a combination of the monoclonal antibodies 2G3 and 21F12 (1:1000 dilution). Goat anti-mouse antibody (1:2500 dilution) conjugated to infrared dye was used to visualize Aβ bands using a LI-COR Odyssey Near Infrared Imaging system.
Electrophysiology.
In vivo electrophysiology was performed as described previously (Klyubin et al., 2008; Freir et al., 2011). Animal experiments were licensed by the Department of Health and Children, Ireland. Adult male Wistar rats were anesthetized with urethane (1.5 g/kg, i.p.). Single pathway recordings of field EPSPs were made from the stratum radiatum in the CA1 area of the dorsal hippocampus in response to stimulation of the ipsilateral Schaffer collateral/commissural pathways. Test EPSPs were evoked at a frequency of 0.033 Hz and at a stimulation intensity adjusted to give an EPSP amplitude of 50% of maximum. The high-frequency stimulation (HFS) protocol for inducing LTP consisted of 10 trains of 20 stimuli, interstimulus interval of 5 ms, and an intertrain interval of 2 s. The intensity was increased to give an EPSP of 75% of maximum amplitude during the HFS. LTP is expressed as the mean ± SEM percentage of baseline field EPSP amplitude recorded over at least a 30 min baseline period. Similar results were obtained when EPSP slope rather than amplitude was measured.
For statistical analysis, EPSP amplitudes were grouped into 10 min epochs. Standard one-way ANOVA was used to compare the magnitude of LTP between multiple groups, followed by post hoc Tukey's tests. Unpaired Student's t tests were used for two-group comparisons. p < 0.05 was considered statistically significant.
To inject samples into the rat brain, a cannula was implanted in the lateral cerebral ventricle (coordinates: 1 mm lateral to the midline and 4 mm below the surface of the dura) just before electrode implantation. Injections (10–8 μl over 6–8 min) were made via a Hamilton syringe connected to the internal cannula. Control human myeloma IgG4 κ antibody was obtained from Sigma (catalog #I4639). An interval of 1.5 h or more was left between the implantation procedure and the intracardiac injection of 0.6 ml of PRN100 or buffer (25 mm NaAc, 75 mm NaCl, pH 5.5) under urethane anesthesia. Intracardiac injections were made via a 23 ga needle inserted directly into rat heart.
Histology and imaging.
Brains were fixed in 10% buffered formal saline, immersed in 98% formic acid for 1 h, and paraffin wax embedded after formalin postfixation. Sections of 4 μm nominal thickness were stained with H&E to visualize basic structures and immunostained for additional markers (microglia: Iba-1 antibody, Wako; myelinated fiber tracts: SMI-94R, Covance; cleaved Caspase-3: Asp175, Cell Signaling Technology) on Ventana Discovery XT automated biomarker platforms using EZ Prep Solution and antigen retrieval with Cell Conditioning Solution Plus (Ventana). Immunostainings were visualized using the DAB Map Detection Kit (Ventana). Sections were counterstained with hematoxylin and coverslipped on a Leica CV5030 coverslipper. Slides were digitized on a Leica SCN400 slide scanner, visualized on the SlidePath Digital Image Hub. Regions of interest were captured with a screen capture tool, followed by arranging captures in Adobe Photoshop.
Collection of plasma and CSF.
A cisternal puncture method was used for CSF sampling under anesthesia. After exposing the atlanto-occipital membrane, CSF was collected from the cisterna magna. Whole blood was obtained from the anesthetized animal by decapitation, collected into Eppendorf tubes containing ∼30 μl of 0.5 m EDTA, and inverted several times before being centrifuged for 10 min at 1000 × g. Plasma and CSF were stored frozen at −80°C.
PRN100 detection ELISA.
ELISA plates were coated with recombinant murine PrP91-231 (2.5 μg/ml) in 50 mm sodium carbonate buffer, pH 9.6, washed with PBS (0.05% Tween 20), and blocked with Superblock T20 (Thermo Scientific). Sera (1:2000) and CSF (1:5) diluted in PBS (2% milk powder) were incubated and washed with PBS (0.05% Tween 20) before detecting with anti-human Fc-specific HRP conjugated antibody (1:25,000) in PBST (0.05% Tween 20; Sigma) coupled to a 3,3′,5,5′-tetramethylbenzidine (BD Biosciences) substrate. Absorbance at 450 nm was read using a Tecan Sunrise plate reader with concentrations of PRN100 calculated relative to purified standard curves diluted in equivalent buffer and control fluid.
Evans blue assay.
Anesthetized rats were infused with a 2% solution of Evans blue (2 ml/kg, intracardiac). Five minutes after the treatment, the animals were killed by intracardiac perfusion with PBS and brains were harvested and preserved in formaldehyde.
Results
PRN100 retains key binding properties of ICMS18
As part of a program to develop a monoclonal anti-PrP antibody for passive immunotherapy of prion infection and disease, we have developed a fully humanized version of ICSM18, a high-affinity mouse monoclonal antibody effective in treating murine prion infection (White et al., 2003; Freir et al., 2011; Klohn et al., 2012). An IgG4 isotype was engineered as a clinical candidate to minimize unwanted Fc effector function and was designated PRN100. Both ICSM18 and PRN100 recognized, and successfully immunoprecipitated, all isoforms of PrP found in human brain (Fig. 1A). This suggests that PRN100 has the potential to target the same PrP species as ICSM18 in vivo and could block the Aβ-induced disruption of LTP caused by PrP-dependent assemblies. PRN100 bound avidly to high-density recombinant murine PrP with an apparent dissociation constant (50 ± 10 pm) comparable to that (40 ± 20 pm) for ICSM18 (Fig. 1B, n = 4). Therefore, the efficacious dose of PRN100 should be similar to ICSM18, but with reduced probability of Fc effector-related toxicity. These data indicate that PRN100 has retained key PrP-binding properties of ICSM18.
Figure 1.

The humanized monoclonal antibody PRN100 displays similar PrP-binding properties to its murine parent, ICSM18. A, Both ICSM18 and PRN100 efficiently immunoprecipitate all major PrP glycoforms from human brain homogenate. B, Representative binding isotherm of PRN100 (blue) and ICSM18 (orange) show a similar high-affinity interaction to surface-bound recombinant murine PrP91-231.
Peripheral infusion of PRN100 blocks AD brain-induced inhibition of LTP
We then tested whether PRN100 could prevent the synaptic plasticity-disrupting action of soluble AD brain extract in live rats, as we have described for ICSM18 (Freir et al., 2011). After intracerebroventricular injection of buffer alone, a robust LTP could be evoked using an HFS protocol. However, injection of a water-soluble AD brain extract rich in Aβ species, including SDS-stable dimers (Fig. 2A), fully inhibited LTP within 3 h (Fig. 2B). Importantly, brain extract immunodepleted of Aβ did not inhibit LTP (n = 5, data not shown). When 30 μg of PRN100 was injected 30 min before the AD brain extract, robust LTP was also observed, indicating that PRN100 can block the disruptive effect of Aβ found in AD brain. An isotype control human IgG4 (IgG4 kappa purified from human myeloma plasma) failed to prevent the toxic effect. Neuropathological analysis showed no signs of PRN100-induced neuronal apoptosis, microglial activation, or demyelination (Fig. 3).
Figure 2.
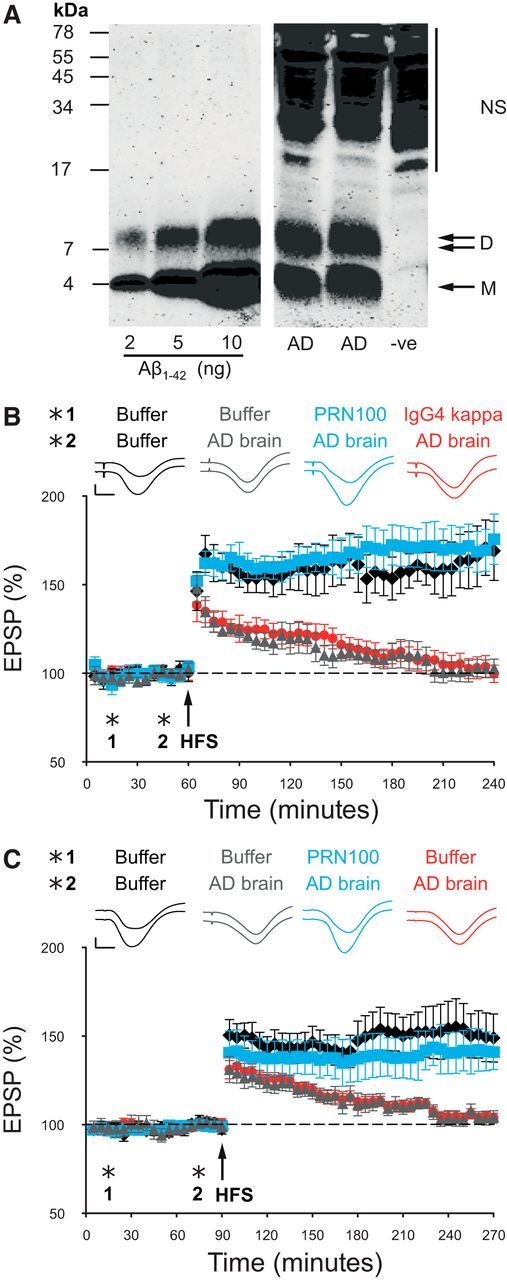
PRN100 blocks soluble AD brain (A) extract-induced inhibition of hippocampal LTP when infused centrally (B) or peripherally (C). A, Soluble AD brain extract contained 3.8 ng/ml Aβ monomers and 2.4 ng/ml Aβ dimers as measured by sensitive immunoprecipitation/Western blotting technique. Bands migrating at a molecular weight consistent for Aβ monomer (M) and SDS-stable dimer (D) are seen in the AD TBS extract. Synthetic Aβ1-42 standards loaded for protein quantification are shown on the left. Nonspecific immunoreactive bands (NS) migrating >17 kDa can also be seen in the negative control lane (−ve). B, Synaptic field potentials were recorded in vivo from the CA1 area of anesthetized male Wistar rats. In buffer-injected rats (1: first injection 10 μl, i.c.v.; 2: second injection 8 μl 30 min later) HFS 15 min after the second injection triggered persistent and stable LTP (black diamonds, 169 ± 17% at 3 h after tetanus, n = 5). In contrast, significant inhibition of LTP was observed after injection of 8 μl of Aβ-containing brain extract in animals preinjected with buffer (gray triangles, 103 ± 5%, n = 5, p < 0.05) or 30 μg of an IgG4 isotype control antibody (IgG4kappa, red circles, 100 ± 5%, n = 5, p < 0.05). However, injection of PRN100 (30 μg) 30 min before injection of soluble AD brain extract prevented the inhibition of LTP (blue squares, 176 ± 14%, n = 5). C, In buffer-injected rats (1: first injection 600 μl, intracardiac; 2: second injection 8 μl, i.c.v. 30 min later) HFS 15 min after the second injection triggered persistent and stable LTP (black diamonds, 149 ± 14% at 3 h after tetanus, n = 6). Intracardiac injection of buffer followed by intracerebroventricular infusion of AD brain fully inhibited LTP (red circles, 104 ± 3%, n = 6, p < 0.05). Intracardiac injection of 6 mg of PRN100 reversed this effect (blue squares, 141 ± 10%, n = 5), showing that peripheral administration of PRN100 blocks AD brain extract-derived Aβ synaptotoxicity. The PRN100 measurements were conducted blind (gray triangles vs blue squares). Insets show representative EPSP traces. Top trace corresponds to 5 min before HFS and the bottom to 3 h after HFS. Horizontal bar, 5 ms; vertical bar, 1 mV.
Figure 3.

ICV injection of PRN100 did not cause overt neuronal damage up to 4 h after administration. Shown is the immunohistochemical analysis of hippocampi from PRN100-injected (A–C) and isotype control-injected (D–F) rats. H&E or immunohistochemical (Iba-1 or SMI-94) staining (A, D) shows no evident neuronal damage, demyelination (C, F), or microglial activation in PRN-100 treated rats (B) compared with isotype control-treated animals (E). Scale bar, 100 μm.
For peripherally administered PRN100 to be therapeutically active against either established neuroinvasive prion infection or AD, it will need to cross the blood–brain barrier (BBB) and target PrP in relevant regions of the brain such as the hippocampus (Roettger et al., 2013). Pharmacokinetic studies are ongoing to determine the dosing regime required to obtain therapeutic levels in the CNS, which will require sufficient dosing to overcome target-mediated drug disposition, because PrP is also widely expressed in the periphery (Linden et al., 2008). However, as a proof of principle, we injected a single 6 mg dose of PRN100 via the intracardiac route 1 h before intracerebroventricular injection of AD brain extract and monitored the effect on LTP induction. Remarkably, such intravascular infusion of PRN100 also prevented inhibition of LTP, suggesting that PRN100 was able to cross the BBB and target PrP (Fig. 2C). As with direct intracerebroventricular injection, there was no histologically detectable neurotoxicity (Fig. 4). We collected blood and CSF from the rats used in the study and quantified PRN100 concentrations. ELISA detection, performed in a blinded fashion, correctly identified all rats with PRN100 in their blood and CSF. Concentrations of 1.4 ± 0.7 μm PRN100 (210 ± 110 μg/ml) were detected in serum and 1.7 ± 3.4 nm (0.26 ± 0.51 μg/ml) concentrations were found in CSF (equivalent to ∼0.1% of that detected in blood), confirming that sufficient PRN100 can enter the CNS even after a single dose (n = 5) to achieve a pharmacodynamic effect. A non-BBB penetrant dye, Evans blue (Kozler and Pokorný, 2003), was injected via the intracardiac route 2 h after electrode implantation to test for BBB integrity and was not detected at the electrode site or elsewhere in the brain (data not shown).
Figure 4.

Intracardiac injection of PRN100 did not cause overt neuronal damage up to 14 h after administration. Shown is the immunohistochemical analysis of whole brains from PRN100-injected (A–I) and buffer control-injected (J–R) rats. H&E staining (A, B, J, K) shows no sign of neuronal damage or demyelination (E, F, N, O). There is no increase in microglia activation in PRN-100 rats (C, D) compared with buffer control-injected animals (L, M) or cleaved Caspase-3 (green arrow; G–I vs P–R). Scale bars, 100 μm for red zoomed panels and 10 μm for green zoomed panels.
Discussion
The potential for anti-PrP antibodies to prevent Aβ synaptotoxicity in AD has been established previously. However, no fully humanized anti-PrP antibody potentially applicable to AD patients has been reported. We show that PRN100 retains key properties of the parent murine antibody, ICSM18, and can block Aβ-induced inhibition of LTP when injected directly into the brain without inducing overt acute neurotoxicity. It would be interesting to determine whether PRN100 can also reverse behavioral deficits seen in AD rodent models, although maintaining long-term dosing may be difficult if antidrug antibodies develop and the PrP dependence of toxicity varies between models and at different time points. Furthermore, it was unknown whether such antibodies could cross the BBB in sufficient quantities to target PrP in disease-relevant areas of the brain such as the hippocampus. We have now shown that, after a single injection into the rat blood stream, PRN100 can indeed reach therapeutically active concentrations even when subnanomolar concentrations are detected in the CSF. We could not reliably visualize PRN100 in fixed rat brain parenchyma using immunohistochemistry due to diffuse background staining. The concentrations observed in the CSF are well below any reported toxic concentrations (Solforosi et al., 2004; Sonati et al., 2013).
Recently, Sonati et al. (2013) have reported acute toxicity associated with some, but not other, anti-PrP antibodies from their POM series (Polymenidou et al., 2008). Toxicity was observed principally in organotypic slice cultures from, and limited in vivo studies in, transgenic mice with high levels of PrP overexpression (Sonati et al., 2013). There was an inconsistent association with epitope. Toxicity was seen at very high local concentrations after direct injection into brain parenchyma and is of questionable relevance to any likely human therapeutic approach.
Although all therapeutics will be toxic at sufficiently high acute or chronic dosing, formal safety studies have been performed as part of the evaluation of PRN100 before an application to UK regulators for a clinical trial authorization (in the primary indication for which PRN100 was developed, Creutzfeldt-Jakob disease [CJD]). Subject to an acceptable risk: benefit evaluation, it is envisaged that first-in-human studies would be in patients with sporadic CJD, a uniformly fatal condition with a mean clinical duration of 4 months. A single ascending dose safety study has been performed in cynomolgus macaques (Covance) to underpin this study and showed no clinical or pathological evidence of CNS toxicity at intravenous doses up to and including 200 mg/kg (achieving peak serum concentrations in excess of 5 mg/ml), well in excess of those proposed for initial human studies.
Several fully penetrant inherited forms of prion disease are caused by single amino acid substitutions in the Aβ-binding epitope and many are found throughout the C terminus of the structured domain, but none are known in the helix-1 region (Collinge, 2001). Therefore, using antibodies exclusively targeting helix-1 of PrP may be a superior therapeutic strategy to reduce PrP-dependent Aβ toxicity rather than using antibodies with nonlinear epitopes or those that directly target the Aβ binding site itself.
Should proposed clinical studies of PRN100 in prion disease (a rapidly fatal, untreatable condition) demonstrate adequate safety characteristics and pharmacokinetic parameters in humans, it may be appropriate to consider experimental medicine studies in early AD to determine whether PRN100 administration has acute effects on memory and learning, which would suggest a potential role for blocking PrP-Aβ interactions in AD therapeutics.
Footnotes
This work was funded by the UK Medical Research Council (J.C.), Science Foundation Ireland and the European Union Framework 7 Project MEMOLOAD (M.J.R.), and the Health Research Board of Ireland (I.K.). We thank P. Seubert and D. Schenk for 2G3 and 21F12; Ray Young for preparation of figures; Angela Richard-Londt and Sarah Lyall for assistance with histology; Mark Batchelor for preparation of recombinant protein; Patrick Hextall, Simon Mead, and Caroline Potter for helpful discussion; and Werner Frings for leading the cynomolgus macaques PK study.
J.C. is a shareholder and director of D-Gen Limited, an academic spin-out company in the field of prion diagnosis, decontamination, and therapeutics. D.M.W. is a member of the scientific advisory board of Cognition Therapeutics, Inc., and is a consultant to Eisai Co., Ltd. The remaining authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Angal S, King DJ, Bodmer MW, Turner A, Lawson AD, Roberts G, Pedley B, Adair JR. A single amino-acid substitution abolishes the heterogeneity of chimeric mouse/human (Igg4) antibody. Mol Immunol. 1993;30:105–108. doi: 10.1016/0161-5890(93)90432-B. [DOI] [PubMed] [Google Scholar]
- Antonyuk SV, Trevitt CR, Strange RW, Jackson GS, Sangar D, Batchelor M, Cooper S, Fraser C, Jones S, Georgiou T, Khalili-Shirazi A, Clarke AR, Hasnain SS, Collinge J. Crystal structure of human prion protein bound to a therapeutic antibody. Proc Natl Acad Sci U S A. 2009;106:2554–2558. doi: 10.1073/pnas.0809170106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer's disease brain-derived amyloid-β-mediated inhibition of LTP in vivo Is prevented by immunotargeting cellular prion protein. J Neurosci. 2011;31:7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, Williams A. Amyloid-β-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem. 2011;286:37955–37963. doi: 10.1074/jbc.M111.248724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béland M, Roucou X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J Neurochem. 2012;120:853–868. doi: 10.1111/j.1471-4159.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 2010;11:130. doi: 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between prion protein and toxic amyloid-β assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GS, Hill AF, Joseph C, Hosszu L, Power A, Waltho JP, Clarke AR, Collinge J. Multiple folding pathways for heterologously expressed human prion protein. Biochim Biophys Acta. 1999;1431:1–13. doi: 10.1016/S0167-4838(99)00038-2. [DOI] [PubMed] [Google Scholar]
- Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human-antibody with those from a mouse. Nature. 1986;321:522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- Khalili-Shirazi A, Summers L, Linehan J, Mallinson G, Anstee D, Hawke S, Jackson GS, Collinge J. PrP glycoforms are associated in a strain-specific ratio in native PrPSc. J Gen Virol. 2005;86:2635–2644. doi: 10.1099/vir.0.80375-0. [DOI] [PubMed] [Google Scholar]
- Klöhn PC, Farmer M, Linehan JM, O'Malley C, Fernandez de Marco M, Taylor W, Farrow M, Khalili-Shirazi A, Brandner S, Collinge J. PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science. 2012;335:52. doi: 10.1126/science.1215579. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid-β protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozler P, Pokorný J. Altered blood-brain barrier permeability and its effect on the distribution of Evans blue and sodium fluorescein in the rat brain applied by intracarotid injection. Physiol Res. 2003;52:607–614. [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- Nicoll AJ, Collinge J. Preventing prion pathogenicity by targeting the cellular prion protein. Infect Disord Drug Targets. 2009;9:48–57. doi: 10.2174/1871526510909010048. [DOI] [PubMed] [Google Scholar]
- Ohsawa N, Song CH, Suzuki A, Furuoka H, Hasebe R, Horiuchi M. Therapeutic effect of peripheral administration of an anti-prion protein antibody on mice infected with prions. Microbiol Immunol. 2013;57:288–297. doi: 10.1111/1348-0421.12037. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, Hafner-Bratkovic I, Jerala R, Hornemann S, Wuthrich K, Bellon A, Vey M, Garen G, James MN, Kav N, Aguzzi A. The POM monoclonals: A comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One. 2008;3:e3872. doi: 10.1371/journal.pone.0003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roettger Y, Du Y, Bacher M, Zerr I, Dodel R, Bach JP. Immunotherapy in prion disease. Nat Rev Neurol. 2013;9:98–105. doi: 10.1038/nrneurol.2012.258. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sánchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303:1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, Swayampakula M, Rahman MH, Das D, Kav N, Riek R, Liberski PP, James MN, Aguzzi A. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501:102–106. doi: 10.1038/nature12402. [DOI] [PubMed] [Google Scholar]
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- Williams DG, Matthews DJ, Jones T. Humanising antibodies by CDR grafting. In: Roland Kontermann SD, editor. Antibody engineering. Berlin: Springer-Verlag; 2010. pp. 319–339. [Google Scholar]
- Zahn R, von Schroetter C, Wüthrich K. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 1997;417:400–404. doi: 10.1016/S0014-5793(97)01330-6. [DOI] [PubMed] [Google Scholar]