

Abstract
Myc and Mad family proteins play opposing roles in the control of cell growth and proliferation. We have visualized the subcellular locations of complexes formed by Myc/Max/Mad family proteins using bimolecular fluorescence complementation (BiFC) analysis. Max was recruited to different subnuclear locations by interactions with Myc versus Mad family members. Complexes formed by Max with Mxi1, Mad3, or Mad4 were enriched in nuclear foci, whereas complexes formed with Myc were more uniformly distributed in the nucleoplasm. Mad4 was localized to the cytoplasm when it was expressed separately, and Mad4 was recruited to the nucleus through dimerization with Max. The cytoplasmic localization of Mad4 was determined by a CRM1-dependent nuclear export signal located near the amino terminus. We compared the relative efficiencies of complex formation among Myc, Max, and Mad family proteins in living cells using multicolor BiFC analysis. Max formed heterodimers with the basic helix-loop-helix leucine zipper (bHLHZIP) domain of Myc (bMyc) more efficiently than it formed homodimers. Replacement of two amino acid residues in the leucine zipper of Max reversed the relative efficiencies of homo- and heterodimerization in cells. Surprisingly, Mad3 formed complexes with Max less efficiently than bMyc, whereas Mad4 formed complexes with Max more efficiently than bMyc. The distinct subcellular locations and the differences between the efficiencies of dimerization with Max indicate that Mad3 and Mad4 are likely to modulate transcription activation by Myc at least in part through distinct mechanisms.
The Myc/Max/Mad network of transcription factors regulates many cellular functions, including proliferation, differentiation, and apoptosis. This network comprises the Myc family of nuclear proto-oncoproteins (c-, N-, and L-Myc), Max, and the Mad family of proteins (Mad1, Mxi1, Mad3, Mad4, and Mnt/Rox). These proteins are a subgroup of basic helix-loop-helix leucine zipper (bHLHZIP) family transcription regulators and can form dimers in multiple combinations through interactions mediated by the helix-loop-helix leucine zipper dimerization interface (9, 13, 29). Different members of the Myc/Max/Mad family have distinct biological functions; Myc proteins promote cell proliferation, whereas Mad family proteins limit proliferation (16, 28). Genes encoding Myc family proteins are mutated or deregulated in many types of cancer, whereas Mad family proteins can inhibit cell transformation (25, 42). The balance between Myc and Mad family proteins is an important determinant of cell proliferation.
Myc and Mad family proteins form dimers with Max, and dimerization with Max is essential for the regulatory functions of Myc and Mad family proteins. Members of the Myc and Mad families are generally not able to form homodimers or heterodimers with each other. The opposing functions of Myc and Mad family proteins are thought to be mediated at least in part by competition for dimerization with Max (2, 26). Myc and Mad1 have been shown to compete for complex formation with Max at specific DNA recognition sites (3). However, the relative affinities of different Myc and Mad family proteins for dimerization with Max have not been determined. Moreover, the dimers formed by different combinations of Myc/Max/Mad family proteins have not been directly visualized in living cells.
The structural basis for the difference between the dimerization specificities of Myc and Max has been investigated using a variety of in vitro approaches. The dimerization specificities of Myc and Max are determined primarily by their leucine zippers (1, 9, 30, 32). The X-ray crystal structures of Max-Max, Mad1-Max, and the bHLHZIP domain of Myc-Max dimers have provided insight into the molecular basis for the differences in dimerization specificity (7, 13, 33). The dimerization specificities are determined in part by electrostatic interactions between amino acid residues at the e and g positions in the coiled coil and by hydrophobic interactions between amino acids at the a and d positions (1, 40, 41). It is, however, not known if the determinants of dimerization specificity in vitro also control the dimerization preferences of the proteins in the normal cellular environment.
Different Myc/Max/Mad family proteins exhibit distinct subnuclear distributions (46). Max is relatively uniformly distributed in the nucleoplasm, whereas Myc, Mad1, Mxi1, and Mad3 are enriched in nuclear foci. The regions of Mad1 and Mxi1 required for the enrichment in foci are located in different parts of the two proteins that have no apparent sequence similarity. Coexpression of Max with Myc, Mad1, or Mad3 has different effects on their localization depending on the ratio of expression vectors introduced into the cell (46). When a larger amount of the Max expression vector is transfected, both proteins adopt a relatively uniform distribution. However, when a smaller amount of the Max expression vector is transfected, both proteins are enriched in subnuclear foci. It is not clear whether direct interactions between the proteins cause these changes in protein localization or whether they are indirect consequences of the distinct functional effects of these proteins in the cell.
The mechanisms that regulate dimer formation among Myc/Max/Mad proteins have not been fully characterized. Dimerization is presumably controlled both by the relative levels of protein expression and by the relative efficiencies of dimerization between different family members. Max is stable and ubiquitously expressed, whereas both Myc and Mad family proteins are rapidly degraded and their synthesis is regulated in response to extracellular stimuli. Members of the Myc family are primarily expressed in proliferating cells, whereas Mad family proteins are often expressed in terminally differentiated cells (2, 21, 35). Myc and Mad family proteins are also coexpressed in many cell types (20, 21, 27, 35). Thus, the competition between Myc and Mad family proteins for dimerization with Max may control the balance between cell proliferation and differentiation.
Myc-Max and Mad-Max have been proposed to act as a molecular switch that engages two alternate transcriptional states of the cell (3). Dimers formed between different Myc/Max/Mad family proteins can bind to DNA sequence elements in the promoters of many genes and have opposite effects on their transcriptional activities. Myc-Max can activate many promoters, whereas Mad-Max can repress many of the same promoters (3, 21, 28, 34, 37, 47). Mad1-Max heterodimers have been shown to replace Myc-Max complexes at the hTERT and cyclin D2 promoters during HL-60 cell differentiation (6, 45). Thus, the competition between Myc and Mad family proteins is likely to be an important determinant of promoter occupancy in the cell.
The opposite transcriptional activities of Myc-Max and Mad-Max dimers are determined at least in part through interactions with different cellular cofactors. Myc-Max heterodimers can recruit complexes containing histone acetyltransferases to promoter regions, whereas Mad-Max heterodimers can recruit complexes containing histone deacetylases (6, 17, 24, 31, 39). The competition between Myc and Mad family proteins for dimerization with Max can therefore determine the nature of the multiprotein complexes that are recruited to promoters containing E-box regulatory elements.
Many transcription factors can associate with different structurally related interaction partners using the same contact interface. The competition among alternative interaction partners is likely to be an important determinant of the specificity of these interactions in the cell. The competition among Myc/Max/Mad family proteins for binding to the same DNA recognition element has been investigated in cell extracts (38). Interactions among these proteins have also been investigated by coprecipitation from cell extracts and by reconstitution of complexes using purified recombinant proteins (28, 29, 38). These approaches do not allow determination of the relative efficiencies of complex formation in the cell or the subcellular locations of those complexes. We set out to directly visualize interactions among Myc/Max/Mad family proteins in order to elucidate the subcellular localization and selectivity of dimer formation in living cells.
MATERIALS AND METHODS
Construction of plasmid vectors.
To construct mammalian expression vectors for bimolecular fluorescence complementation (BiFC) analysis, the sequences encoding enhanced yellow fluorescent protein containing amino acids 1 to 172 EYFP(1-172) (the N-terminal fragment of YFP [YN]) and enhanced cyan fluorescent protein containing amino acids 1 to 172 ECFP(1-172) (the C-terminal fragment of CFP [CN]) as well as EYFP(173-238) (YC) and ECFP(155-238) (CC) were cloned into pCMV-HA (Clontech) and pCMV-FLAG2 (Sigma). The sequences encoding human c-Myc1-439 (Myc), Myc318-439 (bMyc), MycΔ407-432 (MycΔZIP), human Max, MaxΔ28-36 (MaxΔBR), MaxQ91A,N92A (Max91,92), human Mxi1, mouse Mad3, and mouse Mad4 (clone MG194125) were fused to the N-terminal ends of YN, CN, YC, CC, YFP, and CFP.
In Mad4 mutants Mad4Δ1-16, Mad4Δ6-10, Mad4Δ6-9, Mad4L6A, Mad4L6,7A, Mad4L6-9A, and Mad4L6-10A, the residues indicated were deleted or replaced by alanines. In the nuclear export signal (NES)-Mad3 chimera, residues 1 to 10 of Mad3 were replaced by residues 1 to 8 of Mad4. In MaxAAALA, the positively charged residues 143 to 145 and 147 of Max were replaced by alanines. The corresponding sequences were then fused to the N-terminal ends of YFP, CFP, YN, and YC.
Cell culture and fluorescence imaging of living cells.
COS-1, NIH 3T3, 293, and U937 cells were maintained under conditions recommended by the American Type Culture Collection. Cells grown in six-well plates to 30 to 50% confluence were transfected with 0.25 to 0.5 μg of the plasmids expressing the proteins indicated in each experiment using Fugene 6. Transfected cells were incubated at 37°C for 24 h and then switched to 30°C for 0 to 24 h to promote fluorophore maturation. The cells were observed by fluorescence microscopy 6 to 48 h posttransfection using a Nikon TE300 inverted fluorescence microscope with a charge-coupled device camera controlled by SimplePCI software (C-Imaging Inc.). YFP fluorescence emission was measured at 535 ± 15 nm during excitation at 500 ± 10 nm (Y filters). CFP fluorescence was measured at 470 ± 15 nm during excitation at 436 ± 5 nm (C filters).
Simultaneous visualization of protein interactions using multicolor fluorescence complementation analysis.
For multicolor BiFC analysis, COS-1 cells were transfected with plasmids encoding three different fusion proteins (e.g., bMycYN, Mad4CN, and MaxCC), which results in the formation of two BiFC complexes (e.g., bMycYN-MaxCC and Mad4CN-MaxCC) with distinct spectral characteristics. As a control, each pair of proteins was also expressed in separate cells. To image the fluorescence emissions of these complexes, we used two filter sets: (i) Y filters with an excitation wavelength of 500 ± 10 nm and an emission wavelength of 535 ± 15 nm that detect YN-CC complexes and (ii) C filters with an excitation wavelength of 436 ± 5 nm and an emission wavelength of 470 ± 15 nm that detect CN-CC complexes. The emission and excitation light was separated using a dichroic mirror with transmission windows for 450- to 490-nm-wavelength and 520- to 590-nm-wavelength light. Data were collected using a Nikon TE300 inverted fluorescence microscope equipped with a Hamamatsu charge-coupled device camera. There was less than 2% overlap between the signals from YN-CC and CN-CC bimolecular fluorescent complexes. The fluorescence intensities of individual cells were quantified using semiautomated feature recognition software (SimplePCI). The background signal in an area with no cells was subtracted from all values. The fluorescence intensity of YN-CC complexes was plotted against the intensity of CN-CC complexes for each cell. Between 100 and 300 cells were quantified for each combination of proteins examined. The level of expression of each fusion protein was quantified by Western blotting using antibodies directed against the FLAG and hemagglutinin (HA) epitopes (Sigma and Santa Cruz Biotechnology). Controls were performed to ensure that differences in fluorescence intensity were not caused by differences in transfection efficiency (using CFP fusions), promoter competition (using empty cytomegalovirus [CMV] expression vectors), or expression levels (measured by Western blotting).
Immunofluorescence.
U937 cells were grown on lysine-coated glass slides and treated with 1.6 × 10−8 M 12-O-tetradecanoylphobol-13-acetate (TPA) for 24 h. The slides were washed with phosphate-buffered saline (PBS), fixed with cold PBS containing 3.7% formaldehyde, permeabilized with PBS containing 3.7% formaldehyde and 0.5% Triton X-100, and incubated with primary rabbit antibody against Mad4 (Santa Cruz Biotechnology) at 4°C overnight. Secondary Alexa594-conjugated anti-rabbit antibody (Molecular Probes) was used to visualize the primary antibody. The cells were imaged using filters with an excitation wavelength of 560 ± 20 nm and an emission wavelength of 690 ± 40 nm.
RESULTS
Visualization of Myc/Max/Mad family dimerization in living cells.
To investigate dimerization among Myc/Max/Mad family proteins in living cells, we tested dimerization by all combinations of these proteins using BiFC analysis. The BiFC approach is based on the formation of a fluorescent complex when two fragments of a fluorescent protein are brought together by an interaction between proteins fused to the fragments (18). Complementary fragments of YFP (YN and YC) were fused to the carboxyl-terminal ends of Myc, Max, Mxi1, Mad3, and Mad4. Plasmids encoding all combinations of the fusion proteins were transfected into COS-1 cells, and the cells were monitored by fluorescence microscopy (Fig. 1). Fluorescence was observed within 6 to 24 h after transfection in cells expressing Max fused to YN or YC together with Myc, Max, Mxi1, Mad3, or Mad4 fused to the complementary fragment. The average fluorescence intensity of the cells was between 3 and 15% of the average fluorescence intensity of cells that expressed the same proteins fused to intact YFP. Identical results were obtained when the two YFP fragments were exchanged between the interaction partners (i.e., Mad3YN-MaxYC and MaxYN-Mad3YC). No fluorescence was detected in cells transfected with plasmids encoding YN fused to Myc, Max, Mxi1, Mad3, or Mad4 and YC lacking a fusion or vice versa. Thus, formation of the bimolecular fluorescent complex required specific dimerization between Myc or Mad proteins with Max. No fluorescence was observed in cells that expressed any combination of Myc, Mxi1, or Mad3 fused to YN and YC, suggesting that these proteins do not form complexes in cells. Max can therefore dimerize with all of the Myc and Mad family proteins tested, whereas Myc and Mad family proteins do not associate with each other in living cells, consistent with the results from studies in vitro and in two-hybrid assays in yeast (9, 11).
FIG. 1.

Visualization of interactions among Myc/Mad/Max family proteins in living cells. (A to E) The proteins indicated in each panel were coexpressed in COS-1 cells, and the fluorescence emissions of the cells were imaged 6 to 24 h after transfection. The images are representative of greater than 90% of the fluorescent cells in each population. (F to J) The Myc/Mad/Max family members indicated in each panel fused to full-length CFP were expressed in COS-1 cells, and the fluorescence emissions of the cells were imaged 12 h after transfection. (K) Mad4 fused to full-length CFP was coexpressed with unmodified Max in COS-1 cells, and fluorescence was visualized 12 h after transfection. (L to O) Max fused to full-length YFP was coexpressed with unmodified Myc (L), Mxi1 (M), Mad3 (N), or Mad4 (O) in COS-1 cells, and the fluorescence emissions of the cells were imaged 12 h after transfection. The diagrams to the right of the images represent the experimental strategies used.
Max is recruited to different subnuclear locations through interactions with different partners.
The subcellular distributions of dimers formed by Max with different Myc/Max/Mad family proteins were distinct. Max homodimers exhibited a reticular pattern, and Myc-Max heterodimers exhibited a granular pattern, both of which were distributed throughout the nucleoplasm (Fig. 1A and B). In contrast, Mxi1-Max, Mad3-Max, and Mad4-Max heterodimers were enriched in distinct nuclear foci (Fig. 1C, D, and E). The number and size of the foci were not affected by the level of protein expression or the time after transfection. The patterns of fluorescence complementation were consistent in more than 90% of the cells in each population. Equal amounts of the plasmids encoding each protein were transfected into the cells, and the bHLHZIP domains were expressed at comparable levels, as determined by Western blot analysis. Similar patterns of fluorescence complementation were observed in three different cell lines (COS-1, 293, and NIH 3T3). Max was therefore recruited to different subnuclear compartments through interactions with different dimerization partners.
We compared the distributions of the dimers with the distributions of the individual proteins fused to full-length YFP or CFP (Fig. 1F to J). Expression of Myc fused to YFP or CFP produced a speckled pattern that was less uniform than that observed for Myc-Max dimers (Fig. 1G). Mxi1 and Mad3 fused to YFP or CFP were enriched in subnuclear foci that were similar in appearance to those observed for the dimers that they formed with Max (Fig. 1H and I). Mad4 fused to CFP was localized to the cytoplasm when expressed separately, whereas Mad4-Max heterodimers were localized to subnuclear foci (Fig. 1J and E). Thus, the subnuclear distributions of Mxi1-Max and Mad3-Max heterodimers were determined primarily by Mxi1 and Mad3. In contrast, the localization of Mad4-Max complexes was determined by the combined effects of Mad4 and Max. We confirmed that the YFP and CFP fusions did not alter the subcellular distributions of the proteins by determining the distributions of the unmodified proteins by indirect immunofluorescence (data not shown).
To determine whether bimolecular complex formation affected dimer localization, we cotransfected equal amounts of a plasmid encoding Max fused to YFP or CFP and plasmids encoding Myc and Mad family proteins lacking fusions. Coexpression of different dimerization partners resulted in localization of Max to different subnuclear compartments in the absence of bimolecular complex formation (Fig. 1L to O). Likewise, coexpression of Max with Mad4CFP resulted in localization of Mad4CFP to nuclear foci (Fig. 1K). Thus, the relocalization of Myc/Max/Mad family proteins upon heterodimer formation did not require bimolecular complex formation.
The formation of Myc/Max/Mad complexes and their subnuclear localization do not require DNA binding.
Studies of the kinetics of DNA binding by Max in vitro suggest that dimerization occurs subsequent to DNA binding by the monomers (23). We examined the effects of mutations in Myc and Max on dimer formation and localization. Dimers formed by the bHLHZIP domain of Myc and Max exhibited a uniform distribution in the nucleoplasm (Fig. 2A). A deletion in the basic region of Max (MaxΔBR) that eliminates DNA binding in vitro (36) had no detectable effect on the efficiency of fluorescence complementation or on the subcellular localization of the bMyc-Max complex (Fig. 2B). This deletion also did not alter the localization of complexes formed by MaxΔBR with Mad family proteins. Thus, DNA binding was not required for dimerization or for the subnuclear localization of Myc/Max/Mad family dimers in living cells. To confirm that fluorescence complementation between Myc and Max reflected a specific interaction between the proteins, we examined the effect of deletion of the leucine zipper on fluorescence complementation. Deletion of the leucine zipper of bMyc eliminated all detectable fluorescence (Fig. 2C). This deletion had no effect on the level of protein expression or its subcellular localization (Fig. 2D; also data not shown). Thus, fluorescence complementation required a specific dimerization interface between the interaction partners as shown previously for the bZIP domains of Fos and Jun (18).
FIG. 2.

Effects of mutations in the basic region and leucine zipper on bimolecular fluorescent complex formation between bMyc and Max. (A to C) The proteins indicated in each panel were coexpressed in COS-1 cells, and the fluorescence emissions of the cells were imaged 24 h after transfection. (D) Western blot analysis of the levels of protein expression. Cells corresponding to panels A (lane 1) and C (lane 2) that expressed the proteins indicated above the lanes were harvested, and the cell extracts were analyzed by Western blotting using anti-FLAG (detects MaxYN) and anti-HA (detects bMycYC and bMycΔZIPYC) antibodies.
To determine whether the nuclear foci occupied by Mxi1-Max, Mad3-Max, and Mad4-Max correspond to previously characterized nuclear structures, we compared their locations with markers for known nuclear structures using indirect immunofluorescence. The Mxi1-Max, Mad3-Max, and Mad4-Max foci did not colocalize with splicing factor speckles, PML bodies, or Cajal bodies (see Fig. S1 in the supplemental material). Therefore, these foci did not correspond to any of these previously characterized nuclear structures.
Mad4 is exported from the nucleus via a CRM1-dependent pathway.
In contrast to the other Myc/Max/Mad family proteins examined here, Mad4 was localized to the cytoplasm when expressed alone (Fig. 1J). To determine whether the cytoplasmic localization was caused by active export of Mad4 from the nucleus, we treated COS-1 cells expressing Mad4CFP with leptomycin B (LMB), an inhibitor of CRM1-dependent nuclear export (14, 43). In untreated cells, Mad4CFP was exclusively cytoplasmic in the majority of cells (Fig. 3A and C). LMB treatment resulted in at least partial nuclear localization of Mad4CFP in virtually all cells (Fig. 3B and C). Similar results were obtained from experiments using 293 cells (data not shown). These results suggest that the localization of Mad4 was controlled by active nuclear export. LMB treatment had no effect on the localization of other Mad family members studied.
FIG.3.

Mapping the sequence determinants of Mad4 localization. Mad4CFP was expressed in COS-1 cells, and the cells were imaged without LMB treatment (A) or after LMB treatment (1-h incubation with 20 ng of LMB per ml) (B). (C) The subcellular distribution of Mad4CFP was quantified in the absence and presence of LMB. N, exclusively nuclear; C, exclusively cytoplasmic; N+C, partially nuclear and cytoplasmic. (D) Alignment of the N-terminal sequences of Mad family proteins and known nuclear export signals. hp120ctn, human p120 catenin; MAPKK, mitogen-activated protein kinase kinase; TFIIIA, transcription factor IIIA; PKI, protein kinase I. (E to H) Mapping of the Mad4 sequences required for nuclear export. The proteins indicated in each panel were expressed in COS-1 cells, and the fluorescence emissions of the cells were imaged 12 h after transfection. (I) Quantification of the subcellular distributions of Mad4 mutants in COS-1 cells. (J to L) Transfer of the Mad4 nuclear export signal to another Mad family protein. The proteins indicated in each panel were expressed in 293 cells, and the fluorescence emissions of the cells were imaged 12 h after transfection. (M) Visualization of endogenous Mad4 in TPA-treated U937 cells by indirect immunofluorescence. (N) Quantification of the subcellular distributions of Mad4CFP, Mad3CFP, and NES-Mad3CFP in 293 cells.
To identify the peptide motif that determined the difference in localization between Mad4 and other Mad family proteins, we compared their primary sequences. The N-terminal sequence of Mad4 differs from those of other Mad family proteins, and resembles previously identified nuclear export signals (Fig. 3D). To examine the role of this sequence in the nuclear export of Mad4, we constructed N-terminal deletion derivatives of Mad4 fused to CFP and examined their localization in COS-1 cells. Deletion of residues 1 to 16 resulted in nuclear localization of Mad4 in a majority of cells (Fig. 3E and I). Replacement of the run of leucines with alanines (Mad4L6-10A and Mad4L6-9A) or deletion of these leucines (Mad4Δ6-10 and Mad4Δ6-9) had a similar effect (Fig. 3F and I). Replacement of two of the leucines (Mad4L6,7A) also resulted in nuclear localization in most cells (Fig. 3G and I), whereas replacement of a single leucine at position 6 (Mad4L6A) caused partial nuclear accumulation (Fig. 3H). Mad4 therefore contained an N-terminal nuclear export signal.
To ascertain whether the nuclear export signal of Mad4 could alter the localization of other Mad family proteins, we replaced the N-terminal sequence of Mad3 with that of Mad4 and examined the localization of the chimeric protein fused to CFP. The Mad4 export signal caused partially cytoplasmic localization of the chimera in a majority of cells and exclusively cytoplasmic localization in a significant subpopulation (Fig. 3L and N). Similar results were obtained from experiments using either COS-1 or 293 cells. The amino-terminal segment of Mad4 was therefore sufficient for nuclear export of other Mad family proteins.
There was no apparent difference in Mad4 localization when Mad4 was expressed at different levels or at different times after transfection. Nevertheless, the localization of the exogenously expressed Mad4 may differ from that of endogenous Mad4. Western blot analysis demonstrated that Mad4 was induced in U937 cells in response to TPA treatment (data not shown). We therefore examined the localization of endogenous Mad4 in TPA-treated U937 cells using indirect immunofluorescence (Fig. 3M). Mad4 was predominantly cytoplasmic in greater than 90% of the cells. The cytoplasmic localization of transiently expressed Mad4CFP therefore reflects the properties of endogenous Mad4 in differentiated U937 cells.
To determine whether the nuclear export signal of Mad4 affected complex formation with Max or the localization of Mad4-Max heterodimers, we examined interactions between the mutated Mad4 derivatives and Max using the BiFC assay. All of the mutated Mad4 derivatives formed bimolecular fluorescent complexes with Max (Fig. 4A to D). The subnuclear distributions of these complexes were more uniform and exhibited nuclear foci in only a minority of cells (Fig. 4C). The amino-terminal segment of Mad4 that contained the nuclear export signal therefore was not required for complex formation with Max but affected the subnuclear distribution of Mad4-Max heterodimers.
FIG. 4.

Effects of the mutations in the nuclear export and import signals of Mad4 and Max on dimer localization. The proteins indicated in each panel were expressed in COS-1 cells, and the fluorescence emissions of the cells were imaged 24 h after transfection.
A nuclear localization signal has been identified in the C-terminal region of Max (22). To determine whether this sequence affected the formation or localization of Mad4-Max heterodimers, we examined the effects of mutating this sequence (RKKLR to AAALA). MaxAAALACFP exhibited a slightly diminished efficiency of nuclear localization, resulting in a low level of cytoplasmic fluorescence (Fig. 4E). Heterodimers formed by MaxAAALAYN with Mad4YC were exclusively cytoplasmic in a majority of cells and at least partially cytoplasmic in all cells (Fig. 4F). We also examined the effects of deleting the basic region of Max on its localization and dimerization with Mad4. Deletion of the basic region reduced the efficiency of nuclear localization of MaxΔBRCFP slightly, resulting in a level of cytoplasmic fluorescence that was comparable to that observed for MaxAAALACFP (Fig. 4G). Nevertheless, heterodimers formed by MaxΔBRYN with Mad4YC were exclusively nuclear in virtually all cells (Fig. 4H). Thus, the RKKLR-to-AAALA mutation selectively altered the distribution of Mad4-Max heterodimers and had little effect on the localization of Max alone. In contrast, deletion of the basic region had a small effect on the localization of Max alone and no detectable effect on the distribution of Mad4-Max heterodimers. These results are consistent with the hypothesis that dimerization with Max does not mask the nuclear export signal of Mad4 but that the nuclear import signals of Max can overcome the nuclear export signal of Mad4 in the Mad4-Max heterodimer.
Simultaneous visualization of multiple Myc/Max/Mad family complexes in the same cell.
The subnuclear distributions of complexes formed by different Myc/Max/Mad family proteins were distinct when expressed in different cells. To investigate whether these complexes affected the localization of each other, we compared their distributions in the same cell. We have developed a multicolor fluorescence complementation approach for the simultaneous visualization of interactions between different proteins in the same cell (19). This multicolor BiFC approach is based on complementation between fragments of different fluorescent proteins (i.e., YFP and CFP) that produce bimolecular complexes with distinct spectral characteristics. The spectral differences between bimolecular complexes formed by fragments of different fluorescent proteins enable visualization of the subcellular sites of interactions between different proteins in the same cell.
To compare the subnuclear locations of complexes formed between different Myc/Max/Mad family proteins in the same cell, we coexpressed each combination of alternative interaction partners (i.e., Max and bMyc) fused to YN and CN (the N-terminal fragment of CFP) together with a shared interaction partner (i.e., Max) fused to CC (the C-terminal fragment of CFP). When MaxYN and bMycCN were coexpressed with MaxCC, the MaxYN-MaxCC and bMycCN-MaxCC complexes exhibited perfect colocalization in the nucleus (Fig. 5A). Identical results were obtained when the fragments of fluorescent proteins were exchanged between bMyc and Max (data not shown). We also compared the distributions of Mad4YN-MaxCC and Mad3CN-MaxCC complexes in the same cell. These complexes exhibited perfect colocalization in nuclear foci of all cells examined (Fig. 5B). Thus, both bMyc-Max and Max-Max as well as Mad3-Max and Mad4-Max complexes colocalized at the resolution of light microscopy when expressed in the same cells.
FIG.5.

Multicolor BiFC analysis of subnuclear sites of interactions between different members of the Myc/Mad/Max family in the same cells. The proteins indicated to the left of each set of images were coexpressed in COS-1 cells. The diagrams to the left of the images describe the experimental strategy. The YN-CC complexes were visualized using 500-nm-wavelength excitation and 535-nm-wavelength emission filters (shown in green), and the CN-CC complexes were visualized using 436-nm-wavelength excitation and 470-nm-wavelength emission filters (shown in red). The images were superimposed to compare the distributions of the complexes in the same cell.
Myc/Max/Mad family proteins can affect the localization of each other independent of dimerization and DNA binding.
The subnuclear distribution of bMyc-Max was markedly different from those of Mad3-Max and Mad4-Max when expressed in different cells (Fig. 1B, D, and E). To determine whether these complexes affected the localization of each other, we examined their distributions when coexpressed in the same cells. bMycYN-MaxCC was relocalized to nuclear foci in cells that coexpressed either Mad3CN-MaxCC or Mad4CN-MaxCC (Fig. 5C and D). In these cells, bMycYN-MaxCC was partially colocalized with Mad3CN-MaxCC or Mad4CN-MaxCC. Complexes formed by Mad3 and Mad4 with Max therefore altered the subnuclear distribution of bMyc-Max complexes.
We looked at whether the effects of Myc/Max/Mad family proteins on the localization of each other were affected by bimolecular fluorescent complex formation. We coexpressed Mad family proteins fused to CFP with bMycYN-MaxCC heterodimers or MaxYN-MaxCC homodimers (see Fig. S2A to D in the supplemental material). Mxi1CFP and Mad3CFP colocalized with both bMycYN-MaxCC as well as MaxYN-MaxCC in a majority of the cells. The coexpression of Mxi1CFP or Mad3CFP relocalized bMycYN-MaxCC to nuclear foci (see Fig. S2A in the supplemental material). In contrast, the coexpression of MaxYN-MaxCC induced a uniform distribution of Mxi1CFP and Mad3CFP in the nucleoplasm (see Fig. S2B in the supplemental material). Mad4CFP coexpression also induced bMycYN-MaxCC localization to nuclear foci in a subset of cells, whereas the coexpression of MaxYN-MaxCC induced a uniform distribution of Mad4CFP in the nucleoplasm (see Fig. S2C and D in the supplemental material). The plasmids encoding all proteins were transfected at equal concentrations. bMycYN-MaxCC was also localized to nuclear foci in cells that expressed unmodified Mad family proteins, whereas CFP expression had no effect on bMycYN-MaxCC localization (data not shown). Mad family proteins that did not form bimolecular fluorescent complexes therefore altered the localization of bMycYN-MaxCC heterodimers and were themselves relocalized in cells that contained MaxYN-MaxCC homodimers.
We examined whether DNA binding or dimerization affected the relocation of Myc in cells that coexpressed Mad family proteins (see Fig. S2E to H in the supplemental material). MaxΔBRYN-MycYC was relocalized to nuclear foci in cells that expressed Mxi1CFP or Mad3CFP, whereas it was distributed throughout the nucleoplasm alone and in cells that expressed Mad4CFP (see Fig. S2E in the supplemental material; also data not shown). MycCFP was relocalized to nuclear foci in cells that expressed Mxi1YFP or Mad3YFP in the absence of exogenously expressed Max (see Fig. S2F in the supplemental material). MycCFP was also relocalized to nuclear foci in a majority of cells that were cotransfected with unmodified Mad3 (data not shown). Deletion of the leucine zipper of Myc did not prevent MycΔZIPCFP relocalization to nuclear foci in cells that coexpressed MxiYFP or Mad3YFP (see Fig. S2G in the supplemental material; also data not shown). Conversely, the coexpression of either Mad3YFP or unmodified Mad3 did not alter the uniform distribution of bMycCFP in the nucleoplasm (see Fig. S2H in the supplemental material). Consequently, the relocalization of Myc in cells that coexpressed Mad3 or Mxi1 did not require either DNA binding or dimerization but did require Myc sequences outside the bHLHZIP region.
To ascertain the specificity of Myc recruitment to nuclear foci in cells that coexpressed Mad family proteins, we examined the effects of Mxi1 and Mad3 on bFos-bJun heterodimer localization. Coexpression of Mxi1CFP or Mad3CFP had no detectable effect on bFosYN-bJunYC localization (data not shown). Thus, Myc, Max, and Mad family proteins specifically affected the localization of each other in the cell.
Analysis of the relative efficiencies of complex formation using multicolor BiFC.
The relative amounts of dimers formed by different combinations of Myc/Max/Mad family proteins are determined by the local concentrations of the proteins and the relative dimerization efficiencies between different members of the family. We quantified the relative efficiencies of complex formation between different family members by visualizing the competition between alternative interaction partners using multicolor BiFC analysis (Fig. 6) (19). Two alternative interaction partners fused to fragments of different fluorescent proteins (e.g., bMycYN and MaxCN) were coexpressed with a limiting amount of a shared interaction partner fused to a complementary fragment (e.g., MaxCC). The cells were imaged using filters that distinguish the fluorescence of YN-CC and CN-CC complexes. The fluorescence intensities produced by these complexes in each of more than 100 individual cells were plotted. The slope of this plot reflects the relative efficiencies of complex formation by the alternative interaction partners. Comparison of the slopes for different combinations of proteins can provide information about their dimerization preferences in living cells (Fig. 7).
FIG. 6.

Quantitation of the relative efficiencies of complex formation using multicolor BiFC analysis. Cells that express two alternative interaction partners (i.e., bMycYN and MaxCN) together with a limiting concentration of a shared partner (i.e., MaxCC) were imaged using filters that distinguish emissions from the two bimolecular fluorescent complexes (i.e., bMycYN-MaxCC shown in green and MaxCN-MaxCC shown in red). The fluorescence intensities of the complexes were measured in >100 cells, and the bMycYN-MaxCC intensities were plotted as a function of MaxCN-MaxCC intensities.
FIG. 7.

Visualization of the competition for dimerization by alternative interaction partners in living cells. (A to G) The proteins indicated above each graph were coexpressed in COS-1 cells in pairs (green and red) and in three-way competition (yellow). The fluorescence emissions corresponding to the spectrally distinct YN-CC and CN-CC complexes were measured in each cell and were plotted for 100 to 250 cells for each combination of proteins. The line shows the linear regression fit to the fluorescence intensities of cells that expressed all three proteins. The 95% confidence intervals for the regression lines are shown as dotted lines. The slope of the linear regression reflects the relative dimerization preferences of the alternative interaction partners used in each experiment. (H) Western blot analysis of the levels of protein expression in the cells whose fluorescence is shown in panels F and G. Plasmids encoding Mad4CN and MaxCC (lane 1), Mad3CN and MaxCC (lane 2), bMycYN and MaxCC (lane 3), Mad4CN, bMycYN, and MaxCC (lane 4), or Mad3CN, bMycYN, and MaxCC (lane 5) were cotransfected into COS-1 cells. Cell lysates were analyzed by Western blot analysis using antibodies that recognize the FLAG (detects bMycYN, Mad3CN, and Mad4CN) or HA (detects MaxCC) epitope (α-FLAG and α-HA, respectively). The lysates were obtained from the same cells that were used to measure the fluorescence intensities shown in panels F and G.
Multicolor BiFC analysis allows comparison of the efficiencies of complex formation between alternative interaction partners, providing that the fragments of the fluorescent proteins do not alter the selectivity of protein interactions. To establish a reference that would allow normalization for any effect of the fluorescent protein fragments on the relative efficiencies of complex formation, we measured the fluorescence intensities of bimolecular fluorescent complexes formed by fragments of different fluorescent proteins fused to the same interaction partners. We compared the fluorescence intensities of complexes formed by MaxYN versus MaxCN with MaxCC when they were expressed separately and when they were expressed in the same cells (Fig. 7A). The average fluorescence intensities of MaxYN-MaxCC and MaxCN-MaxCC complexes were reduced by about 50% when they were expressed together in the same cells compared to their fluorescence intensities when expressed separately. This is consistent with equal efficiencies of competition by MaxYN and MaxCN for dimerization with the limiting amount of MaxCC in each cell. The fluorescence intensities of MaxYN-MaxCC and MaxCN-MaxCC in individual cells exhibited a linear relationship (slope = 0.98). This relationship provides a calibration standard for determination of the relative efficiencies of dimerization with different interaction partners. The deviation from perfect linearity (95% confidence interval = 0.05) likely reflects variation in the relative expression levels of the alternative interaction partners in individual cells and provides a measure of the uncertainty of the result.
Max favors heterodimerization with bMyc over homodimerization in cells.
We compared the efficiencies of Max-Max homodimer and bMyc-Max heterodimer formation by cotransfection of equal amounts of plasmids encoding bMycYN and MaxCN with a limiting amount of the plasmid encoding MaxCC into COS-1 cells (Fig. 7B). When bMycYN-MaxCC and MaxCN-MaxCC were expressed separately, they produced relative fluorescence intensities similar to those observed for MaxYN-MaxCC and MaxCN-MaxCC expressed in separate cells (compare the fluorescence intensities produced by the pairs of proteins shown in green and red in Fig. 7B and A). When bMycYN and MaxCN were coexpressed with MaxCC, the relationship between the fluorescence intensities produced by bMycYN-MaxCC and MaxCN-MaxCC was significantly different (slope = 1.3; 95% confidence interval = 0.08) from that observed for MaxYN-MaxCC and MaxCN-MaxCC (compare the fluorescence intensities produced by the three-way competitions shown in yellow in Fig. 7B and A). Since the fluorescence intensities of the complexes were not significantly different when they were expressed separately, the total amount of complexes formed was likely not affected by the dimerization efficiencies. In contrast, the difference between their fluorescence intensities when they were expressed together was likely due to unequal efficiencies of competition for the limiting amount of the shared interaction partner. Thus, bMycYN competes more efficiently than MaxCN for complex formation with MaxCC in living cells.
To determine whether the difference between bMycYN and MaxCN dimerization with MaxCC was caused by the difference between the fluorescent protein fragments (YN versus CN), we examined the effect of exchanging these fragments between bMyc and Max. The MaxYN and bMycCN proteins were expressed both separately and in the same cells together with a limiting amount of MaxCC (Fig. 7C). When expressed separately, the relative fluorescence intensities of MaxYN-MaxCC and bMycCN-MaxCC were comparable to those observed for MaxYN-MaxCC and MaxCN-MaxCC. When MaxYN and bMycCN were expressed in the same cells with a limiting concentration of MaxCC, the relationship between the fluorescence intensities produced by MaxYN-MaxCC and bMycCN-MaxCC was significantly different (slope = 0.7; 95% confidence interval = 0.04) from that observed for MaxYN-MaxCC and MaxCN-MaxCC (compare Fig. 7C and A). The shift in the slope of the relationship between YN-CC and CN-CC fluorescence intensities was opposite for bMycYN-MaxCC and MaxCN-MaxCC versus MaxYN-MaxCC and bMycCN-MaxCC.
The similar fluorescence intensities produced by bMycCN-MaxCC and MaxCN-MaxCC when expressed separately suggest that the proteins were expressed at similar levels. To confirm that the levels of protein expression were similar, we compared their expression by Western blot analysis using antibodies that recognize the same epitope on both proteins. The levels of bMycCN and MaxCN expression were within 5% of each other relative to the constant levels of MaxYN and MaxCC expression in these cells (data not shown). Thus, Max favored heterodimerization with bMyc over homodimerization in living cells, regardless of which fluorescent protein fragments were fused to the interaction partners. The fluorescence intensities of complexes formed by full-length Myc were much lower that those of complexes formed by the bHLHZIP domain, likely reflecting the lower level of expression of full-length Myc. It was therefore not possible to compare the efficiencies of dimerization using full-length Myc.
Mutations in the leucine zipper alter the relative efficiencies of bMyc-Max heterodimer and Max homodimer formation in cells.
The leucine zipper has been proposed to be the main determinant of the dimerization preferences of Myc/Max/Mad family proteins on the basis of the results of biophysical studies and X-ray crystallographic analysis (7, 13, 32, 33). We investigated the effects of mutations in the leucine zipper of Max (Q91A and N92A) that are predicted to alter dimerization selectivity (33) on the relative efficiencies of Myc-Max and Max-Max dimerization in cells. First, we examined the effects of these mutations on the relative efficiencies of fluorescence complementation by YN-CC and CN-CC fused to Max homodimers (Fig. 7D). The relationship between the fluorescence intensities of MaxYN-Max91,92CC and MaxCN-Max91,92CC expressed in the same cells (slope = 0.98; 95% confidence interval = 0.06) was similar to that observed for the wild-type proteins (compare Fig. 7D and A). Therefore, the mutations did not alter the relative efficiencies of bimolecular complex formation by different fluorescent protein fragments fused to the same interaction partners. Next, we examined whether the mutations affected the relative efficiencies of Max homodimer formation and heterodimerization with bMyc (Fig. 7E). The relationship between the fluorescence intensities of bMycYN-Max91,92CC and MaxCN-Max91,92CC expressed in the same cells (slope = 0.5; 95% confidence interval = 0.05) was significantly different from the relationship observed for bMycYN-MaxCC and MaxCN-MaxCC (compare Fig. 7E and B). Thus, the Q91A and N92A mutations in Max significantly reduced the efficiency of bMyc-Max heterodimer formation relative to Max homodimer formation in living cells.
bMyc outcompetes Mad3, but Mad4 outcompetes bMyc for dimerization with Max.
Mad family proteins are thought to counteract the effects of Myc in cells in large part by competition for dimerization with Max. We examined the relative efficiencies of Max dimerization with Myc, Mad3, and Mad4. When expressed in separate cells, bMycYN-MaxCC and Mad3CN-MaxCC produced fluorescence intensities that were comparable to those observed for bMycYN-MaxCC and MaxCN-MaxCC. However, when expressed in the same cells, the average fluorescence intensity of bMycYN-MaxCC was virtually unchanged, whereas the fluorescence intensity of Mad3CN-MaxCC was markedly reduced (by 78%). The relationship between the fluorescence intensities of bMycYN-MaxCC and Mad3CN-MaxCC expressed in the same cells (slope = 3; 95% confidence interval = 0.5) was significantly different from that observed for bMycYN-MaxCC and MaxCN-MaxCC (compare Fig. 7F and B). Similar results were obtained when we compared the relative efficiencies of Mad3CN-MaxCC heterodimer and MaxYN-MaxCC homodimer formation (data not shown). Mad3 therefore competed less efficiently for dimerization with Max than bMyc or Max did.
We also compared the efficiencies of Mad4CN-MaxCC and bMycYN-MaxCC heterodimerization (Fig. 7G). When expressed in separate cells, bMycYN-MaxCC and Mad4CN-MaxCC produced relative fluorescence intensities that were comparable to those observed for bMycYN-MaxCC and MaxCN-MaxCC. In contrast to the competition between bMycYN and Mad3CN, the average fluorescence intensity of bMycYN-MaxCC was markedly reduced (by 67%) whereas that of Mad4CN-MaxCC was enhanced (by 118%) when the complexes were expressed in the same cells. The relationship between the fluorescence intensities of bMycYN-MaxCC and Mad4CN-MaxCC (slope = 0.14; 95% confidence interval = 0.06) was shifted in the direction opposite that observed for bMycYN-MaxCC and Mad3CN-MaxCC (compare Fig. 7G and F). Therefore, Mad4 competed more efficiently for dimerization with Max than bMyc did.
The fluorescence intensities of complexes formed by Mad3CN-MaxCC and Mad4CN-MaxCC were comparable to those observed for MaxCN-MaxCC and bMycCN-MaxCC when expressed separately, suggesting that the proteins were expressed at comparable levels. To compare the levels of these proteins when they were expressed in the same cells, we examined their expression by Western blot analysis using antibodies that recognize the same epitope in each of the proteins (Fig. 7H). The levels of Mad3CN and Mad4CN expression were comparable to that of bMycYN expressed in the same cells and were not affected by bMycYN coexpression. Thus, Mad4 forms complexes with Max more efficiently than bMyc, whereas Mad3 forms complexes with Max less efficiently than bMyc in living cells.
DISCUSSION
The combinatorial regulation of transcription requires that individual transcription factors have many alternative interaction partners and that they selectively interact with different proteins in different cell types and in response to different extracellular stimuli. Interactions with alternative partners can influence the target gene specificities and transcriptional activities of individual transcription factors. One characteristic that reflects the functions of transcription factor complexes is their subcellular localization. Determination of the subcellular locations of complexes formed with alternative interaction partners can therefore provide insight into the effects of interactions with alternative partners on transcription factor function.
Interactions among Myc/Max/Mad family proteins regulate cellular responses to signals that either stimulate or inhibit cell proliferation. We found that Max was recruited to different subnuclear locations by interactions with Myc versus Mad family members. The distinct subnuclear distributions of dimers formed by Max with Myc versus Mad family proteins did not require DNA binding by the complexes. Myc, Max, and Mad family proteins also affected the subnuclear distributions of each other through mechanisms that did not require dimerization. Myc and Max have been shown to form heterotetramers in vitro (7, 8, 33). However, no BiFC was observed by any combination of Myc, Mxi1, or Mad3 fusions to YN and YC in the presence or absence of unmodified Max (data not shown). Moreover, the relocalization of Myc by Mad family proteins did not require the leucine zipper but did require sequences outside the bHLHZIP domain of Myc. It is therefore likely that interactions with other cellular proteins also contribute to the effects of Myc, Max, and Mad family proteins on the localization of each other.
The subcellular localization of transcription factors can regulate their transcriptional activities (12, 15, 44). Most of the Myc and Mad family proteins examined here were nuclear, and the dimers they formed with Max exhibited distributions that were similar to those observed when the proteins when expressed alone. In contrast, Mad4 was localized to the cytoplasm when it was expressed alone, and Mad4 was recruited to the nucleus by dimerization with Max. Endogenous Mad4 was localized to the cytoplasm in TPA-treated U937 cells. We have been unable to detect the other endogenous Mad family proteins using indirect immunofluorescence in any of the cell lines that we have examined. It is therefore possible that the localization that we observe using transiently expressed proteins differs from that of the endogenous proteins.
The cytoplasmic localization of Mad4 in COS-1 cells was determined by CRM1-dependent export from the nucleus. The Mlx and MondoA bHLHZIP proteins are also exported from the nucleus via CRM1-dependent pathways (5). The nuclear export signal of Mad4 was located near the amino terminus in a region that is not conserved in other Mad family proteins. This signal is immediately adjacent to a sequence that mediates interactions with the Sin3 transcriptional corepressor that is conserved in all Mad family proteins (10). It is therefore possible that interactions of Mad4 with CRM1 and Sin3 affect each other. This provides a potential mechanism for coordination of the regulation of transcriptional repression and nuclear export.
The opposite transcriptional activities and effects on cell growth of Myc versus Mad family transcription factors suggest that they counteract the effects of each other by competition for shared interaction partners in the cell. One such shared interaction partner is the Max protein, which can form heterodimers with both families, and is necessary for DNA binding and transcription regulation by all family members examined. The multicolor BiFC approach enables analysis of the competition between alternative interaction partners in living cells (19). The relative efficiencies of complex formation can be determined by comparing the relative fluorescence intensities of spectrally distinct bimolecular fluorescent complexes formed through interactions between alternative partners in the same cells. Comparison of the relative efficiencies of complex formation between bMyc-Max and Max-Max demonstrated that Max forms heterodimers with bMyc more efficiently than homodimers (Fig. 8). This is consistent with the higher stability of bMyc-Max heterodimers than Max homodimers in vitro (32). Moreover, mutation of residues 91 and 92 in the leucine zipper of Max (Q91R and N92R) affected the relative efficiencies of bMyc-Max heterodimer and Max homodimer formation in vitro (33). Replacing the same residues with alanines (Q91A and N92A) also altered the relative efficiencies of complex formation by bMyc-Max heterodimers and Max homodimers in cells (Fig. 7). These results suggest that the dimerization preferences of bMyc and Max determined in vitro apply also in the normal cellular environment. These observations also corroborate the validity of the multicolor BiFC assay for the measurement of the relative dimerization preferences among alternative interaction partners in living cells.
FIG. 8.
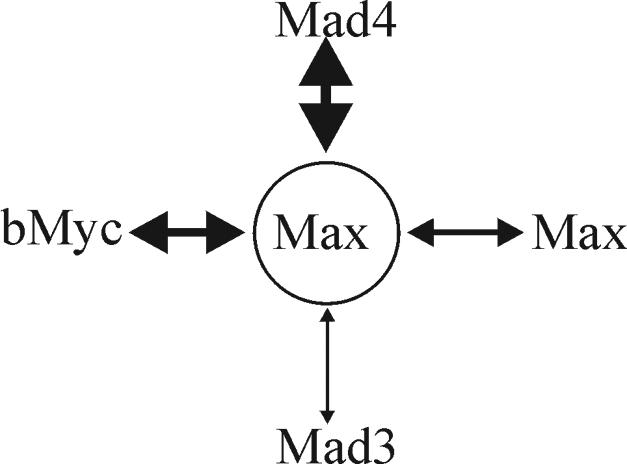
Schematic diagram representing the relative efficiencies of Max dimerization with Mad4, bMyc, Max, and Mad3 in living cells. The thickness of the double-headed arrow connecting each pair of proteins indicates their relative efficiency of complex formation.
Myc and Mad family proteins are thought to counteract the effects of each other at least in part through competition for dimerization with Max (Fig. 8). In living cells, Mad3 competed less efficiently for heterodimer formation with Max than bMyc did. In contrast, Mad4 competed more efficiently for heterodimerization with Max than bMyc did. Mad3 is therefore predicted to be a less efficient repressor than Mad4 if the efficiency of repression is determined solely by the efficiency of competition for dimerization with Max. Alternatively, repression by Mad3 may be mediated by other mechanisms, such as the relocalization of Myc-Max heterodimers.
The marked difference between the efficiencies of Mad3-Max and Mad4-Max dimerization in living cells is likely to be due to a difference between the leucine zippers of the proteins. Two amino acid residues in the leucines zippers of Myc/Max/Mad family proteins have been proposed to be critical for dimerization specificity on the basis of results of X-ray crystallographic analysis (33). One of these amino acid residues is conserved between Mad4 (Glu122) and other Mad family proteins but is replaced by an amino acid residue of opposite charge (Lys126) in Mad3. This substitution may be responsible for the lower efficiency of Mad3-Max dimerization compared with Mad4-Max in the BiFC assay. The leucine zippers of human, rat, and mouse Mad3 contain either arginine or lysine at this position, suggesting that the difference between the efficiencies of Mad3 and Mad4 dimerization with Max is conserved among mammalian Mad family proteins.
Bimolecular fluorescent complex formation is essentially irreversible in vitro (18). Thus, the BiFC assay does not reflect a true equilibrium among the interaction partners. Nevertheless, unmodified interaction partners can compete with bimolecular fluorescent complex formation at equimolar concentrations in vitro (18). The bZIP domains of Fos and Jun fused to fragments of different fluorescent proteins formed dimers with identical efficiencies in vitro (19). It is therefore likely that the relative efficiencies of complex formation in the multicolor BiFC assay reflect the relative efficiencies of interactions between the unmodified proteins in living cells.
The quantitative visualization of protein interactions in living cells provides the potential for the identification of modulators of those interactions in the normal cellular environment. Factors that can modulate the relative efficiencies of Myc-Max and Mad-Max interactions could provide leads for the development of pharmacological agents for anticancer therapy. Previous studies using the fluorescence resonance energy transfer assay in vitro have shown that small-molecule antagonists of Myc-Max heterodimerization can be identified by using fluorescence-based assays (4). The multicolor BiFC assay provides the potential for the development of high-throughput screens for agents that alter interactions between specific proteins in the normal cellular context. Moreover, this approach allows screening for agents that either alter the selectivity of interactions in cells or selectively affect only one of several interactions. These characteristics are likely to make the multicolor BiFC assay a powerful approach for the identification of both natural and artificial modulators of protein interactions in living cells.
Supplementary Material
Acknowledgments
We are grateful to Yurii Chinenov, Lars Larsson, Barry Wolf, Daniel Wechsler, Don Ayer, Chi Dang, Peter Vogt, and Bob Eisenman for helpful discussions and constructive criticisms of the manuscript. We thank Martin Thompson for assistance with the construction of Myc and Mad family fusion proteins. We thank Daniel Wechsler (University of Michigan) for plasmids encoding Myc, Max, Mxi1, Mad3, and Mad4.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Amati, B., M. W. Brooks, N. Levy, T. D. Littlewood, G. I. Evan, and H. Land. 1993. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 72:233-245. [DOI] [PubMed] [Google Scholar]
- 2.Ayer, D. E., and R. N. Eisenman. 1993. A switch from Myc:Max to Mad:Max heterocomplexes accompanies monocyte/macrophage differentiation. Genes Dev. 7:2110-2119. [DOI] [PubMed] [Google Scholar]
- 3.Ayer, D. E., L. Kretzner, and R. N. Eisenman. 1993. Mad: a heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell 72:211-222. [DOI] [PubMed] [Google Scholar]
- 4.Berg, T., S. B. Cohen, J. Desharnais, C. Sonderegger, D. J. Maslyar, J. Goldberg, D. L. Boger, and P. K. Vogt. 2002. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 99:3830-3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Billin, A. N., A. L. Eilers, K. L. Coulter, J. S. Logan, and D. E. Ayer. 2000. MondoA, a novel basic helix-loop-helix-leucine zipper transcriptional activator that constitutes a positive branch of a Max-like network. Mol. Cell. Biol. 20:8845-8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouchard, C., O. Dittrich, A. Kiermaier, K. Dohmann, A. Menkel, M. Eilers, and B. Luscher. 2001. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 15:2042-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brownlie, P., T. Ceska, M. Lamers, C. Romier, G. Stier, H. Teo, and D. Suck. 1997. The crystal structure of an intact human Max-DNA complex: new insights into mechanisms of transcriptional control. Structure 5:509-520. [DOI] [PubMed] [Google Scholar]
- 8.Dang, C. V., M. McGuire, M. Buckmire, and W. M. Lee. 1989. Involvement of the ′leucine zipper' region in the oligomerization and transforming activity of human c-myc protein. Nature 337:664-666. [DOI] [PubMed] [Google Scholar]
- 9.Davis, L. J., and T. D. Halazonetis. 1993. Both the helix-loop-helix and the leucine zipper motifs of c-Myc contribute to its dimerization specificity with Max. Oncogene 8:125-132. [PubMed] [Google Scholar]
- 10.Eilers, A. L., A. N. Billin, J. Liu, and D. E. Ayer. 1999. A 13-amino acid amphipathic alpha-helix is required for the functional interaction between the transcriptional repressor Mad1 and mSin3A. J. Biol. Chem. 274:32750-32756. [DOI] [PubMed] [Google Scholar]
- 11.Estojak, J., R. Brent, and E. A. Golemis. 1995. Correlation of two-hybrid affinity data with in vitro measurements. Mol. Cell. Biol. 15:5820-5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabbro, M., and B. R. Henderson. 2003. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp. Cell Res. 282:59-69. [DOI] [PubMed] [Google Scholar]
- 13.Ferre-D'Amare, A. R., G. C. Prendergast, E. B. Ziff, and S. K. Burley. 1993. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 363:38-45. [DOI] [PubMed] [Google Scholar]
- 14.Fornerod, M., M. Ohno, M. Yoshida, and I. W. Mattaj. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051-1060. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-κB puzzle. Cell 109(Suppl.):S81-S96. [DOI] [PubMed] [Google Scholar]
- 16.Grandori, C., S. M. Cowley, L. P. James, and R. N. Eisenman. 2000. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16:653-699. [DOI] [PubMed] [Google Scholar]
- 17.Hassig, C. A., T. C. Fleischer, A. N. Billin, S. L. Schreiber, and D. E. Ayer. 1997. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 89:341-347. [DOI] [PubMed] [Google Scholar]
- 18.Hu, C. D., Y. Chinenov, and T. K. Kerppola. 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9:789-798. [DOI] [PubMed] [Google Scholar]
- 19.Hu, C. D., and T. K. Kerppola. 2003. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21:539-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hurlin, P. J., K. P. Foley, D. E. Ayer, R. N. Eisenman, D. Hanahan, and J. M. Arbeit. 1995. Regulation of Myc and Mad during epidermal differentiation and HPV-associated tumorigenesis. Oncogene 11:2487-2501. [PubMed] [Google Scholar]
- 21.Hurlin, P. J., C. Queva, P. J. Koskinen, E. Steingrimsson, D. E. Ayer, N. G. Copeland, N. A. Jenkins, and R. N. Eisenman. 1995. Mad3 and Mad4: novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 14:5646-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato, G. J., W. M. Lee, L. L. Chen, and C. V. Dang. 1992. Max: functional domains and interaction with c-Myc. Genes Dev. 6:81-92. [DOI] [PubMed] [Google Scholar]
- 23.Kohler, J. J., S. J. Metallo, T. L. Schneider, and A. Schepartz. 1999. DNA specificity enhanced by sequential binding of protein monomers. Proc. Natl. Acad. Sci. USA 96:11735-11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laherty, C. D., W. M. Yang, J. M. Sun, J. R. Davie, E. Seto, and R. N. Eisenman. 1997. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 89:349-356. [DOI] [PubMed] [Google Scholar]
- 25.Lahoz, E. G., L. Xu, N. Schreiber-Agus, and R. A. DePinho. 1994. Suppression of Myc, but not E1a, transformation activity by Max-associated proteins, Mad and Mxi1. Proc. Natl. Acad. Sci. USA 91:5503-5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larsson, L. G., M. Pettersson, F. Oberg, K. Nilsson, and B. Luscher. 1994. Expression of mad, mxi1, max and c-myc during induced differentiation of hematopoietic cells: opposite regulation of mad and c-myc. Oncogene 9:1247-1252. [PubMed] [Google Scholar]
- 27.Luo, Q., E. Harmon, B. G. Timms, and L. Kretzner. 2001. Novel expression patterns of the myc/max/mad transcription factor network in developing murine prostate gland. J. Urol. 166:1071-1077. [PubMed] [Google Scholar]
- 28.Luscher, B. 2001. Function and regulation of the transcription factors of the Myc/Max/Mad network. Gene 277:1-14. [DOI] [PubMed] [Google Scholar]
- 29.Luscher, B., and L. G. Larsson. 1999. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation. Oncogene 18:2955-2966. [DOI] [PubMed] [Google Scholar]
- 30.Marchetti, A., M. Abril-Marti, B. Illi, G. Cesareni, and S. Nasi. 1995. Analysis of the Myc and Max interaction specificity with lambda repressor-HLH domain fusions. J. Mol. Biol. 248:541-550. [DOI] [PubMed] [Google Scholar]
- 31.McMahon, S. B., M. A. Wood, and M. D. Cole. 2000. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol. Cell. Biol. 20:556-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muhle-Goll, C., M. Nilges, and A. Pastore. 1995. The leucine zippers of the HLH-LZ proteins Max and c-Myc preferentially form heterodimers. Biochemistry 34:13554-13564. [DOI] [PubMed] [Google Scholar]
- 33.Nair, S. K., and S. K. Burley. 2003. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112:193-205. [DOI] [PubMed] [Google Scholar]
- 34.O'Hagan, R. C., N. Schreiber-Agus, K. Chen, G. David, J. A. Engelman, R. Schwab, L. Alland, C. Thomson, D. R. Ronning, J. C. Sacchettini, P. Meltzer, and R. A. DePinho. 2000. Gene-target recognition among members of the myc superfamily and implications for oncogenesis. Nat. Genet. 24:113-119. [DOI] [PubMed] [Google Scholar]
- 35.Queva, C., P. J. Hurlin, K. P. Foley, and R. N. Eisenman. 1998. Sequential expression of the MAD family of transcriptional repressors during differentiation and development. Oncogene 16:967-977. [DOI] [PubMed] [Google Scholar]
- 36.Reddy, C. D., P. Dasgupta, P. Saikumar, H. Dudek, F. J. Rauscher III, and E. P. Reddy. 1992. Mutational analysis of Max: role of basic, helix-loop-helix/leucine zipper domains in DNA binding, dimerization and regulation of Myc-mediated transcriptional activation. Oncogene 7:2085-2092. [PubMed] [Google Scholar]
- 37.Schreiber-Agus, N., L. Chin, K. Chen, R. Torres, G. Rao, P. Guida, A. I. Skoultchi, and R. A. DePinho. 1995. An amino-terminal domain of Mxi1 mediates anti-Myc oncogenic activity and interacts with a homolog of the yeast transcriptional repressor SIN3. Cell 80:777-786. [DOI] [PubMed] [Google Scholar]
- 38.Sommer, A., K. Bousset, E. Kremmer, M. Austen, and B. Luscher. 1998. Identification and characterization of specific DNA-binding complexes containing members of the Myc/Max/Mad network of transcriptional regulators. J. Biol. Chem. 273:6632-6642. [DOI] [PubMed] [Google Scholar]
- 39.Sommer, A., S. Hilfenhaus, A. Menkel, E. Kremmer, C. Seiser, P. Loidl, and B. Luscher. 1997. Cell growth inhibition by the Mad/Max complex through recruitment of histone deacetylase activity. Curr. Biol. 7:357-365. [DOI] [PubMed] [Google Scholar]
- 40.Soucek, L., M. Helmer-Citterich, A. Sacco, R. Jucker, G. Cesareni, and S. Nasi. 1998. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 17:2463-2472. [DOI] [PubMed] [Google Scholar]
- 41.Tchan, M. C., K. J. Choy, J. P. Mackay, A. T. Lyons, N. P. Bains, and A. S. Weiss. 2000. Interfacial asparagine residues within an amide tetrad contribute to Max helix-loop-helix leucine zipper homodimer stability. J. Biol. Chem. 275:37454-37461. [DOI] [PubMed] [Google Scholar]
- 42.Wechsler, D. S., C. A. Shelly, C. A. Petroff, and C. V. Dang. 1997. MXI1, a putative tumor suppressor gene, suppresses growth of human glioblastoma cells. Cancer Res. 57:4905-4912. [PubMed] [Google Scholar]
- 43.Wolff, B., J. J. Sanglier, and Y. Wang. 1997. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4:139-147. [DOI] [PubMed] [Google Scholar]
- 44.Xiao, Z., R. Latek, and H. F. Lodish. 2003. An extended bipartite nuclear localization signal in Smad4 is required for its nuclear import and transcriptional activity. Oncogene 22:1057-1069. [DOI] [PubMed] [Google Scholar]
- 45.Xu, D., N. Popov, M. Hou, Q. Wang, M. Bjorkholm, A. Gruber, A. R. Menkel, and M. Henriksson. 2001. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc. Natl. Acad. Sci. USA 98:3826-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin, X., M. F. Landay, W. Han, E. S. Levitan, S. C. Watkins, R. M. Levenson, D. L. Farkas, and E. V. Prochownik. 2001. Dynamic in vivo interactions among Myc network members. Oncogene 20:4650-4664. [DOI] [PubMed] [Google Scholar]
- 47.Zervos, A. S., J. Gyuris, and R. Brent. 1993. Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell 72:223-232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}