

Abstract
Behavioral and physiological coupling between energy balance and fluid homeostasis is critical for survival. The orexigenic hormone ghrelin has been shown to stimulate the secretion of the osmoregulatory hormone vasopressin (VP), linking nutritional status to the control of blood osmolality, although the mechanism of this systemic crosstalk is unknown. Here, we show using electrophysiological recordings and calcium imaging in rat brain slices that ghrelin stimulates VP neurons in the hypothalamic paraventricular nucleus (PVN) in a nutritional state-dependent manner by activating an excitatory GABAergic synaptic input via a retrograde neuronal–glial circuit. In slices from fasted rats, ghrelin activation of a postsynaptic ghrelin receptor, the growth hormone secretagogue receptor type 1a (GHS-R1a), in VP neurons caused the dendritic release of VP, which stimulated astrocytes to release the gliotransmitter adenosine triphosphate (ATP). ATP activation of P2X receptors excited presynaptic GABA neurons to increase GABA release, which was excitatory to the VP neurons. This trans-neuronal–glial retrograde circuit activated by ghrelin provides an alternative means of stimulation of VP release and represents a novel mechanism of neuronal control by local neuronal–glial circuits. It also provides a potential cellular mechanism for the physiological integration of energy and fluid homeostasis.
Keywords: astrocyte, ATP, ghrelin, hypothalamus, paraventricular nucleus, vasopressin
Introduction
Ghrelin is often referred to as the hunger hormone, as it is the only known nutritional signal from the gut to the brain that is orexigenic. Ghrelin is released during negative energy balance and strongly activates feeding behavior by acting on the growth hormone secretagogue receptor (GHS-R) in the hypothalamus (Kojima et al., 1999; Cummings et al., 2001; Sugino et al., 2002b). Blood levels of ghrelin increase in anticipation of an expected meal (Cummings et al., 2001; Sugino et al., 2002a, b) and during periods of fasting (Tschöp et al., 2000; Asakawa et al., 2001), and decrease after food ingestion (Tschöp et al., 2000; Cummings et al., 2001; Sugino et al., 2002a, b). Several studies revealed that ghrelin stimulates vasopressin (VP) release by activating VP neurons in the hypothalamus (Nakazato et al., 2001; Ishizaki et al., 2002; Lawrence et al., 2002), suggesting that ghrelin may modulate fluid homeostatic function by controlling VP release. Indeed, ghrelin has been shown to affect drinking behavior and blood osmolality (Kozaka et al., 2003; Tachibana et al., 2006; Hashimoto et al., 2007; Mietlicki et al., 2009) and may serve as a central integrative signal linking energy homeostasis with fluid homeostasis.
Energy balance and fluid homeostasis are closely related, as evidenced by the tight coupling between eating and drinking behaviors. (Fitzsimons and Le Magnen, 1969; Bellisle and Le Magnen, 1981). Chronic dehydration leads to a state of anorexia, or decreased food intake (Watts et al., 1999; Watts, 1999), presumably because solid food ingestion under conditions of high blood osmolality would be harmful or even fatal to the organism (Watts and Boyle, 2010). When an animal is food deprived and engaged in food-seeking behavior, it is critical that the animal's blood osmolality be maintained at a level at which the animal can tolerate solid food intake in preparation for the ingestion of food when it is found. Water ingestion is decreased during food deprivation (Amlal et al., 2001; Wilke et al., 2005), possibly to maintain circulating concentrations of critical minerals. Thus, ghrelin-induced VP release and increased water retention under these conditions are an important mechanism in maintaining blood osmolality at a physiological set point that can tolerate food ingestion.
How ghrelin activates VP neurons and VP secretion, however, has not been elucidated. Ghrelin-expressing axon terminals and GHS-R1a expression have been demonstrated in the paraventricular nucleus (PVN), indicating that the PVN is a site of ghrelin action (Guan et al., 1997; Cowley et al., 2003; Zigman et al., 2006; Harrold et al., 2008). Understanding the modulation of VP neurons by ghrelin may reveal an underlying central mechanism by which ghrelin can integrate different homeostatic functions. In the present study, we used patch-clamp electrophysiological and calcium imaging methods in acute slices to study the ghrelin regulation of synaptic inputs to VP neurons. We demonstrate a ghrelin stimulation of excitatory GABA inputs to VP neurons that is dependent on the nutritional state of the animal and that is mediated via a novel retrograde circuit that involves trans-neuronal–glial signaling.
Materials and Methods
Animals.
We used 5- to 9-week-old male transgenic Wistar rats expressing a VP-eGFP fusion protein or wild-type Wistar rats according to a protocol approved by the Tulane University Institutional Animal Care and Use Committee and in compliance with United States Public Health Service guidelines We established the transgenic rat colony from founders obtained from Dr. Yoichi Ueta of the University of Occupational and Environmental Health in Kitakyushu, Japan (Ueta et al., 2005). Wild-type Wistar rats were purchased from Harlan Laboratory and were allowed to acclimate to their environment in the vivarium for at least 1 week before experiments. The fasted groups were prepared by removing food from the cage for 24 h. All rats had ad libitum access to water.
Slice preparation.
Rats were deeply anesthetized by isoflurane inhalation (VetOne, Meridian, ID) and decapitated in a rodent guillotine. The brain was carefully removed and transferred within 2 min of decapitation to cold aCSF (1°C-2°C). The aCSF was composed of the following (in mm): 140 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 2.4 CaCl2, 11 glucose, and 5 HEPES, pH adjusted to 7.2–7.4 with NaOH and oxygenated with 100% O2. The hypothalamus was blocked, and the block was glued to the chuck of a vibrating microtome with the rostral side face up. We collected two or three 300-μm-thick coronal hypothalamic slices containing the PVN and bisected them along the midline. The hemi-slices were allowed to recover in oxygenated aCSF at room temperature for at least 1 h before being used for electrophysiological recordings or calcium imaging.
Electrophysiological recording methods.
Slices were transferred to the recording chamber, which was continuously perfused with oxygenated aCSF maintained at 30°C. Whole-cell patch-clamp electrodes were made from borosilicate glass (1.65 μm outer diameter, 1.2 μm inner diameter; KG-33; King Precision Glass) with a horizontal micropipette puller (P-97, Sutter Instruments) and had a tip resistance of 3–6 mΩ. The patch electrode solution contained the following (in mm): 120 K-gluconate, 10 KCl, 1 NaCl, 1 MgCl2, 1 CaCl2, 10 EGTA, 2 Mg-adenosine triphosphate (ATP), 0.3 Na-GTP, and 10 HEPES, pH adjusted to 7.3 with KOH. For recordings of GABAergic PSCs, a high-[Cl−] electrode solution was used, containing the following (in mm): 120 CsCl, 2 MgCl2, 1 CaCl2, 11 EGTA, 4 ATP-Mg salt, and 30 HEPES, pH adjusted to 7.2 with CsOH and osmolarity adjusted to 300 mOsmol with d-sorbitol. For whole-cell voltage-clamp recordings, cells were held at a holding potential of −60 mV. Magnocellular neuroendocrine cells in the PVN were identified based on their morphology, their location within the nucleus, and their transient outward rectification generated with intracellular current injection (Tasker and Dudek, 1991; Luther and Tasker, 2000). Vasopressin (VP)-expressing neurons were distinguished from oxytocin neurons by their GFP fluorescence. Before the start of experiments, all recordings were allowed to stabilize for at least 3 min after the whole-cell configuration was attained. For miniature PSC recordings, TTX (0.5–1 μm) was added to the aCSF to block voltage-gated sodium channels. To record GABAergic PSCs, we included the ionotropic glutamate receptor agonists DNQX (15 μm) and dl-2-amino-5-phosphonopentanoic acid (dl-AP5, 50 μm) in the aCSF. To isolate glutamatergic PSCs, we included the GABAA receptor antagonist bicuculline methiodide (10 μm) or picrotoxin (50 μm) in the aCSF. For loose-seal cell-attached patch-clamp recordings, pipettes with a bigger tip size (pipette resistance = 1–3 mΩ) were used to avoid the spontaneous formation of a high-resistance seal; spiking activity was recorded at the resting potential with 0 pA of current injection. Only loose-seal recordings with a stable baseline and seal resistance of ≤40 mΩ were considered acceptable. Because the majority of cells recorded in the loose-seal cell-attached patch configuration were quiescent in the control condition, we increased the extracellular [K+] to 10 mm in these recordings to depolarize cells. The high-K+ aCSF was composed of the following (in mm): 133 NaCl, 10 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 2.4 CaCl2, 11 glucose, and 5 HEPES, pH adjusted to 7.2–7.4 with NaOH. The high-K+ solution did not cause a significant change in the GABA current reversal potential (EGABA), as reported previously (Haam et al., 2012). To measure EGABA in cells, we used the gramicidin-perforated patch recording method. The gramicidin patch solution was prepared by first dissolving gramicidin in DMSO at a concentration of 0.05 mg/μl as a stock solution, and then the stock solution was diluted in the K-gluconate patch solution to a final concentration of 100 μg/ml. The EGABA was obtained by plotting the current-voltage relation of evoked GABAergic synaptic currents at different holding potentials, as described previously (Haam et al., 2012).
Calcium imaging.
After a stabilization period, the slices were bulk loaded with the calcium indicator Rhod-2 AM (Life Technologies). The Rhod-2 AM stock solution was reconstituted in 40 μl of DMSO and stored at −20°C until it was applied to brain slices to record calcium signals in glia. The stock solution of Rhod-2 AM was used within a week to avoid loss of cell loading capacity. The dye loading dish was prepared using a 30-mm-diameter culture plate insert (Millipore) that was placed into a 35 mm dish. A total of 1 ml of aCSF was then added both inside and outside of the plate insert before one or two hemi-slices were moved onto the plate insert. A total of 2–5 μl of Rhod-2 AM stock solution were then dropped directly onto the PVN area of each hemi-slice. The incubation was performed in the dark at room temperature for 1 h, during which 100% O2 was continuously supplied to the dish. After the incubation, the brain hemi-slices were washed for 30 min to 2 h in dye-free medium to remove any dye nonspecifically associated with cell surfaces and to allow deesterification of the intracellular acetoxymethyl (AM). Fluorescence images were obtained using an intensified CCD camera (QuantumEM CCD) at 1 frame/5 s and were captured on a PC using IP Laboratory 4.0 software (Scanalytics). We used 555 nm for excitation of Rhod-2 AM dye and quantified the fluorescence intensity of each cell using MBF ImageJ software (National Institutes of Health), which allowed for analysis of regions of interest located in multiple individual cells. To calculate a drug effect, we averaged the fluorescence intensity of the last 2 min (24 frames) of the drug application period and compared it with that of the last 2 min of the preceding control period.
Drug application.
We stored the following drugs as stock solutions in frozen aliquots (−20°C) and dissolved the stock solutions in aCSF to the final concentration immediately before experiments: n-octanoylated ghrelin peptide (R&D Systems) (10 and 100 nm), TTX (R&D Systems) (0.5–1 μm), the GHS-R1a antagonist d-[Lys-3] GHRP-6 (R&D Systems) (5 μm), DNQX (R&D Systems) (15 μm), DL-AP5 (R&D Systems) (50 μm), bicuculline methiodide (R&D Systems) (10–50 μm), picrotoxin (R&D Systems) (50 μm); Manning compound (R&D Systems) (1 μm), l-NAME (R&D Systems) (100 μm), TNP-ATP (R&D Systems) (10 μm), SR49059 (R&D Systems) (10 μm), and fluorocitric acid (Sigma-Aldrich) (100 μm). To prepare the fluorocitric acid solution, we precipitated barium by adding 2–3 drops of 0.1 m Na2SO4 to a fluorocitrate barium salt solution (8.26 mg of fluorocitrate barium salt in 1 ml of 0.1 m HCl). We then added 2 ml of 0.1 m Na2HPO4 before centrifuging the solution at 1000 g for 5 min. Only the suspension was separated and added to 100 ml aCSF. We included the G-protein inhibitor GDP-β-S (Sigma-Aldrich) (1 mm) in a patch solution that did not contain GTP. All the drugs were reconstituted with autoclaved deionized distilled water and aliquoted for single use. ATP-γ-S (R&D Systems) (100 μm) and arginine VP (R&D Systems) (20 μm) were applied via a puff pipette (8–20 psi, 30 s) connected to a Picospritzer II microinjector (Parker Hannifin). All peptide-based aliquots were stored at −20°C and were used within 3 months of preparation.
Immunohistochemistry for GHS-R1a.
Eight- to 12-week-old wild-type Wistar rats (n = 3) were deeply anesthetized with ketamine (50 mg/kg)/xylazine (5 mg/kg) and intracardially perfused with heparinized PBS followed by 4% paraformaldehyde in 0.1 m PBS. After sufficient perfusion, brains were dissected out of the cranium and postfixed in 4% PFA in PBS overnight. The brains were then incubated in 20% sucrose in PBS overnight, and 30-μm-thick brain sections were obtained using a cryostat (CM3050, Leica). The sections were incubated in 5% normal goat serum containing 0.1% Triton X for 1 h on a shaker and then treated with the primary antibody to GHS-R1a (H-001–62, 1:2000, Phoenix Pharmaceuticals) (Abizaid et al., 2006; Jiang et al., 2008) overnight at 4°C. The sections were washed with 0.1 m PBS five times and then treated with the goat anti-rabbit secondary antibody conjugated to AlexaFluor-546 (A-11010, 1:400, Invitrogen) for 1 h at room temperature. After rinsing five times with PBS, the sections were mounted and coverslipped using Vectashield mounting medium (Vector Laboratory), and were imaged on a laser confocal microscope (Zeiss LSM510, Carl Zeiss). The primary and secondary antibodies were omitted as controls, and no staining was observed. The GHS-R1a antibody has also been tested previously for specificity in GHSR knock-out mice (Abizaid et al., 2006). Finally, a GHS-R1a antibody blocking peptide was used in preabsorbtion controls (human growth hormone secretagogue receptor type 1A [Cys0] (330–366), Phoenix Pharmaceuticals).
Statistical analysis.
The analysis of synaptic currents was performed using Mini Analysis 6.0.9 software (Synaptosoft) and Clampfit 9.2 (Molecular Devices). Data were analyzed using the Student's paired t test for two-group comparisons and the one-way or two-way repeated-measures ANOVA followed by the Tukey's test for multiple-group comparisons. For the datasets that did not follow a normal distribution, determined by the Shapiro–Wilk test, we used the Mann–Whitney U test for between-cell comparisons and the Wilcoxon signed-rank test for within-cell comparisons. All the electrophysiological data were expressed as mean ± SEM. p < 0.05 was considered significant.
Results
To examine the effect of ghrelin on glutamatergic and GABAergic synaptic inputs to VP neurons of the PVN, we used various patch-clamp recording techniques in acute slices of rat hypothalamus. To differentiate VP neurons from PVN oxytocinergic magnocellular neurons and parvocellular neurons, we targeted enhanced GFP-expressing neurons in the lateral magnocellular division of the PVN of slices from transgenic rats that express a VP-GFP fusion protein under the control of the VP promoter. The expression of GFP protein in these rats is specific to VP neurons (Ueta et al., 2005; Haam et al., 2012). Lateral magnocellular neurons lacking GFP fluorescence were identified as putative oxytocin neurons.
Ghrelin increases synaptic inputs to VP neurons
Previous studies showed that the actions of ghrelin are potentiated during fasting due to increased levels of expression of the ghrelin receptor, GHS-R, in the hypothalamus (Kim et al., 2003). We recorded the effect of ghrelin on spontaneous postsynaptic currents (sPSCs) in GFP-positive VP neurons and GFP-negative, putative oxytocin neurons in the PVN of ad libitum-fed and 24 h-fasted VP-GFP rats in the whole-cell patch-clamp configuration. VP neurons showed a feeding-status-dependent increase in the frequency of inward sPSCs in response to a 10 min bath application of ghrelin (100 nm) (Fig. 1A,B). VP neurons from fasted rats showed a potentiated response to ghrelin (133.6 ± 7.8% of baseline, n = 18, Wilcoxon signed-rank test, W = −171, z = 3.724, degrees of freedom = 18, p < 0.01) compared with VP neurons from ad libitum fed rats (112.4 ± 6.1% of baseline, n = 12, Student's paired t test, t(11) = −2.293, p < 0.05) (Fig. 1C,D). The effect of a lower concentration of ghrelin (10 nm) on the sPSC frequency in VP neurons from 24 h-fasted rats was significantly smaller (112.2 ± 5.6% of baseline, n = 9, Student's paired t test, t(8) = −2.431, p < 0.05) and was comparable with the effect of the higher ghrelin concentration in VP neurons from ad libitum-fed rats (Fig. 1D). Whereas the effect of the 10 min application of ghrelin did not recover back to baseline after 20 min of washout of the drug, the effect of a shorter application was reversible after a 15 min washout. Thus, a 5 min bath application of ghrelin (100 nm) to slices from fasted rats caused a comparable increase in sPSC frequency (134.4 ± 15.0% of baseline, n = 11, one-way repeated-measures ANOVA followed by Tukey's test, F(2,17) = 3.819, p < 0.05) in VP neurons, which recovered nearly to baseline within 15 min of washout (113.5 ± 11.5%, n = 8, one-way repeated-measures ANOVA followed by Tukey's test, F(2,17) = 3.819, p = 0.41) (data not shown). There was no significant effect of ghrelin on sPSC amplitude or decay time (amplitude, 99.7 ± 4.6% of baseline, n = 18, Wilcoxon signed-rank test, W = 17, z = 0.37, degrees of freedom = 18, p = 0.73; decay time, 101.9 ± 3.8% of baseline, n = 18, Student's paired t test, t(17) = −0.407, p = 0.69), suggesting that the site of ghrelin action was presynaptic to the VP neurons. Ghrelin had no significant effect on sPSCs in GFP-negative, putative oxytocin neurons (sPSC frequency: 99.0 ± 13.3% of baseline, n = 4 from 24 h-fasted rats, Student's paired t test, t(3) = −0.454, p = 0.68) (data not shown).
Figure 1.
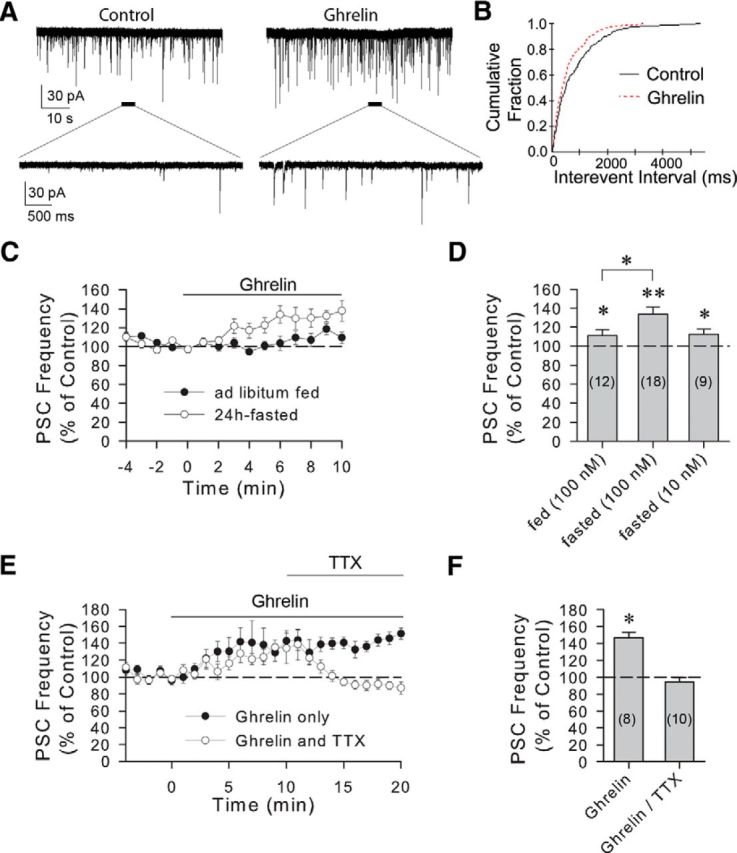
Ghrelin-induced increase in sPSC frequency in VP neurons. A, Whole-cell recording of a VP-GFP-expressing cell in the PVN of a 24 h-fasted rat showing sPSCs before (Control) and during a 10 min bath application of ghrelin (Ghrelin, 100 nm). B, Cumulative frequency plots of sPSC intervals from the same cell showed a shift toward shorter interevent intervals in ghrelin, indicating an increase in sPSC frequency. C, Time course of mean normalized frequencies of sPSCs showing the effect of ghrelin (100 nm) on sPSC frequency in PVN VP neurons from fed and fasted rats. An increase in sPSC frequency was seen in response to ghrelin, which was potentiated in fasted rats (open circles, n = 18) compared with fed rats (filled circles, n = 12). D, Summary of the dose-dependent effects of ghrelin on mean PSC frequency, as a percentage of baseline, in VP neurons from ad libitum fed and 24 h-fasted rats. E, Time course of normalized mean sPSC frequencies in PVN VP cells from 24 h-fasted rats showing the spike dependence of the ghrelin-induced increase in PSC frequency. The ghrelin-induced increase in sPSC frequency did not return to the baseline during 20 min of ghrelin application (Ghrelin), indicating that the ghrelin effect does not desensitize within 20 min (n = 8). The ghrelin-induced increase in sPSC frequency was reversed by the introduction of TTX (0.5–1 μm) into the perfusion bath (TTX, n = 10). F, Summary of mean normalized PSC frequencies at 17–20 min of ghrelin application in the absence (Ghrelin) and in the presence of TTX (Ghrelin/TTX). Blocking spike generation with TTX completely reversed the ghrelin effect on synaptic inputs to VP neurons. Numerals in parentheses in this and the following figures represent the numbers of cells included in the mean ± SEM. *p < 0.05. **p < 0.01.
The ghrelin-induced increase in synaptic inputs is spike-dependent
The fact that ghrelin caused a significant change in PSC frequency without causing a change in PSC amplitude or decay time suggested that the site of ghrelin action is presynaptic. We therefore tested whether the ghrelin effect is dependent on spike generation in presynaptic cells by blocking action potential generation with the voltage-gated sodium channel blocker TTX. After 10 min of ghrelin application to VP neurons from 24 h-fasted rats, TTX (0.5–1 μm) completely reversed the ghrelin-induced increase in sPSC frequency (one-way repeated-measures ANOVA, F(2,17) = 8.318, p < 0.01 followed by multiple comparisons: ghrelin: 141.3 ± 10.5% of baseline, n = 10, p < 0.05; ghrelin in TTX: 94.9 ± 4.9% of baseline, n = 9, p = 0.11) (Fig. 1E,F). The TTX effect on sPSC frequency was specific to the ghrelin-induced increase because TTX had no significant effect on sPSC frequency in VP neurons in the absence of ghrelin (98.8 ± 6.2% of baseline, n = 6, Wilcoxon signed-rank test, W = −7, z = −0.734, degrees of freedom = 6, p = 0.34). Consistent with this, TTX application before ghrelin application completely blocked the ghrelin-induced increase in sPSC frequency in VP neurons from 24 h-fasted rats. Indeed, in TTX, ghrelin caused a small, but significant, decrease in the frequency of miniature PSCs (84.7 ± 2.6% of baseline, n = 18, Wilcoxon signed-rank test, W = −165, z = −3.593, degrees of freedom = 18, p < 0.01), without affecting miniature PSC amplitude (101.2 ± 2.5% of baseline, n = 18, Student's paired t test, t(17) = −0.424, p = 0.68) or decay time (105.5 ± 3.7% of baseline, n = 18, Wilcoxon signed-rank test, W = 81, z = 1.764, degrees of freedom = 18, p = 0.08) (data not shown).
The ghrelin effect is specific to GABA inputs
We next determined which type of synaptic input was facilitated by ghrelin by pharmacologically isolating glutamatergic and GABAergic PSCs. In the presence of the GABAA receptor antagonists bicuculline methiodide (10 μm) or picrotoxin (50 μm), ghrelin (100 nm) had no significant effect on the frequency of glutamatergic sPSCs in VP cells from either ad libitum-fed rats (86.1 ± 8.3% of baseline, n = 9, Student's paired t test, t(8) = 1.872, p = 0.10) or 24 h-fasted rats (100.3 ± 8.5%, n = 7, Student's paired t test, t(6) = 0.0926, p = 0.93) (Fig. 2A,B). In the presence of the glutamate receptor antagonists DNQX (15 μm) and DL-AP5 (50 μm), ghrelin (100 nm) caused an increase in the frequency of GABAergic sPSCs recorded in VP cells (152.8 ± 16.4% of baseline, n = 16, Wilcoxon signed-rank test, W = 106, Z = 2.741, degrees of freedom = 16, p < 0.01) (Fig. 2C,D), but had no significant effect on GABAergic sPSCs in putative oxytocin neurons (108.6 ± 18.3% of baseline, n = 7, Student's paired t test, t(6) = 0.206, p = 0.84) from 24-h-fasted rats. The percentage increase in the frequency of isolated GABAergic sPSCs (52.8 ± 16.4%) in VP neurons was greater than that of unidentified sPSCs (33.6 ± 7.8%), probably because the unidentified sPSC frequency values also included glutamatergic sPSCs, which were unresponsive to ghrelin application and which diluted, therefore, the ghrelin effect on GABAergic sPSCs. The ghrelin effect on GABAergic sPSCs was also spike-dependent because it was abolished by TTX (one-way repeated-measures ANOVA, F(2,8) = 5.738, p < 0.05 followed by Tukey's test: ghrelin, 166 ± 27.7% of baseline, n = 5, p < 0.05; ghrelin in TTX, 92.1 ± 11.0% of baseline, n = 5, p = 0.91). Ghrelin had no significant effect on the GABAergic sPSC amplitude (104.4 ± 3.5% of baseline, n = 15, Wilcoxon signed-rank test, W = 62, z = 1.761, degrees of freedom = 15, p = 0.08) or decay time (104.8 ± 3.3% of baseline, n = 15, Student's paired t test, t(14) = −0.897, p = 0.38).
Figure 2.

Ghrelin modulation of GABAergic sPSCs in VP neurons. A, Time course of mean normalized frequencies of glutamatergic sPSCs recorded in the presence of the GABAA receptor antagonist bicuculline or picrotoxin in PVN VP neurons from ad libitum fed (n = 9) and 24 h-fasted rats (n = 7). Ghrelin (100 nm) had no significant effect on the frequency of glutamatergic sPSCs. B, Summary of mean normalized glutamatergic sPSC frequency response to ghrelin (100 nm) in the presence of GABAA receptor antagonists in VP neurons from ad libitum fed and fasted rats. C, Time course of mean normalized frequency of GABAergic sPSCs recorded in the presence of the glutamate receptor antagonists DNQX and AP-5 in PVN VP neurons from ad libitum fed and fasted rats. Ghrelin (100 nm) caused an increase in the frequency of GABAergic sPSCs in VP cells from fasted, but not fed, rats. D, Summary of mean normalized GABA sPSC frequency response to ghrelin (100 nm) in the presence of glutamate receptor antagonists in VP neurons from ad libitum fed and fasted rats. **p < 0.01.
The ghrelin effect requires activation of postsynaptic ghrelin receptors
Of the two identified ghrelin receptors, GHS-R1a and GHS-R1b, GHS-R1a has been shown to mediate most of the known physiological effects of ghrelin (Howard et al., 1996; McKee et al., 1997; Asakawa et al., 2003; Sun et al., 2004; Esler et al., 2007). To examine whether the ghrelin-induced facilitation of synaptic inputs to VP neurons is mediated by GHS-R1a, we preapplied the GHS-R1a antagonist [d-Lys]-GHRP-6. This antagonist has been shown to effectively block ghrelin-induced food intake, weight gain, and gastric emptying, as well as the ghrelin-induced changes in feeding behavior and gastrointestinal motility (Nakazato et al., 2001; Asakawa et al., 2003; Fujino et al., 2003; Wang et al., 2007). Bath application of [d-Lys]-GHRP-6 (5 μm) for 5 min had no significant effect on sPSC frequency by itself (105.5 ± 4.3% of baseline, n = 9, Student's paired t test, t(8) = −1.740, p = 0.12) but completely blocked the ghrelin-induced increase in sPSC frequency (97.4 ± 7.5% of baseline, 92.2 ± 4.7% compared with [D-lys]-GHRP-6 alone, n = 9, one-way repeated-measures ANOVA, F(2,15) = 0.532, p = 0.60) (Fig. 3A,B), suggesting that the ghrelin-induced facilitation of synaptic inputs to VP neurons is mediated by GHS-R1a.
Figure 3.
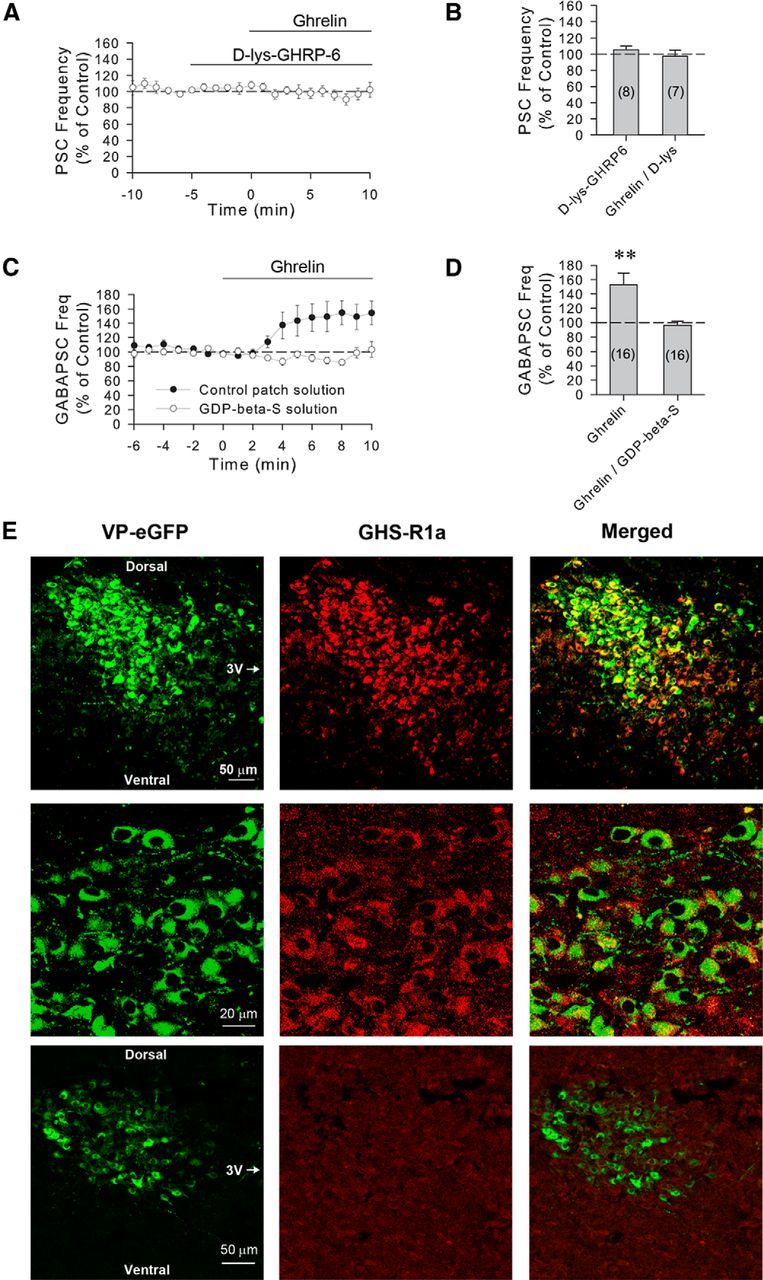
The ghrelin modulation of GABA synaptic inputs to VP neurons is mediated by postsynaptic ghrelin receptors. A, Time course of mean normalized frequency of sPSC frequencies, as a percentage of baseline, showing that the increase in sPSC frequency caused by ghrelin (100 nm) is blocked by the ghrelin GHS-1a receptor antagonist d-lys-GHRP-6 in VP neurons from 24 h-fasted rats (n = 8). B, Summary of effect of d-lys-GHRP-6 and d-lys-GHRP-6 plus ghrelin application on the mean sPSC frequency, as a percentage of baseline. C, Time course of mean normalized GABAergic sPSC frequencies in PVN VP neurons from fasted rats with and without G-protein blockade. Intracellular application of the G-protein blocker GDP-β-S (1 mm) via the patch electrode solution (n = 16) blocked the ghrelin-induced increase in GABAergic sPSC frequency seen in control recordings (n = 16), indicating a postsynaptic G-protein-dependent mechanism that mediates the ghrelin effect in VP cells. D, Summary of mean normalized GABAergic sPSC frequencies, as a percentage of baseline, with and without postsynaptic G-protein blockade. Blocking postsynaptic G-protein activity with intracellular GDP-β-S application blocked the ghrelin-induced increase in GABAergic sPSCs in VP neurons. E, Confocal micrographs of a section of the PVN showing eGFP-labeled VP neurons under green emission filters (VP-eGFP) and GHS-R1a-immunolabeled neurons under red emission filters (GHS-R1a), and the overlay (Merged) of the two images showing eGFP-GHS-R1a double labeling at low (top row) and high magnifications (middle row). Bottom row, Preabsorption control, showing a low-magnification confocal micrograph of a section of the PVN in which the primary antibody had been preabsorbed with a synthetic GHS-R1a peptide. Dorsal, ventral, and medial (third ventricle [3V]) aspects are indicated for orientation. **p < 0.01.
The presynaptic stimulatory effect of ghrelin on GABA release implies either the activation of ghrelin receptors located presynaptically or the activation of postsynaptic ghrelin receptors and the release of a retrograde messenger that alters presynaptic release. We next tested whether the ghrelin effect is mediated by the G-protein-coupled GHS-R1a located postsynaptically in the VP neurons. When GDP-β-S (1 mm) was included in the patch electrode to inhibit G-protein activity selectively in the recorded VP neurons, the ghrelin-induced increase in GABAergic PSC frequency was blocked on average across all the cells tested (96.0 ± 5.9% of baseline, n = 16, Student's paired t test, t(15) = 1.602, p = 0.13) (Fig. 3C,D). The ghrelin-induced increase in GABAergic PSC frequency was maintained, however, in 25% of the VP neurons tested (i.e., 4 of the 16 cells showed an increase in the frequency of GABAergic PSCs, ranging from 20% to 44%).
Previous studies have shown that ghrelin receptors are expressed in PVN neurons (Zigman et al., 2006; Harrold et al., 2008). We examined whether GHS-R1a receptors are expressed in VP neurons in the PVN using immunofluorescence labeling with an antiserum to the GHS-R1a in VP-GFP neurons (Abizaid et al., 2006; Jiang et al., 2008). We found clear double labeling of the GFP-expressing VP neurons with the GHS-R1a antibody in sections of PVN from ad libitum fed rats visualized using confocal microscopy (Fig. 3E). Omitting the primary or secondary antibodies and a preabsorption control of the primary antibody specificity with a synthetic GHS-R1a peptide showed no GHS-R1a immunostaining (Fig. 3E).
Together, these findings suggest that the ghrelin-induced facilitation of GABA synaptic currents in VP neurons is mediated by GHS-R1a receptors expressed in the VP neurons and that their activation leads to the release of a retrograde messenger from the VP cells to mediate the presynaptic facilitation of synaptic GABA release.
The ghrelin effect requires dendritic release of VP
Nitric oxide (NO) is well known as a retrograde messenger that increases GABA synaptic inputs to neurons in the PVN (Bredt et al., 1990; Bains and Ferguson, 1997b; Di et al., 2009), including in PVN parvocellular neuroendocrine cells via a TTX-sensitive mechanism (Bains and Ferguson, 1997a). To test for the NO dependence of the ghrelin facilitation of GABA release, we applied the NO synthase inhibitor l-NAME (100 μm) 10 min before coapplication of ghrelin (100 nm) with l-NAME. l-NAME failed to block the increase in the frequency of GABAergic PSCs caused by ghrelin (136.0 ± 17.7% of baseline, n = 15, Wilcoxon signed-rank test, W = 72, z = 2.045, degrees of freedom = 15, p < 0.05) (Fig. 4A), indicating that the ghrelin effect on GABA release is not dependent on NO.
Figure 4.

The ghrelin-induced increase in synaptic GABA inputs to VP neurons is mediated by the retrograde release of VP. A, Changes in mean normalized GABAergic sPSC frequencies caused by ghrelin in the presence of nitric oxide synthase and VP V1a blockers. Blockade of nitric oxide synthesis by l-NAME failed to block the ghrelin effect, whereas blockade of VP V1a receptors with SR49059 abolished the ghrelin-induced increase in GABA sPSC frequency. B, Whole-cell recording of synaptic currents in a PVN VP neuron showing an increase in GABAergic sPSC frequency in response to puff application of VP (20 μm, 30 s) in a slice from a 24 h-fasted rat. C, Summary of the mean GABAergic sPSC frequencies in VP neurons before (Control) and after application of VP. *p < 0.05.
Previous studies have shown that the dendritic release of VP modulates synaptic inputs to VP neurons by acting as a retrograde and/or autocrine messenger (Kombian et al., 1997; Gouzènes et al., 1998; Hirasawa et al., 2003). Interestingly, VP has been shown to increase GABA input to PVN neurons by acting at V1a receptors, an effect that, similar to the ghrelin effect described here, is sensitive to blockade by TTX (Hermes et al., 2000). To examine whether VP mediates the retrograde signaling induced by ghrelin, we preapplied the selective V1a receptor antagonist SR49059 (10 μm) for 10 min before the coapplication of ghrelin (100 nm). The V1a antagonist alone had no significant effect on the GABAergic sPSC frequency in VP neurons (95.6 ± 4.2% of baseline, n = 13, Student's paired t test, t(12) = 0.388, p = 0.70), but abolished the ghrelin-induced increase in the frequency of GABAergic sPSCs (103.1 ± 9.8% of baseline, 110.2 ± 12.9% compared with SR49059, n = 13, one-way ANOVA on ranks, χ(2)2 = 0.118, p = 0.94) (Fig. 4A). This implicated dendritically released VP as the retrograde messenger required for the ghrelin modulation of GABAergic synaptic inputs to the VP neurons. To confirm that VP causes an increase in GABAergic synaptic inputs to VP neurons, we applied VP via puff application close to the recorded VP neurons. VP (20 μm, 30 s) caused an increase in the GABAergic sPSC frequency in VP neurons (165% of baseline, from 1.44 ± 0.24 Hz to 2.38 ± 0.35 Hz, n = 10, Student's paired t test, t(9) = −2.45, p < 0.05) (Fig. 4B,C).
Ghrelin activates astrocytes via dendritic release of VP
That the ghrelin-induced increase in GABA inputs to VP neurons requires spiking in presynaptic neurons suggested that the retrograde VP messenger must stimulate action potential generation distally in the upper axon or the somatic/dendritic region of the presynaptic GABA neurons, or that it activates an intermediate that can signal at a distance from the VP neuron dendrites. Neurons signal to astrocytes via dendritic release of neurotransmitters (Bernardinelli et al., 2011), and astrocytes have been shown to signal to spatially defined sets of neurons via gliotransmitter release (Bushong et al., 2002; Halassa et al., 2007). Astrocytes play a critical role in the synaptic regulation of magnocellular neuroendocrine cells in the PVN and SON (Oliet et al., 2001; Hussy, 2002; Boudaba et al., 2003; Gordon et al., 2005; Panatier et al., 2006; Di et al., 2013). In addition, glia have been shown to express V1a receptors, and activation of these receptors induces an increase in astrocytic Ca2+ levels, raising the possibility that glia may receive signals from VP neurons and relay information to presynaptic neurons (Yamazaki et al., 1997; Chen et al., 2000; Zhao and Brinton, 2003; Syed et al., 2007). To test the hypothesis that astrocytes serve as an intermediate relay in the ghrelin-induced retrograde modulation of GABAergic synaptic inputs to VP neurons, we first examined whether ghrelin stimulates a calcium response in astrocytes using the glia-specific calcium fluorophore Rhod-2AM. After bulk loading of Rhod-2AM in our hypothalamic slices, bath application of ghrelin induced an elevation of calcium levels in glial cells in the PVN (F/F0 = 126.7 ± 4.6% of baseline, n = 53, Wilcoxon signed-rank test, W = 1139, z = 5.042, degrees of freedom = 53, p < 0.01) (Fig. 5A–C). The ghrelin-induced calcium response in astrocytes was characterized by an increase in the baseline calcium level, the number of calcium transients, or both. In the absence of ghrelin, no change in the baseline calcium level was observed during 30 min of imaging (F/F0 = 100.0 ± 1.8% of baseline, n = 89, Wilcoxon signed-rank test, W = −633, z = −1.295, degrees of freedom = 89, p = 0.196). Preapplication of an antagonist to the oxytocin receptor (OTR) and VP V1a receptor, the Manning compound (1 μm), blocked the ghrelin-induced increase in astrocytic calcium and caused ghrelin to elicit a small, but significant, decrease in intracellular calcium concentration (F/F0 = 96.5 ± 1.2% of baseline, n = 58, Wilcoxon signed-rank test, W = −711, z = −2.752, degrees of freedom = −58, p < 0.01) (Fig. 5C). This indicated that ghrelin stimulates an increase in intracellular calcium in astrocytes via an OTR/V1a receptor-dependent mechanism. In addition, we tested for a VP-induced calcium response in astrocytes; bath application of VP (200 nm) also caused an increase in the intracellular calcium concentration in astrocytes, measured with the Rhod-2AM indicator (F/F0 = 114.8 ± 3.4% of baseline, n = 55, Wilcoxon signed-rank test, W = 1004, z = 4.206, degrees of freedom = 55, p < 0.01) (Fig. 5C). The ghrelin-induced increase in calcium levels in astrocytes was also blocked when slices were preincubated for 2 h in the glial cell metabolism blocker fluorocitric acid (FCA, 100 μm), and ghrelin caused a small but significant decrease in calcium levels (F/F0 = 93.8 ± 0.9% of baseline, n = 47, Wilcoxon signed-rank test, W = −956, z = −5.058, degrees of freedom = 47, p < 0.01) (Fig. 5C), suggesting that the ghrelin-induced elevation of astrocytic calcium requires intact glial metabolic activity. Together, these imaging data suggested that ghrelin stimulates a calcium signal in PVN astrocytes via the activation of OTR or V1a receptors.
Figure 5.

The ghrelin-induced increase in synaptic GABA inputs to VP neurons is mediated by astrocytes. A, Standard fluorescence images of a Rhod-2 AM-loaded slice of PVN from a 24 h-fasted rat showing calcium levels in astrocytes before (Control) and after the bath application of ghrelin (Ghrelin, 100 nm). B, Calcium measurements of the ghrelin-induced increases in calcium levels in the 7 glial cells (1–7) labeled with Rhod-2 AM in the PVN brain slice shown in A. C, Summary of mean normalized fluorescence in PVN astrocytes. Bath application of ghrelin caused a significant increase in intracellular calcium (Ghrelin); the ghrelin response was blocked with the V1a receptor antagonist Manning compound (Ghrelin/Manning); bath application of VP also caused a significant increase in intracellular calcium (VP) in Rhod-2 AM-loaded cells; preincubation of the slices in the gliotoxin FCA blocked the ghrelin-induced increase in astrocytic calcium (Ghrelin/FCA). D, Time course of the mean normalized GABAergic sPSC frequency response to ghrelin in the presence and absence of FCA in PVN VP neurons from 24 h-fasted rats. Pretreatment of PVN brain slices with FCA blocked the ghrelin-induced increase in GABAergic PSC frequency (n = 16). E, Summary of the mean normalized GABAergic sPSC frequency response to ghrelin in VP neurons in the absence (Ghrelin) and presence of FCA (Ghrelin/FCA). F, Summary of the mean normalized GABAergic sPSC frequency response to VP in VP neurons in the absence (VP) and presence of FCA (VP/FCA). FCA blocked the VP-induced increase in GABAergic PSC frequency in VP neurons. *p < 0.05. **p < 0.01.
We next asked whether the ghrelin-induced activation of PVN astrocytes is required for the ghrelin facilitation of GABAergic synaptic inputs to VP neurons by testing whether the blockade of astrocyte metabolic activity disrupts the ghrelin-induced increase in GABAergic sPSCs. Preincubation of slices from fasted rats in FCA (100 μm) for 2–4 h before ghrelin application completely abolished the ghrelin-induced increase in GABAergic sPSC frequency in VP neurons (101.5 ± 11.8% of baseline, n = 15, Wilcoxon signed-rank test, W = −10, z = −0.284, degrees of freedom = 15, p = 0.90) (Fig. 5D,E), indicating that glia play a critical role in the ghrelin modulation of synaptic GABA inputs. Because the ghrelin effect is dependent on the dendritic release of VP, we also tested whether the VP-induced increase in GABAergic inputs to VP neurons depends on glial function. In the presence of FCA (100 μm), puff application of VP (20 μm, 30 s) failed to cause an increase in GABAergic PSC frequency in VP neurons (89.1 ± 14.2% of baseline, n = 7, Student's paired t test, t(6) = 1.237, p = 0.26) (Fig. 5F). Together, these findings suggested an astrocytic relay in the ghrelin-induced retrograde VP signaling to presynaptic GABA neurons.
Activation of presynaptic GABA neurons by ATP gliotransmission
We next investigated how glial cells stimulate the presynaptic GABA neurons to increase GABA release onto the VP neurons. ATP is a well-known excitatory gliotransmitter that activates adjacent neurons via actions at ionotropic P2X and/or metabotropic P2Y receptors (Zhang et al., 2003; Bowser and Khakh, 2004). Previous studies have shown that activation of P2X receptors facilitates GABA release in different parts of the brain (Hugel and Schlichter, 2000; Jameson et al., 2008; Vavra et al., 2011). It has also been shown that P2X receptors are expressed in the SON and PVN and that activation of P2X receptors stimulates VP neurons (Day et al., 1993; Vulchanova et al., 1996; Troadec et al., 1998; Gomes et al., 2009; Vavra et al., 2011). Therefore, we next examined whether purinergic P2X receptors mediate the ghrelin-induced increase in GABAergic synaptic input to VP neurons. Bath application of the P2X receptor antagonist TNP-ATP (10 μm) alone had no significant effect on the GABAergic sPSC frequency (86.5 ± 8.3% of baseline, n = 9, Wilcoxon signed-rank test, W = −31, z = −1.836, degrees of freedom = 9, p = 0.074) but blocked the ghrelin-induced increase in sPSC frequency (88.2 ± 12.9% of baseline, 100.2 ± 10.0% of TNP-ATP, n = 9, one-way ANOVA on ranks, χ2(2) = 2.889, p = 0.28) (Fig. 6A,B). Therefore, the ghrelin-induced modulation of synaptic GABA inputs to VP neurons is dependent on P2X receptor activation. We also tested whether the activation of purinergic receptors mimics the ghrelin-induced increase in GABAergic synaptic inputs to VP neurons. We used the nonhydrolyzable form of ATP, ATP-γ-S, which is not hydrolyzed to adenosine and lacks, therefore, the ability to activate adenosine receptors. Puff application of ATP-γ-S (100 μm) caused a significant increase in GABAergic sPSC frequency in VP neurons (from 1.95 ± 0.22 Hz to 8.83 ± 2.01 Hz, n = 7, Student's paired t test, t(6) = −3.317, p < 0.05) (Fig. 6C,D). Similar to the ghrelin effect, the ATP-γ-S-induced increase in GABAergic inputs to VP neurons was sensitive to spike generation because it was suppressed by TTX (n = 7, two-way repeated-measures ANOVA, significant interaction between ATP-γ-S and TTX, F(1,6) = 24.089, p < 0.01). The effect of ATP-γ-S on GABA PSC frequency in TTX did not reach statistical significance (0.94 ± 0.14 Hz in TTX; 3.20 ± 1.02 Hz in ATP-γ-S/TTX, n = 7, Tukey's test following the two-way ANOVA, p = 0.24) (Fig. 6D). The ATP-γ-S-mediated increase in GABAergic sPSC frequency was maintained after pretreatment of the slices with the glial toxin FCA (from 1.17 ± 0.21 Hz to 12.06 ± 2.62 Hz, n = 9, Student's paired t test, t(8) = −4.213, p < 0.01) (Fig. 6D), indicating that the effect of ATP on GABA synaptic inputs to VP neurons is downstream from glial activation.
Figure 6.

The ghrelin-induced increase in GABA synaptic inputs to VP neurons is dependent on ATP P2X receptor activation. A, Time course of the mean normalized GABAergic sPSC frequency response to ghrelin in the presence of an ATP P2X receptor antagonist. Preapplication of the selective ATP P2X receptor antagonist TNP-ATP blocked the ghrelin effect on GABA PSC frequency (n = 9) in VP neurons from 24 h-fasted rats. B, Summary of mean normalized GABAergic sPSC frequencies in the P2X antagonist (TNP-ATP) and the P2X antagonist plus ghrelin (Ghrelin/TNP-ATP). C, Whole-cell recording of GABAergic synaptic currents in a VP neuron from a 24 h-fasted rat showing an increase in GABAergic PSCs in response to puff application of the ATP agonist ATPγS. D, Summary of mean normalized GABAergic sPSC frequencies in VP neurons showing the increase in GABA sPSC frequency in the purinergic agonist (ATPγS, n = 7), in the purinergic agonist in the presence of TTX (ATPγS/TTX, n = 7), and in the purinergic agonist in the presence of the gliotoxin (ATPγS/FCA, n = 9). The effect of purinergic receptor activation was not blocked by inhibiting astrocytic activity with FCA but was blocked by inhibiting spike generation with TTX. *p < 0.05. **p < 0.01.
Ghrelin activates VP neurons
We next tested whether the stimulation of GABAergic synaptic inputs to VP neurons by ghrelin is capable of activating the VP neurons. Previously, we showed that GABA acts as an excitatory signal in VP neurons, but not in oxytocin neurons, because of an altered Cl− homeostasis in the VP neurons (Haam et al., 2012). We first determined whether the GABA reversal potential (EGABA) was altered in VP neurons during fasting with gramicidin-perforated patch-clamp recordings, which maintain the native intracellular Cl− concentration during recordings. The EGABA recorded in VP neurons from 24 h-fasted rats was not significantly different from that in VP neurons from ad libitum-fed rats (24 h-fasted: −33.2 ± 2.8 mV, n = 3; ad libitum fed: −33.1 ± 3.4 mV, n = 15, Student's unpaired t test, t(16) = −0.005753, p = 0.99), indicating that GABA is also excitatory in VP neurons from fasted rats. Next, to examine the ghrelin effect on the spiking activity of VP neurons without disturbing intracellular ionic concentrations, we used loose-seal cell-attached patch recordings, which are extracellular recordings that maintain the membrane integrity and prevent current from the patch electrode from influencing the membrane potential of the recorded cells. Consistent with the effect of ghrelin on GABAergic synaptic inputs, the ghrelin effect on spiking was feeding status-dependent, causing a robust increase in spiking frequency in VP neurons in 24 h-fasted rats (201.6 ± 33.6% of baseline, n = 12, Student's paired t test, t(11) = −3.862, p < 0.01) (Fig. 7A–C), but a weaker increase in spiking frequency that did not reach statistical significance in ad libitum-fed rats (151.4 ± 11.1% of baseline, n = 6, Student's paired t test, t(5) = −1.541, p = 0.18) (Fig. 7C). As in our previous study (Haam et al., 2012), blockade of GABAA receptors with bath application of bicuculline methiodide (50 μm) reduced the basal spiking frequency in VP neurons (control: 1.34 ± 0.28 Hz, n = 12; bicuculline: 0.17 ± 0.07 Hz, n = 14, Mann–Whitney U Test, U = 19.5, T = 226.5, small number = 12, big number = 14, p < 0.01) (Fig. 7D), indicating that tonic excitatory GABAergic synaptic activity contributes significantly to the basal firing activity of VP cells. The ghrelin-induced increase in spiking frequency in VP neurons from fasted rats was dependent on GABAA receptors (n = 14, two-way repeated-measures ANOVA, significant interaction between ghrelin and bicuculline, F(1,24) = 14.142, p < 0.01) The ghrelin effect was abolished by blocking GABAA receptors (bicuculline: 0.17 ± 0.07 Hz; bicuculline + ghrelin: 0.28 ± 0.08 Hz, n = 14, Tukey's test following the two-way ANOVA, control vs ghrelin in the presence of bicuculline, p = 0.556) (Fig. 7D), indicating that the ghrelin-induced increase in spiking activity in VP neurons is mediated by excitatory GABAergic synaptic inputs.
Figure 7.

Ghrelin elicited an increase in spiking in VP cells. A, Spiking activity in a VP neuron from a 24 h-fasted rat recorded extracellularly with the loose-seal cell-attached patch technique. Ghrelin (100 nm) elicited an increase in spiking. B, Distribution plot of changes in spike frequency in individual cells before (Control) and during ghrelin application (Ghrelin) (n = 12). C, Summary of mean normalized spike frequency responses to ghrelin in PVN VP neurons from ad libitum fed and 24 h-fasted rats. D, Summary of mean spike frequency responses to ghrelin in VP cells from fasted rats in the absence (n = 14) and presence of the GABAA receptor antagonist bicuculline (n = 12). Blocking GABAA receptors decreased the basal spike frequency and blocked the ghrelin-induced increase in spike frequency. *p < 0.05. n.s., Not significant.
Discussion
Our findings suggest that ghrelin stimulates VP neurons by engaging a novel retrograde neuronal–glial circuit that activates presynaptic GABA neurons. According to this model (Fig. 8), ghrelin triggers the dendritic release of VP, which activates astrocytes to release ATP, which in turn excites upstream GABA neurons and stimulates an excitatory GABAergic input back to the VP neurons, completing a retrograde neuronal–glial autoregulatory circuit.
Figure 8.

Proposed model of ghrelin modulation of GABA synaptic inputs to VP neurons. Ghrelin binding to the GHS-R1a receptor in VP neurons induces the dendritic release of VP, which activates V1a receptors on neighboring astrocytes and elicits an astrocytic calcium response. Activation of astrocytes causes the release of the gliotransmitter ATP, which activates ionotropic P2X receptors on presynaptic GABA neurons, stimulating action potential generation in the GABA neurons and resulting in an increase in GABAergic synaptic inputs to the VP neurons.
Ghrelin activates an excitatory GABA input to VP neurons
Activation of GABA synaptic inputs is an effective way to increase the excitability of VP neurons because GABAergic synapses comprise >50% of the total synapses in the PVN (Theodosis et al., 1986; Decavel and Van den Pol, 1990; El Majdoubi et al., 1997) and, as we recently reported, GABA is excitatory in VP neurons (Haam et al., 2012). Our observations here confirm those findings. The ghrelin modulation of VP neurons is novel as it synaptically activates these neurons by increasing an excitatory GABAergic synaptic input without affecting glutamatergic inputs, providing evidence for the regulation of an excitatory GABA input that stimulates the VP neurons. Previous studies have shown that central ghrelin administration induces Fos expression in PVN VP neurons (Nakazato et al., 2001; Lawrence et al., 2002) and increases VP release into the blood (Ishizaki et al., 2002), which, because the ghrelin effect on VP neurons is specific to GABA inputs, supports an excitatory role for GABA in these cells (Haam et al., 2012). The recruitment of excitatory GABAergic circuits by ghrelin may represent a secondary mode of stimulation of the VP system, thereby reserving the excitatory glutamatergic circuits for the primary osmoregulatory responses of the VP neurons because osmotic stimulation engages glutamate inputs to these cells (Richard and Bourque, 1995; Sladek et al., 1995; Onaka and Yagi, 2001; Kawasaki et al., 2006). Ghrelin had no effect on either the glutamate or the GABA inputs to oxytocin neurons. The origin of the GABAergic synaptic inputs to PVN VP neurons activated by ghrelin is not known, but is likely to be in the peri-PVN hypothalamic area, consistent with previous findings (Decavel and Van den Pol, 1990; Thellier et al., 1994; Vrang et al., 1995; Boudaba et al., 1996), because these inputs were activated in a spike-dependent manner in slices, in which projected afferents are cut.
Glia mediate ghrelin-induced synaptic modulation
The ghrelin effect on GABA input to VP neurons is unique in that it is TTX-sensitive, glia-dependent, and mediated by postsynaptic ghrelin receptors. Because ghrelin did not elicit a change in postsynaptic membrane conductances, the TTX sensitivity of the response indicates that ghrelin increases spiking in presynaptic GABA neurons, and suggests the somata/dendrites of GABA neurons as sites of action. However, postsynaptic G-protein blockade and GHS-R1a immunofluorescence suggested that postsynaptic ghrelin receptors mediate the ghrelin effect, raising the question as to how the activation of ghrelin receptors in VP neurons excites presynaptic GABA neurons. Surprisingly, blockade of glial metabolism and glial calcium imaging indicated that astrocytes are activated by ghrelin and necessary for the VP neuron response to ghrelin and suggest, therefore, that glia relay the postsynaptic ghrelin signal to the presynaptic GABA neurons. Previous studies have shown that GHS-R1a activation causes an increase in intracellular calcium (Howard et al., 1996; McKee et al., 1997). Dendritic release of VP is dependent on calcium signaling (Di Scala-Guenot et al., 1987; Ludwig et al., 1995), suggesting that the dendritic release of VP is mediated by ghrelin-induced calcium signaling in the VP neurons.
Our data demonstrate that glial cells respond to the dendritic release of VP. Dendritically released VP has been shown to act as a retrograde messenger to modulate synaptic inputs to VP neurons (Hirasawa et al., 2003; Song et al., 2007), but the mechanism underlying this modulation had not been elucidated. We show that glia respond to dendritically released VP by increasing intracellular calcium levels, which is critical for the ghrelin-induced synaptic modulation. The activated glia then release the gliotransmitter ATP to activate P2X receptors on upstream GABA neurons. The dendritic release of VP, unlike axonal release, does not require depolarization (Di Scala-Guenot et al., 1987; Ludwig et al., 1995). The glia-mediated activation of excitatory synaptic inputs, therefore, may be an efficient means to stimulate axonal VP release. Dendritically released VP may also induce behavioral responses via central VP receptors, enabling ghrelin to regulate both systemic physiological and central behavioral VP actions.
Our study demonstrates, therefore, that astrocytes play a critical role in the ghrelin modulation of synaptic inputs to VP neurons by integrating into a retrograde circuit between the postsynaptic and presynaptic neurons. This retrograde signaling is triggered by the dendritic release of VP and is transmitted to upstream GABA neurons via the glial release of ATP.
Ghrelin activates VP neurons
We show here that ghrelin can stimulate VP neurons in the hypothalamus directly by acting at postsynaptic GHS-R1a receptors. Although ghrelin is synthesized predominantly in the stomach, it is well established that the ghrelin regulation of feeding behavior is mediated primarily by its central actions (Wren et al., 2000, 2001; Nakazato et al., 2001; Lawrence et al., 2002). How peripheral ghrelin activates neurons in the CNS remains controversial. Specialized ghrelin transporters can transport blood-borne ghrelin across the blood–brain barrier into the brain (Banks et al., 2002). Vagal afferents have also been shown to transmit the hunger signal to the brain (Date et al., 2002), and activation of the vagal nerve stimulates ghrelin-expressing neurons in the hypothalamus (Cowley et al., 2003). Ghrelin peptide expression has been described in the arcuate nucleus, pituitary, and internuclear areas of the hypothalamus (Kojima et al., 1999; Lu et al., 2002; Cowley et al., 2003; Mozid et al., 2003), some of which project to the PVN. Although the ghrelin concentrations we used (10–100 nm) are higher than reported circulating levels (0.5–3.5 nm) (Tschöp et al., 2000; Cummings et al., 2001; Sugino et al., 2002a), ghrelin levels in the brain may reach significantly higher concentrations either by localized blood–brain transport (Banks et al., 2002) or by synaptic release from ghrelin-expressing neurons (Kojima et al., 1999; Lu et al., 2002; Cowley et al., 2003; Mozid et al., 2003). The source of the ghrelin responsible for the activation of VP neurons remains to be determined.
The ghrelin activation of spiking triggered by dendritic VP release might be expected to cause the VP neurons to enter into a positive feedback loop via further spiking-induced VP release from the dendrites. We did not see evidence of this because the response was reversible with washout of the ghrelin (although, interestingly, responses to 10 min ghrelin applications did not reverse after 20 min of washout). This suggests either that axonal and dendritic responses are uncoupled from each other or that there is a break in the retrograde signaling after depolarization of the dendrites (e.g., V1a desensitization). The dissociation of axonal and dendritic peptide release has been demonstrated before in VP and oxytocin neurons (Ludwig et al., 1995; Sabatier et al., 2003).
The ghrelin effect on GABAergic synaptic inputs was potentiated during fasting. Previous studies showed an increase in the sensitivity of hypothalamic neurons to ghrelin during food deprivation (Hewson and Dickson, 2000; Scott et al., 2007) that may be caused by an increase in GHS-R1a expression in the hypothalamus (Kim et al., 2003; Sirotkin et al., 2013). Similarly, GHS-R1a levels in the vagal nerve follow circadian rhythms and are regulated by feeding status (Sato et al., 2007). Thus, the regulation of GHS-R1a expression may be responsible for the feeding status dependence of the ghrelin sensitivity of VP neurons that we observed.
Ghrelin's modulation of VP neurons suggests it may link energy and fluid homeostasis. The close relationship between feeding and drinking behaviors is well documented (Fitzsimons and Le Magnen, 1969; Bellisle and Le Magnen, 1981; Engell, 1988). Drinking behavior is most often expressed during feeding to meet the osmotic requirements of digestion (Fitzsimons and Le Magnen, 1969). The digestion of solid food during dehydration can be detrimental to survival, and high plasma osmolality inhibits feeding behavior (Watts et al., 1999; Watts, 1999). The water retention induced by VP secretion during fasting prepares the fasted animal for food intake by maintaining sufficiently low blood osmolality. In addition, ghrelin inhibits drinking (Hashimoto et al., 2007; Mietlicki et al., 2009), which is in line with VP's antidipsogenic effect and consistent with ghrelin acting through VP release (Pasqualini and Codevilla, 1959; Smith and Mc, 1962; Kozlowski and Szczepnska-Sadowska, 1975; Racotta et al., 1995). The ghrelin-induced, VP-mediated increase in water retention and decrease in water intake during food deprivation may be responsible, therefore, for maintaining a critical hydration state without losing essential minerals and ions, and thus may play a primary role in the survival of the organism. Like VP, ghrelin has also been implicated in the activation of the hypothalamic-pituitary-adrenal axis during stress; and because fasting presents a physiological stress, there may also be an interaction between the two systems that modulates the neuroendocrine stress response.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS042081, the Catherine and Hunter Pierson Endowed Chair in Neuroscience, and the Tulane Bridge Fund. We thank Dr. Bret Smith for assistance with GHS-R1a immunohistochemistry and Dr. Ion Popescu for helpful discussion.
The authors declare no competing financial interests.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, Gao XB, Horvath TL. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlal H, Chen Q, Habo K, Wang Z, Soleimani M. Fasting downregulates renal water channel AQP2 and causes polyuria. Am J Physiol Renal Physiol. 2001;280:F513–F523. doi: 10.1152/ajprenal.2001.280.3.F513. [DOI] [PubMed] [Google Scholar]
- Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Ueno N, Makino S, Fujimiya M, Niijima A, Fujino MA, Kasuga M. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology. 2001;120:337–345. doi: 10.1053/gast.2001.22158. [DOI] [PubMed] [Google Scholar]
- Asakawa A, Inui A, Kaga T, Katsuura G, Fujimiya M, Fujino MA, Kasuga M. Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut. 2003;52:947–952. doi: 10.1136/gut.52.7.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains JS, Ferguson AV. Nitric oxide depolarizes type II paraventricular nucleus neurons in vitro. Neuroscience. 1997a;79:149–159. doi: 10.1016/S0306-4522(96)00670-7. [DOI] [PubMed] [Google Scholar]
- Bains JS, Ferguson AV. Nitric oxide regulates NMDA-driven GABAergic inputs to type I neurones of the rat paraventricular nucleus. J Physiol. 1997b;499:733–746. doi: 10.1113/jphysiol.1997.sp021965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Tschöp M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood–brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther. 2002;302:822–827. doi: 10.1124/jpet.102.034827. [DOI] [PubMed] [Google Scholar]
- Bellisle F, Le Magnen J. The structure of meals in humans: eating and drinking patterns in lean and obese subjects. Physiol Behav. 1981;27:649–658. doi: 10.1016/0031-9384(81)90237-7. [DOI] [PubMed] [Google Scholar]
- Bernardinelli Y, Salmon C, Jones EV, Farmer WT, Stellwagen D, Murai KK. Astrocytes display complex and localized calcium responses to single-neuron stimulation in the hippocampus. J Neurosci. 2011;31:8905–8919. doi: 10.1523/JNEUROSCI.6341-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudaba C, Szabó K, Tasker JG. Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J Neurosci. 1996;16:7151–7160. doi: 10.1523/JNEUROSCI.16-22-07151.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudaba C, Linn DM, Halmos KC, Tasker JG. Increased tonic activation of presynaptic metabotropic glutamate receptors in the rat supraoptic nucleus following chronic dehydration. J Physiol. 2003;551:815–823. doi: 10.1113/jphysiol.2003.042739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci. 2004;24:8606–8620. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao Z, Hertz L. Vasopressin increases [Ca(2+)](i) in differentiated astrocytes by activation of V1b/V3 receptors but has no effect in mature cortical neurons. J Neurosci Res. 2000;60:761–766. doi: 10.1002/1097-4547(20000615)60:6<761::AID-JNR8>3.0.CO%3B2-R. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschöp M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661. doi: 10.1016/S0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–1719. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology. 2002;123:1120–1128. doi: 10.1053/gast.2002.35954. [DOI] [PubMed] [Google Scholar]
- Day TA, Sibbald JR, Khanna S. ATP mediates an excitatory noradrenergic neuron input to supraoptic vasopressin cells. Brain Res. 1993;607:341–344. doi: 10.1016/0006-8993(93)91528-Z. [DOI] [PubMed] [Google Scholar]
- Decavel C, Van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- Di S, Maxson MM, Franco A, Tasker JG. Glucocorticoids regulate glutamate and GABA synapse-specific retrograde transmission via divergent nongenomic signaling pathways. J Neurosci. 2009;29:393–401. doi: 10.1523/JNEUROSCI.4546-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Popescu IR, Tasker JG. Glial control of endocannabinoid heterosynaptic modulation in hypothalamic magnocellular neuroendocrine cells. J Neurosci. 2013;33:18331–18342. doi: 10.1523/JNEUROSCI.2971-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Scala-Guenot D, Strosser MT, Richard P. Electrical stimulations of perifused magnocellular nuclei in vitro elicit Ca2+-dependent, tetrodotoxin-insensitive release of oxytocin and vasopressin. Neurosci Lett. 1987;76:209–214. doi: 10.1016/0304-3940(87)90717-8. [DOI] [PubMed] [Google Scholar]
- El Majdoubi M, Poulain DA, Theodosis DT. Lactation-induced plasticity in the supraoptic nucleus augments axodendritic and axosomatic GABAergic and glutamatergic synapses: an ultrastructural analysis using the disector method. Neuroscience. 1997;80:1137–1147. doi: 10.1016/S0306-4522(97)00193-0. [DOI] [PubMed] [Google Scholar]
- Engell D. Interdependency of food and water intake in humans. Appetite. 1988;10:133–141. doi: 10.1016/0195-6663(88)90064-5. [DOI] [PubMed] [Google Scholar]
- Esler WP, Rudolph J, Claus TH, Tang W, Barucci N, Brown SE, Bullock W, Daly M, Decarr L, Li Y, Milardo L, Molstad D, Zhu J, Gardell SJ, Livingston JN, Sweet LJ. Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology. 2007;148:5175–5185. doi: 10.1210/en.2007-0239. [DOI] [PubMed] [Google Scholar]
- Fitzsimons TJ, Le Magnen J. Eating as a regulatory control of drinking in the rat. J Comp Physiol Psychol. 1969;67:273–283. doi: 10.1037/h0026772. [DOI] [PubMed] [Google Scholar]
- Fujino K, Inui A, Asakawa A, Kihara N, Fujimura M, Fujimiya M. Ghrelin induces fasted motor activity of the gastrointestinal tract in conscious fed rats. J Physiol. 2003;550:227–240. doi: 10.1113/jphysiol.2003.040600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes DA, Song Z, Stevens W, Sladek CD. Sustained stimulation of vasopressin and oxytocin release by ATP and phenylephrine requires recruitment of desensitization-resistant P2X purinergic receptors. Am J Physiol Regul Integr Comp Physiol. 2009;297:R940–R949. doi: 10.1152/ajpregu.00358.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Baimoukhametova DV, Hewitt SA, Rajapaksha WR, Fisher TE, Bains JS. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci. 2005;8:1078–1086. doi: 10.1038/nn1498. [DOI] [PubMed] [Google Scholar]
- Gouzènes L, Desarménien MG, Hussy N, Richard P, Moos FC. Vasopressin regularizes the phasic firing pattern of rat hypothalamic magnocellular vasopressin neurons. J Neurosci. 1998;18:1879–1885. doi: 10.1523/JNEUROSCI.18-05-01879.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, Smith RG, Van der Ploeg LH, Howard AD. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997;48:23–29. doi: 10.1016/S0169-328X(97)00071-5. [DOI] [PubMed] [Google Scholar]
- Haam J, Popescu IR, Morton LA, Halmos KC, Teruyama R, Ueta Y, Tasker JG. GABA is excitatory in adult vasopressinergic neuroendocrine cells. J Neurosci. 2012;32:572–582. doi: 10.1523/JNEUROSCI.3826-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27:6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrold JA, Dovey T, Cai XJ, Halford JC, Pinkney J. Autoradiographic analysis of ghrelin receptors in the rat hypothalamus. Brain Res. 2008;1196:59–64. doi: 10.1016/j.brainres.2007.12.055. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Fujihara H, Kawasaki M, Saito T, Shibata M, Otsubo H, Takei Y, Ueta Y. Centrally and peripherally administered ghrelin potently inhibits water intake in rats. Endocrinology. 2007;148:1638–1647. doi: 10.1210/en.2006-0993. [DOI] [PubMed] [Google Scholar]
- Hermes ML, Ruijter JM, Klop A, Buijs RM, Renaud LP. Vasopressin increases GABAergic inhibition of rat hypothalamic paraventricular nucleus neurons in vitro. J Neurophysiol. 2000;83:705–711. doi: 10.1152/jn.2000.83.2.705. [DOI] [PubMed] [Google Scholar]
- Hewson AK, Dickson SL. Systemic administration of ghrelin induces Fos and Egr-1 proteins in the hypothalamic arcuate nucleus of fasted and fed rats. J Neuroendocrinol. 2000;12:1047–1049. doi: 10.1046/j.1365-2826.2000.00584.x. [DOI] [PubMed] [Google Scholar]
- Hirasawa M, Mouginot D, Kozoriz MG, Kombian SB, Pittman QJ. Vasopressin differentially modulates non-NMDA receptors in vasopressin and oxytocin neurons in the supraoptic nucleus. J Neurosci. 2003;23:4270–4277. doi: 10.1523/JNEUROSCI.23-10-04270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J, Paress PS, Diaz C, Chou M, Liu KK, McKee KK, Pong SS, Chaung LY, Elbrecht A, Dashkevicz M, Heavens R, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974–977. doi: 10.1126/science.273.5277.974. [DOI] [PubMed] [Google Scholar]
- Hugel S, Schlichter R. Presynaptic P2X receptors modulate synaptic GABA release from cultured neonatal rat dorsal horn neurones. Eur J Neurosci. 2000;12:365. [Google Scholar]
- Hussy N. Glial cells in the hypothalamo-neurohypophysial system: key elements of the regulation of neuronal electrical and secretory activity. Prog Brain Res. 2002;139:95–112. doi: 10.1016/S0079-6123(02)39010-1. [DOI] [PubMed] [Google Scholar]
- Ishizaki S, Murase T, Sugimura Y, Kakiya S, Yokoi H, Tachikawa K, Arima H, Miura Y, Oiso Y. Role of ghrelin in the regulation of vasopressin release in conscious rats. Endocrinology. 2002;143:1589–1593. doi: 10.1210/en.143.5.1589. [DOI] [PubMed] [Google Scholar]
- Jameson HS, Pinol RA, Mendelowitz D. Purinergic P2X receptors facilitate inhibitory GABAergic and glycinergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Brain Res. 2008;1224:53–62. doi: 10.1016/j.brainres.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Li LJ, Wang J, Xie JX. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp Neurol. 2008;212:532–537. doi: 10.1016/j.expneurol.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Kawasaki A, Shutoh F, Nogami H, Hisano S. VGLUT2 expression is up-regulated in neurohypophysial vasopressin neurons of the rat after osmotic stimulation. Neurosci Res. 2006;56:124–127. doi: 10.1016/j.neures.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Kim MS, Yoon CY, Park KH, Shin CS, Park KS, Kim SY, Cho BY, Lee HK. Changes in ghrelin and ghrelin receptor expression according to feeding status. Neuroreport. 2003;14:1317–1320. doi: 10.1097/00001756-200307180-00006. [DOI] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Mouginot D, Pittman QJ. Dendritically released peptides act as retrograde modulators of afferent excitation in the supraoptic nucleus in vitro. Neuron. 1997;19:903–912. doi: 10.1016/S0896-6273(00)80971-X. [DOI] [PubMed] [Google Scholar]
- Kozaka T, Fujii Y, Ando M. Central effects of various ligands on drinking behavior in eels acclimated to seawater. J Exp Biol. 2003;206:687–692. doi: 10.1242/jeb.00146. [DOI] [PubMed] [Google Scholar]
- Kozlowski S, Szczepnska-Sadowska E. Antagonistic effects of vasopressin and hypervolemia on osmotic reactivity of the thirst mechanism in dogs. Pflugers Arch. 1975;353:59–65. doi: 10.1007/BF00584511. [DOI] [PubMed] [Google Scholar]
- Lawrence CB, Snape AC, Baudoin FM, Luckman SM. Acute central ghrelin and GH secretagogues induce feeding and activate brain appetite centers. Endocrinology. 2002;143:155–162. doi: 10.1210/endo.143.1.8561. [DOI] [PubMed] [Google Scholar]
- Lu S, Guan JL, Wang QP, Uehara K, Yamada S, Goto N, Date Y, Nakazato M, Kojima M, Kangawa K, Shioda S. Immunocytochemical observation of ghrelin-containing neurons in the rat arcuate nucleus. Neurosci Lett. 2002;321:157–160. doi: 10.1016/S0304-3940(01)02544-7. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Callahan MF, Morris M. Effects of tetrodotoxin on osmotically stimulated central and peripheral vasopressin and oxytocin release. Neuroendocrinology. 1995;62:619–627. doi: 10.1159/000127058. [DOI] [PubMed] [Google Scholar]
- Luther JA, Tasker JG. Voltage-gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2000;523:193–209. doi: 10.1111/j.1469-7793.2000.t01-1-00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee KK, Palyha OC, Feighner SD, Hreniuk DL, Tan CP, Phillips MS, Smith RG, Van der Ploeg LH, Howard AD. Molecular analysis of rat pituitary and hypothalamic growth hormone secretagogue receptors. Mol Endocrinol. 1997;11:415–423. doi: 10.1210/mend.11.4.9908. [DOI] [PubMed] [Google Scholar]
- Mietlicki EG, Nowak EL, Daniels D. The effect of ghrelin on water intake during dipsogenic conditions. Physiol Behav. 2009;96:37–43. doi: 10.1016/j.physbeh.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozid AM, Tringali G, Forsling ML, Hendricks MS, Ajodha S, Edwards R, Navarra P, Grossman AB, Korbonits M. Ghrelin is released from rat hypothalamic explants and stimulates corticotrophin-releasing hormone and arginine-vasopressin. Horm Metab Res. 2003;35:455–459. doi: 10.1055/s-2003-41801. [DOI] [PubMed] [Google Scholar]
- Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S. A role for ghrelin in the central regulation of feeding. Nature. 2001;409:194–198. doi: 10.1038/35051587. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Onaka T, Yagi K. Involvement of N-methyl-d-aspartic acid receptor activation in oxytocin and vasopressin release after osmotic stimuli in rats. J Neuroendocrinol. 2001;13:166–174. doi: 10.1046/j.1365-2826.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Pasqualini RQ, Codevilla A. Thirst-suppressing (antidipsetic) effect of pitressin in diabetes insipidus. Acta Endocrinol (Copenh) 1959;30:37–41. doi: 10.1530/acta.0.0300037. [DOI] [PubMed] [Google Scholar]
- Racotta R, Soto-Mora LM, Palacios E, Quevedo L. Norepinephrine inhibition of water and food intake: comparison with vasopressin effects. Physiol Behav. 1995;57:141–145. doi: 10.1016/0031-9384(94)00218-T. [DOI] [PubMed] [Google Scholar]
- Richard D, Bourque CW. Synaptic control of rat supraoptic neurones during osmotic stimulation of the organum vasculosum lamina terminalis in vitro. J Physiol. 1995;489:567–577. doi: 10.1113/jphysiol.1995.sp021073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier N, Caquineau C, Dayanithi G, Bull P, Douglas AJ, Guan XM, Jiang M, Van der Ploeg L, Leng G. Alpha-melanocyte-stimulating hormone stimulates oxytocin release from the dendrites of hypothalamic neurons while inhibiting oxytocin release from their terminals in the neurohypophysis. J Neurosci. 2003;23:10351–10358. doi: 10.1523/JNEUROSCI.23-32-10351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Nakahara K, Miyazato M, Kangawa K, Murakami N. Regulation of GH secretagogue receptor gene expression in the rat nodose ganglion. J Endocrinol. 2007;194:41–46. doi: 10.1677/JOE-06-0078. [DOI] [PubMed] [Google Scholar]
- Scott V, McDade DM, Luckman SM. Rapid changes in the sensitivity of arcuate nucleus neurons to central ghrelin in relation to feeding status. Physiol Behav. 2007;90:180–185. doi: 10.1016/j.physbeh.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Sirotkin AV, Pavlova S, Tena-Sempere M, Grossmann R, Jiménez MR, Rodriguez JM, Valenzuela F. Food restriction, ghrelin, its antagonist and obestatin control expression of ghrelin and its receptor in chicken hypothalamus and ovary. Comp Biochem Physiol A Mol Integr Physiol. 2013;164:141–153. doi: 10.1016/j.cbpa.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Sladek CD, Fisher KY, Sidorowicz HE, Mathiasen JR. Osmotic stimulation of vasopressin mRNA content in the supraoptic nucleus requires synaptic activation. Am J Physiol. 1995;268:R1034–R1039. doi: 10.1152/ajpregu.1995.268.4.R1034. [DOI] [PubMed] [Google Scholar]
- Smith RW, McCann SM. Alterations in food and water intake after hypothalamic lesions in the rat. Am J Physiol. 1962;203:366–370. doi: 10.1152/ajplegacy.1962.203.2.366. [DOI] [PubMed] [Google Scholar]
- Song Z, Vijayaraghavan S, Sladek CD. ATP increases intracellular calcium in supraoptic neurons by activation of both P2X and P2Y purinergic receptors. Am J Physiol Regul Integr Comp Physiol. 2007;292:R423–R431. doi: 10.1152/ajpregu.00495.2006. [DOI] [PubMed] [Google Scholar]
- Sugino T, Hasegawa Y, Kikkawa Y, Yamaura J, Yamagishi M, Kurose Y, Kojima M, Kangawa K, Terashima Y. A transient ghrelin surge occurs just before feeding in a scheduled meal-fed sheep. Biochem Biophys Res Commun. 2002a;295:255–260. doi: 10.1016/S0006-291X(02)00654-X. [DOI] [PubMed] [Google Scholar]
- Sugino T, Yamaura J, Yamagishi M, Ogura A, Hayashi R, Kurose Y, Kojima M, Kangawa K, Hasegawa Y, Terashima Y. A transient surge of ghrelin secretion before feeding is modified by different feeding regimens in sheep. Biochem Biophys Res Commun. 2002b;298:785–788. doi: 10.1016/S0006-291X(02)02572-X. [DOI] [PubMed] [Google Scholar]
- Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A. 2004;101:4679–4684. doi: 10.1073/pnas.0305930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed N, Martens CA, Hsu WH. Arginine vasopressin increases glutamate release and intracellular Ca2+ concentration in hippocampal and cortical astrocytes through two distinct receptors. J Neurochem. 2007;103:229–237. doi: 10.1111/j.1471-4159.2007.04737.x. [DOI] [PubMed] [Google Scholar]
- Tachibana T, Kaiya H, Denbow DM, Kangawa K, Furuse M. Central ghrelin acts as an anti-dipsogenic peptide in chicks. Neurosci Lett. 2006;405:241–245. doi: 10.1016/j.neulet.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Tasker JG, Dudek FE. Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J Physiol. 1991;434:271–293. doi: 10.1113/jphysiol.1991.sp018469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellier D, Moos F, Richard P, Stoeckel ME. Evidence for connections between a discrete hypothalamic dorsochiasmatic area and the supraoptic and paraventricular nuclei. Brain Res Bull. 1994;34:261–274. doi: 10.1016/0361-9230(94)90063-9. [DOI] [PubMed] [Google Scholar]
- Theodosis DT, Paut L, Tappaz ML. Immunocytochemical analysis of the GABAergic innervation of oxytocin- and vasopressin-secreting neurons in the rat supraoptic nucleus. Neuroscience. 1986;19:207–222. doi: 10.1016/0306-4522(86)90016-3. [DOI] [PubMed] [Google Scholar]
- Troadec JD, Thirion S, Nicaise G, Lemos JR, Dayanithi G. ATP-evoked increases in [Ca2+]i and peptide release from rat isolated neurohypophysial terminals via a P2X2 purinoceptor. J Physiol. 1998;511:89–103. doi: 10.1111/j.1469-7793.1998.089bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- Ueta Y, Fujihara H, Serino R, Dayanithi G, Ozawa H, Matsuda K, Kawata M, Yamada J, Ueno S, Fukuda A, Murphy D. Transgenic expression of enhanced green fluorescent protein enables direct visualization for physiological studies of vasopressin neurons and isolated nerve terminals of the rat. Endocrinology. 2005;146:406–413. doi: 10.1210/en.2004-0830. [DOI] [PubMed] [Google Scholar]
- Vavra V, Bhattacharya A, Zemkova H. Facilitation of glutamate and GABA release by P2X receptor activation in supraoptic neurons from freshly isolated rat brain slices. Neuroscience. 2011;188:1–12. doi: 10.1016/j.neuroscience.2011.04.067. [DOI] [PubMed] [Google Scholar]
- Vrang N, Larsen PJ, Møller M, Mikkelsen JD. Topographical organization of the rat suprachiasmatic–paraventricular projection. J Comp Neurol. 1995;353:585–603. doi: 10.1002/cne.903530409. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Arvidsson U, Riedl M, Wang J, Buell G, Surprenant A, North RA, Elde R. Differential distribution of two ATP-gated channels (P2X receptors) determined by immunocytochemistry. Proc Natl Acad Sci U S A. 1996;93:8063–8067. doi: 10.1073/pnas.93.15.8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dong L, Cheng Y, Zhao P. Effects of ghrelin on feeding regulation and interdigestive migrating complex in rats. Scand J Gastroenterol. 2007;42:447–453. doi: 10.1080/00365520600979567. [DOI] [PubMed] [Google Scholar]
- Watts AG. Dehydration-associated anorexia: development and rapid reversal. Physiol Behav. 1999;65:871–878. doi: 10.1016/s0031-9384(98)00244-3. [DOI] [PubMed] [Google Scholar]
- Watts AG, Boyle CN. The functional architecture of dehydration-anorexia. Physiol Behav. 2010;100:472–477. doi: 10.1016/j.physbeh.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Sanchez-Watts G, Kelly AB. Distinct patterns of neuropeptide gene expression in the lateral hypothalamic area and arcuate nucleus are associated with dehydration-induced anorexia. J Neurosci. 1999;19:6111–6121. doi: 10.1523/JNEUROSCI.19-14-06111.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke C, Sheriff S, Soleimani M, Amlal H. Vasopressin-independent regulation of collecting duct aquaporin-2 in food deprivation. Kidney Int. 2005;67:201–216. doi: 10.1111/j.1523-1755.2005.00071.x. [DOI] [PubMed] [Google Scholar]
- Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA, Bloom SR. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology. 2000;141:4325–4328. doi: 10.1210/en.141.11.4325. [DOI] [PubMed] [Google Scholar]
- Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, Batterham RL, Taheri S, Stanley SA, Ghatei MA, Bloom SR. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50:2540–2547. doi: 10.2337/diabetes.50.11.2540. [DOI] [PubMed] [Google Scholar]
- Yamazaki RS, Chen Q, Schreiber SS, Brinton RD. Localization of V1a vasopressin receptor mRNA expression in cultured neurons, astroglia, and oligodendroglia of rat cerebral cortex. Brain Res Mol Brain Res. 1997;45:138–140. doi: 10.1016/S0169-328X(96)00285-9. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/S0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zhao L, Brinton RD. Vasopressin-induced cytoplasmic and nuclear calcium signaling in embryonic cortical astrocytes: dynamics of calcium and calcium-dependent kinase translocation. J Neurosci. 2003;23:4228–4239. doi: 10.1523/JNEUROSCI.23-10-04228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]