

Abstract
Genes involved in human growth consist of major growth genes and minor growth genes. Major growth genes have fundamental effects on human growth, and their mutations cause growth failure (or overgrowth) which are recognizable as single gene disorders. Minor growth genes exert relative minor additive effects on human growth, and their combination is involved in the development of short (or tall) stature as a multifactorial trait. This review summarizes the current knowledge about the major and the minor growth genes, and refers to the recent molecular approach of identification of the growth genes.
Keywords: genetics, major growth gene, minor growth gene, mutation, susceptibility
Introduction
Growth is a representative quantitative character subject to both genetic and environmental factors. Of these, genetic factors consist of major and minor growth genes (1,2,3). The differentiation between the major and the minor growth genes are conceptual, and there has been no authorized definition. Theoretically, however, the discrimination can be based on the “penetrance”.
Penetrance is defined as the prevalence of abnormal statural growth (e.g., short stature) in patients with a particular genetic variation. For example, if the variance (or standard deviation, SD) of growth remains constant, a genetic variation reducing the stature by 1 SD causes short stature (<–2 SD) in roughly 16% of affected individuals; thus, the penetrance is 16% for short stature (Fig. 1). In this example, only individuals with an original genetic potential below –1 SD show short stature (<–2 SD) in the presence of the genetic variation. This has a significant effect, however. Since short stature (<–2 SD) is exhibited by about 2.3% of individuals in the general population, a genetic variation reducing the stature by 1 SD increases the frequency of short stature with a relative risk of about 7 (16% vs. 2.3%). In this context, a genetic variation reducing the stature by >4 SD leads to short stature in nearly all the affected individuals irrespective of the original height potential. Such a variant, e.g. the FGFR3 mutation leading to achondroplasia (4), can be viewed as a major growth gene with nearly complete penetrance.
Fig. 1.

The proportion of short stature (<–2 SD) in patient populations with gene variations reducing the height by 1 SD, 2 SD, 3 SD, and 4 SD. Provided that the height distribution remains constant, the prevalence of short stature should become the values shown in this figure.
Thus, the major and the minor growth genes are defined as genes whose variations are associated with high and low penetrance, respectively. The former is regarded as a mutation, and the latter as a susceptibility allele. In other words, a gene variation which results in a pathologic situation recognizable as a single gene disorder can be recognized as a major growth gene, and a gene variation which raises the susceptibility to short stature as a multifactorial trait can be regarded as a minor growth gene. Empirically, a genetic variation influencing stature by more than 1–2 SD may cause a pathologic condition that is discernible as a single gene disorder. In this review article, I will summarize the current knowledge regarding the major and the minor growth genes.
Major Growth Gene
Major growth genes exert a fundamental effect on human growth (1), and include the genes for endocrine, skeletal, and malformation disorders. They are relevant to growth failure (or overgrowth) that can be recognized as a single gene disorder. To date, molecular analyses have successfully identified multiple major growth genes.
Growth pattern
The growth pattern of patients with mutant major growth genes is characterized by the reduced height velocity and resultant severe growth deficiency and by the sex difference in the adult height ascribed to the presence or absence of the Y-growth gene (5). Such growth characteristics are well represented by achondroplasia (6). Furthermore, two findings are noteworthy in the growth pattern of achondroplasia. First, the lack of the pubertal growth spurt is consistent with an intrinsic skeletal defect. Since linear growth is limited to approximately 4 cm per year because of bone abnormality, linear skeletal growth cannot respond to the exaggerated GH secretion during puberty. Second, the SD is comparable to that of the normal population. This is consistent with SD being composed of a combination of multiple minor genes that should be identical between the achondroplasia patients and the normal population. In this regard, if the growth deficit caused by each mutation of a major growth gene is variable, the SD should become different between the two groups; in achondroplasia, however, the type of FGFR3 mutation is strictly limited (4), and it is very likely that the degree of growth deficit is quite similar among patients with achondroplasia.
Therapeutic implication
Therapeutic strategies may be possible for mutations of the major growth genes. First, when a specific causative therapy is available, marked "catch-up growth" is expected (7). This phenomenon has been reported in several diseases such as growth hormone and thyroid hormone deficiencies. In such situations, hormone replacement therapy leads to a dramatic improvement in the linear growth. Second, when a therapy is available for a modifying factor, it would mitigate the prognosis of the corresponding disorder. A good example of this would be treatment for SHOX haploinsufficiency (8). Genotype-phenotype correlations indicate that skeletal anomalies referred to as Léri-Weill dyschondrosteosis and growth deficiency are obviously severe in pubertal and adult females with normal ovarian function, implying that the severity of skeletal phenotype largely depends on the skeletal maturing effects of gonadal estrogens (8). Thus, it is reasonable to perform gonadal suppression therapy with GnRH analog in female patients, especially in early maturing girls. While the experience of GnRH therapy remains poor, a good prognosis has been suggested recently (9). Third, the effect of non-specific therapy can be evaluated in terms of mutations. A good example of this would be the effect of GH therapy in Noonan syndrome patients. Since PTPN11 mutations have been identified in roughly 40% of Noonan syndrome patients, Noonan syndrome patients can be divided into mutation positive and negative groups (10). Although the natural statural growth is comparable between the two groups (11), the effect of GH therapy is much better in mutation negative patients (12). This implies that GH therapy should be performed in mutation negative patients. In this context, the similarity in non-GH treated height between the two groups would be due to PTPN11 being related to multiple signal transductions, because, in this case, the relevance of attenuated endogenous GH signaling to short stature remains at a clinically undetectable level. By contrast, since the effect of exogenous GH is primarily mediated by the GH receptor, it would be compromised more drastically in patients with hyperfunctional PTPN11 inhibiting the phosphorylation dependent GH signaling (13).
Heterozygous mutations
Heterozygous mutations of recessive genes can be relevant to clinically discernible short stature. For example, it has been suggested that heterozygosity for the GH1 gene mutation decreases the height by roughly 1 SD (Table 1) (14), so this situation would lead to short stature (<–2.0 SD) in individuals with an original height potential below –1 SD. Thus, heterozygosity for the GH1 gene causes short stature with a penetrance of approximately 16% and arelative risk of about 7.0. Furthermore, heterozygous mutations of the GH receptor gene (GHR) have been identified in many patients with idiopathic short stature, possibly accounting for up to 5% of idiopathic short stature (15). Similar situation may also take place for many major growth genes. Such heterozygosity can have not only diagnostic but also therapeutic implications, because it is assumed that the response to GH therapy is good in patients heterozygous for GH1 mutations and poor in those heterozygous for GHR mutations.
Table 1. Height SD in individuals heterozygous for the GH1 gene mutation and in those homozygous for the normal allele from a large pedigree.

Minor Growth Gene
Minor growth genes have relative minor additive effects on human growth, and may comprise functional polymorphisms of major genes and/or morphogenic genes with small penetrance on growth. They are relevant to the statural distribution and the height correlation of the normal population, and are involved in the development of short (or tall) stature as a multifactorial trait.
Height distribution
The heights in a normal population, in which the major growth genes should stay normal, follow the normal (Gaussian) distribution (1,2,3). This indicates that the height variation is primarily determined by the combination of multiple minor growth genes. Namely, individuals with a similar number of advantageous and disadvantageous minor growth genes have an average height and account for the major part of a given population; by contrast, individuals with multiple advantageous (or disadvantageous) minor growth genes have tall (or short) stature and should be small in number. This leads to the Gaussian distribution of heights.
Height SD remains constant for a long time (16). This implies that the power of expanding variation is quite comparable to that of narrowing variation. Thus, when parents heterozygous for multiple minor growth genes have a child homozygous for plural minor growth genes advantageous or disadvantageous to statural growth, this results in divergence of the SD; by contrast, when the parents homozygous for multiple minor growth genes advantageous or disadvantageous to statural growth have a child heterozygous for plural minor growth genes, this leads to regression of the SD. The latter phenomenon is known as the "regression towards the mean" (2). This concept implies that most, if not all, minor growth genes are associated with advantageous and disadvantageous allelic variants.
Correlation
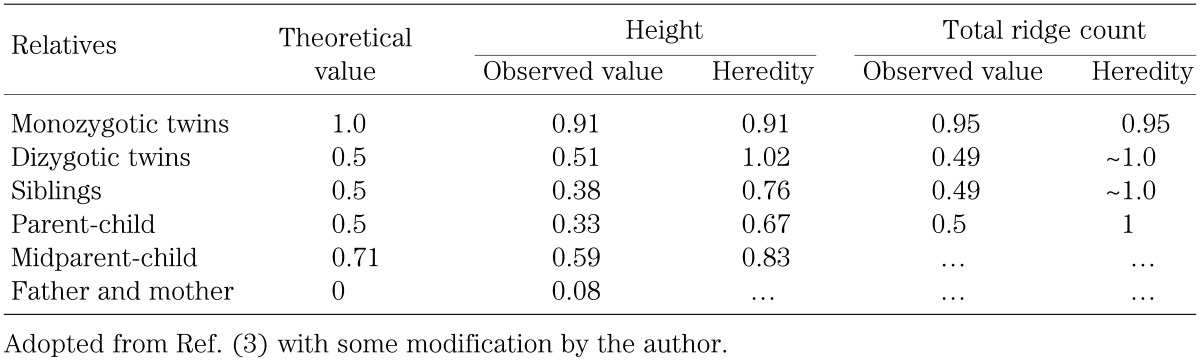
The height correlation among relatives is determined by the number of genes in common (17). For example, the theoretical correlation coefficient is 0.5 between a child and one parent, because they share half of the genes in common. Similarly, the theoretical correlation coefficient is 0.71 (root 2 divided by 2) between a child and mid-parents. In this regard, since the correlation coefficient for height is comparable among normal males, normal females, Turner females, and Klinefelter males (18), this implies the absence of a minor growth gene on the human sex chromosome.
However, the observed correlation coefficient is usually lower than the theoretical correlation coefficient (Table 2), primarily because of the effects of environmental factors. In this context, it is notable that the correlation coefficient for total ridge count, a well studied quantitative character in human, is nearly identical between the theoretical values and the observed values (Table 2). This is consistent with total ridge count being determined in the early fetal life, so that the influence of environmental factors remains minimal for this trait. By contrast, statural growth is subject to environmental factors for about 15–20 years. Here, the well preserved correlation coefficient of dizygotic twins could be due to the similarity of environmental factors. In addition, the low but positive correlation coefficient between parents indicates the presence of assertive mating, which could lead to the "regression towards the mean" and resultant reduced heredity.
Table 2. Correlation coefficient.
The observed height correlation is variable among the growth stages (19). It is low at birth (∼0.2–0.25), primarily due to the birth size being grossly determined by the maternal and placental function, rather than the parental height. It is also low in puberty because of the variable timing of pubertal development. Interestingly, it is lower in childhood (0.4–0.5) than in adulthood (∼0.6) (19). This would primarily be due to the prepubertal height being subject to the maturing tempo: an early maturing child tends to have relative tall stature in childhood and a late maturing child tends to have relative short stature in childhood, even though they have the same adult height (20). This implies that statural minor genes interact with minor maturational genes.
The presence of the high statural correlation offers a theoretical background to height prediction on the basis of the parental height. Indeed, target height (TH, a child’s adult height as predicted from the parental height) and target range (TR, ±2 SD of the TH) have been utilized in the assessment of child’s growth potential (16). For contemporary Japanese, the following equations have been proposed: boys, TH={PH+(MH+13)}÷2; TR=TH±9 cm; girls, TH={(PH-13)+MH}÷2; TR=TH±8 cm, where PH indicates paternal height and MH maternal height (21). TH/TR represents the point/range estimation of the child’ adult height born to the parents with no abnormalities of the major growth genes or growth related factors, and TR indicates the range of adult height variation predicted from the combination of minor growth genes. Thus, several points should be made with respect to the clinical application of TH/TR. First, TH/TR cannot be utilized for a child born to a parent(s) with an abnormal major growth gene or a growth related factor. For example, it does not make sense at all to use TH/TR in the assessment of a child born to a father with achondroplasia and a normal mother. Second, there should be no drastic difference in the environmental factors between the two generations. Drastic changes in environmental factors hves taken place after the World War II (16). Third, since only mean data are utilized for the TH/TR estimation, the accuracy will be different among children depending on the parental height. For example, although TH/TR is the same between a child born to parents of average height (father of 172 cm tall and mother of 159 cm tall) and a child born to tall father (182 cm) and short mother (149 cm), the height variation must be large in the latter case than in the former case
Heredity
Height correlation provides useful information for heredity that represents the relative contribution of genetic factors to the determination of individual height. In short, height heredity is obtained by the ratio between the observed correlation coefficient and the theoretical correlation coefficient (Table 2). In recent Japanese populations, the heredity for height is expected to be ∼80–90%, because of highly improved environmental factors.
Recent Molecular Approach
Recently, a molecular approach has been attempted for common diseases subject to various genetic and environmental factors, such as diabetes mellitus and hypertension. In particular, non-parametric analyses, such as case-control analysis, affected sib-pair analysis, transmission disequilibrium analysis, and quantitative trait loci analysis, have yielded significant results. Similar analyses have also been applied to statural growth.
Association study
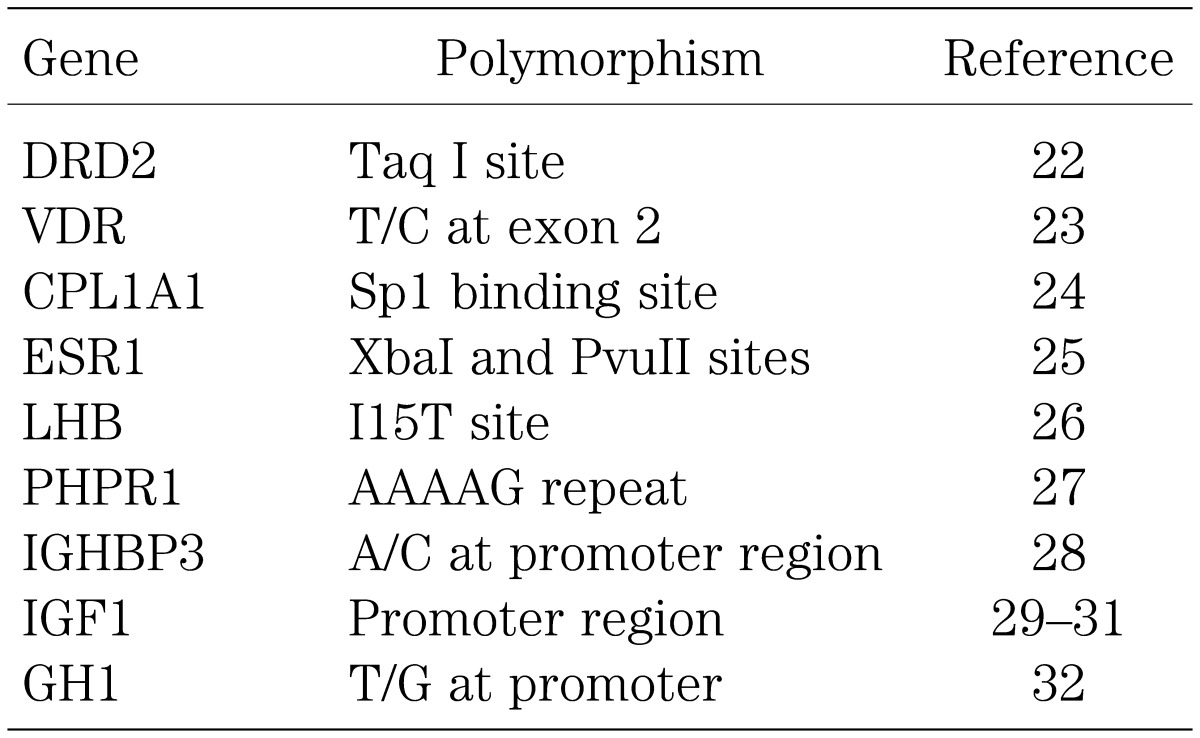
Case-control association studies have been carried out for many polymorphisms of the major growth genes. Consequently, multiple polymorphisms have been suggested as possible growth controlling alleles (Table 3). However, there is as yet no compelling evidence for a positive association. Rather, the previous studies have several critical problems: [1] positive associations have not been reproduced in most of the polymorphisms; [2] the possibility that the positive association has been obtained by stratification has not been excluded; for example, since Rh (–) blood type is more frequent in the English population than in the Japanese population, a strong association would be detected between the ability to speak English language and Rh (–) blood type; however, such an association has no biological significance at all; [3] Bonferroni correction for multiple testing has not been performed frequently; [4] whether genotype frequency follows the Hardy-Weinberg equilibrium has not been studied in many examinations; this is usually performed with Pearson’s Chi-square test with 1 degree of freedom); [5] haplotype analyses and functional studies have not been carried out in many studies, so that it remains possible that a polymorphism with a positive result is not the true functional polymorphism but remains just a marker for the hidden true polymorphism; and [6] most results have not been confirmed by cohort studies.
Table 3. Summary of association studies for height.
Genomewide screen
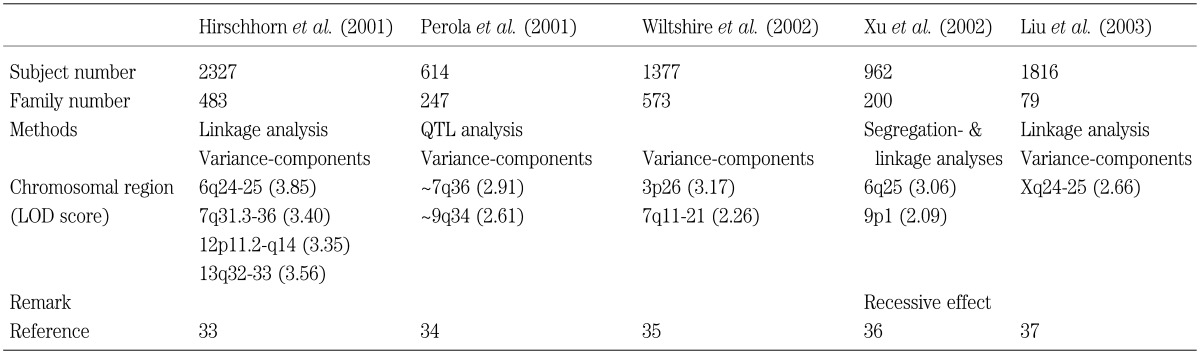
Recently, several genomewide scans have been carried out to identify the growth control genes. To date, five large studies have been done, and the 6q25 and the 7q26 regions have been identified by two studies, thereby exhibiting good candidate regions (Table 4). However, the regions are still very huge in a molecular term, and further localization is necessary. Furthermore, since most studies are based on the height data of individuals with common diseases such as diabetes mellitus and hypertension, the effect of original disorders cannot be excluded. In addition, since most examined individuals have stature within the normal range, the power to identify the statural genes is small.
Table 4. Possible chromosomal regions for the determination of the adultt height indicated by the genomewide screen (Lod score≥2.0).
Summary
Studies on human growth began with auxological analysis in soldiers, and has progressed to extensive examination by molecular methods. Although isolation of growth genes may still be far off, it is expected that recent progress in molecular technology and the accumulation of genome data will enable the discovery of growth genes. Successful isolation of growth genes will facilitate the understanding of human growth biology and the exploration of new therapeutic strategies.
Acknowledgement
This article is supported by a grant for Child Health and Development from the Ministry of Health, Labor, and Welfare (17C-2).
References
- 1.Vogel F, Motulsky AG. Human genetics: Problems and approaches. Berlin/Heidelberg/New York/Tokyo: Springer-Verlag, 1986 [Google Scholar]
- 2.Nassbaum RL, McInnes RR, Willard HF. Genetics in medicine. 6th ed, Philadelphia: WB Saubders, 2001 [Google Scholar]
- 3.Furusho T. Human genetics for genetic clounseling. Tokyo: Shiinko-Igaku, 1985. (in Japanese). [Google Scholar]
- 4.Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 1994; 371: 252–4 doi: 10.1038/371252a0 [DOI] [PubMed] [Google Scholar]
- 5.Ogata T, Matsuo N. The Y-specific growth gene(s): how does it promote the stature? J Med Genet 1997; 34: 323–5 doi: 10.1136/jmg.34.4.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tachibana K, et al. Growth curves in Japanese patients with achondroplasia. Shounikarinsho 1997; 60: 1363–9 [Google Scholar]
- 7.Tanner JM. Catch-up growth in man. Br Med Bull 1981;37: 233–8 [DOI] [PubMed] [Google Scholar]
- 8.Ogata T. SHOX haploinsufficiency: lessons from clinical studies. Cur Opin Endocrinol Diabetes 2002; 9: 13–20. doi: 10.1097/00060793-200202000-00003 [DOI] [Google Scholar]
- 9.Ogata T, Onigata K, Hotsubo T, Matsuo N, Rappold GA. Growth hormone and gonadotropin-releasing hormone analog therapy in haploinsufficiency of SHOX. Endocr J 2001; 48: 317–22 doi: 10.1507/endocrj.48.317 [DOI] [PubMed] [Google Scholar]
- 10.Ogata T, Yoshida R. PTPN11 mutations and genotype-phenotype correlations in Noonan and LEOPARD syndromes. Pediatr Endocrinol Rev 2005; 2: 669–74 [PubMed] [Google Scholar]
- 11.Yoshida R, Hasegawa T, Hasegawa Y, Nagai T, Kinoshita E, Tanaka Y, et al. Protein-tyrosine phosphatase, nonreceptor type 11 mutation analysis and clinical assessment in 45 patients with Noonan syndrome. J Clin Endocrinol Metab 2004; 89: 3359–64 doi: 10.1210/jc.2003-032091 [DOI] [PubMed] [Google Scholar]
- 12.Ferreira LV, Souza SA, Arnhold IJ, Mendonca BB, Jorge AA. PTPN11 (protein tyrosine phosphatase, nonreceptor type 11) mutations and response to growth hormone therapy in children with Noonan syndrome. J Clin Endocrinol Metab 2005; 90: 5156–60 doi: 10.1210/jc.2004-2559 [DOI] [PubMed] [Google Scholar]
- 13.Stofega MR, Herrington J, Billestrup N, Carter-Su C. Mutation of the SHP-2 binding site in growth hormone (GH) receptor prolongs GH-promoted tyrosyl phosphorylation of GH receptor, JAK2, and STAT5B. Mol Endocrinol 2000; 14: 1338–50 doi: 10.1210/me.14.9.1338 [DOI] [PubMed] [Google Scholar]
- 14.Leiberman E, Pesler D, Parvari R, Elbedour K, Abdul-Latif H, Brown MR, et al. Short stature in carriers of recessive mutation causing familial isolated growth hormone deficiency. Am J Med Genet 2000; 90: 188–92 doi: [DOI] [PubMed] [Google Scholar]
- 15.Blair J, Savage M. The GH-IGF-I axis in children with idiopathic short stature. Trends Endocrinol Metab 2002; 13: 325–30 doi: 10.1016/S1043-2760(02)00631-8 [DOI] [PubMed] [Google Scholar]
- 16.Ogata T, Matsuo N, Tamai S, Osano M, Tango T. Target height and target range for the Japanese. Jpn J Paediatr 1990; 94: 1535–40 [Google Scholar]
- 17.Emery EH. Methodorogy in medical genetics. 2nd ed, Edinburgh: Churchill Livingstone, 1986 [Google Scholar]
- 18.Brook CG, Gasser T, Werder EA, Prader A, Vanderschueren-Lodewykx MA. Height correlations between parents and mature offspring in normal subjects and in subjects with Turner’s and Klinefelter’s and other syndromes. Ann Hum Biol 1977; 4: 17–22 doi: 10.1080/03014467700001911 [DOI] [PubMed] [Google Scholar]
- 19.Mueller WH. The genetics of size and shape in children and adults. In: Falkner F, Tanner JM, editors. Human growth 2nd ed, Vol. 3. New York/London: Plenum; 1986. pp 154–68. [Google Scholar]
- 20.Tanner JM, Davies PS. Clinical longitudinal standards for height and height velocity for North American children. J Pediatr 1985; 107: 317–29 doi: 10.1016/S0022-3476(85)80501-1 [DOI] [PubMed] [Google Scholar]
- 21.Kagami M, Tanaka T, Ogata T. Target height and target range for the Japanese: revisited. Abstract for the 37th Annual Meeting for the Japanese Society for Pediatric Endocrinology, Sapporo, 2003 [DOI] [PMC free article] [PubMed]
- 22.Miyake H, Nagashima K, Onigata K, Nagashima T, Takano Y, Morikawa A. Allelic variations of the D2 dopamine receptor gene in children with idiopathic short stature. J Hum Genet 1999; 44: 26–9 doi: 10.1007/s100380050101 [DOI] [PubMed] [Google Scholar]
- 23.Minamitani K, Takahashi Y, Minagawa M, Yasuda T, Niimi H. Difference in height associated with a translation start site polymorphism in the vitamin D receptor gene. Pediatr Res 1998; 44: 628–32 doi: 10.1203/00006450-199811000-00002 [DOI] [PubMed] [Google Scholar]
- 24.Garnero P, Borel O, Grant SF, Ralston SH, Delmas PD. Collagen Ialpha1 Sp1 polymorphism, bone mass, and bone turnover in healthy French premenopausal women: the OFELY study. J Bone Miner Res 1998; 13: 813–7 doi: 10.1359/jbmr.1998.13.5.813 [DOI] [PubMed] [Google Scholar]
- 25.Lorentzon M, Lorentzon R, Backstrom T, Nordstrom P. Estrogen receptor gene polymorphism, but not estradiol levels, is related to bone density in healthy adolescent boys: a cross-sectional and longitudinal study. J Clin Endocrinol Metab 1999; 84: 4597–601 doi: 10.1210/jc.84.12.4597 [DOI] [PubMed] [Google Scholar]
- 26.Raivio T, Huhtaniemi I, Anttila R, Siimes MA, Hagenas L, Nilsson C, et al. The role of luteinizing hormone-beta gene polymorphism in the onset and progression of puberty in healthy boys. J Clin Endocrinol Metab 1996; 81: 3278–82 doi: 10.1210/jc.81.9.3278 [DOI] [PubMed] [Google Scholar]
- 27.Minagawa M, Yasuda T, Watanabe T, Minamitani K, Takahashi Y, Goltzman D, et al. Association between AAAG repeat polymorphism in the P3 promoter of the human parathyroid hormone (PTH)/PTH-related peptide receptor gene and adult height, urinary pyridinoline excretion, and promoter activity. J Clin Endocrinol Mtetab 2002; 87: 1791–6 doi: 10.1210/jc.87.4.1791 [DOI] [PubMed] [Google Scholar]
- 28.Deal C, Ma J, Wilkin F, Paquette J, Rozen F, Ge B, et al. Novel promoter polymorphism in insulin-like growth factor-binding protein-3: correlation with serum levels and interaction with known regulators. J Clin Endocrinol Metab 2001; 86: 1274–80 doi: 10.1210/jc.86.3.1274 [DOI] [PubMed] [Google Scholar]
- 29.Johnston LB, Leger J, Savage MO, Clark AJ, Czernichow P. The insulin-like growth factor-I (IGF-I) gene in individuals born small for gestational age (SGA). Clin Endocrinol 1999; 51: 423–7 doi: 10.1046/j.1365-2265.1999.00803.x [DOI] [PubMed] [Google Scholar]
- 30.Vaessen N, Janssen JA, Heutink P, Hofman A, Lamberts SW, Oostra BA, et al. Association between genetic variation in the gene for insulin-like growth factor-I and low birthweight. Lancet 2002; 359: 1036–7 doi: 10.1016/S0140-6736(02)08067-4 [DOI] [PubMed] [Google Scholar]
- 31.Arends N, Johnston L, Hokken-Koelega A, van Duijn C, et al. Polymorphism in the IGF-I gene: clinical relevance for short children born small for gestational age (SGA). J Clin Endocrinol Metab 2002; 87: 2720–4 doi: 10.1210/jc.87.6.2720 [DOI] [PubMed] [Google Scholar]
- 32.Hasegawa Y, Fujii K, Yamada M, Igarashi Y, Tachibana K, Tanaka T, et al. Identification of novel human GH-1 gene polymorphisms that are associated with growth hormone secretion and height. J Clin Endocrinol Metab 2000; 85: 1290–5 doi: 10.1210/jc.85.3.1290 [DOI] [PubMed] [Google Scholar]
- 33.Hirschhorn JN, Lindgren CM, Daly MJ, Kirby A, Schaffner SF, Burtt NP, et al. Genomewide linkage analysis of stature in multiple populations reveals several regions with evidence of linkage to adult height. Am J Hum Genet 2001; 69: 106–16 doi: 10.1086/321287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perola M, Ohman M, Hiekkalinna T, Leppavuori J, Pajukanta P, Wessman M, et al. Quantitative-trait-locus analysis of body-mass index and of stature, by combined analysis of genome scans of five Finnish study groups. Am J Hum Genet 2001; 69: 117–23 doi: 10.1086/321286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiltshire S, Frayling TM, Hattersley AT, Hitman GA, Walker M, Levy JC, et al. Evidence for linkage of stature to chromosome 3p26 in a large U.K. family data set ascertained for type 2 diabetes. Am J Hum Genet 2002; 70: 543–6 doi: 10.1086/338760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, Bleecker ER, Jongepier H, Howard TD, Koppelman GH, Postma DS, et al. Major recessive gene(s) with considerable residual polygenic effect regulating adult height: Confirmation of genomewide scan results for chromosomes 6, 9, and 12. Am J Hum Genet 2002; 71: 646–50 doi: 10.1086/342216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu YZ, Xu FH, Shen H, Deng H, Liu YJ, Zhao LJ, et al. Confirmation linkage study in support of the X chromosome harbouring a QTL underlying human height variation. J Med Genet 2003; 40: 825–31 doi: 10.1136/jmg.40.11.825 [DOI] [PMC free article] [PubMed] [Google Scholar]