

Abstract
This report concerns control of adrenocortical carcinoma in a 4-yr-old boy by adjuvant mitotane therapy. He presented precocious puberty and was diagnosed with adrenocortical carcinoma. He underwent surgical resection, and adjuvant mitotane therapy was initiated, leading to a final dose of 5.0 g/day. Despite monitoring of the plasma mitotane level, encephalopathy developed 5 mo after initiation. Although he recovered from the encephalopathy, careful follow-up of his growth and development is necessary. On the other hand, he has been free of recurrence and metastases for 3 yr since discontinuation of mitotane. A high dose of mitotane is potentially effective as an adjuvant chemotherapy for adrenocortical carcinoma, although optimal and safe usage needs to be established for children.
Keywords: adrenocortical carcinoma, adjuvant therapy, mitotane, encephalopathy
Introduction
Adrenocortical carcinoma is a rare malignant tumor, especially in children. It has a poor prognosis, and the only curative therapy is complete resection by surgery (1,2,3). Mitotane (o,p’-DDD) has been widely used as an adjuvant chemotherapy in adults, and the importance of monitoring the plasma level has been emphasized (4,5,6). However, its efficacy is controversial, and the optimal therapy for children remains to be established. Its narrow therapeutic range easily leads to several side effects and makes the therapy difficult (4, 6, 7). We herein report a 4-yr-old boy with adrenocortical carcinoma that was controlled by adjuvant therapy with a high dose of mitotane.
Case Report
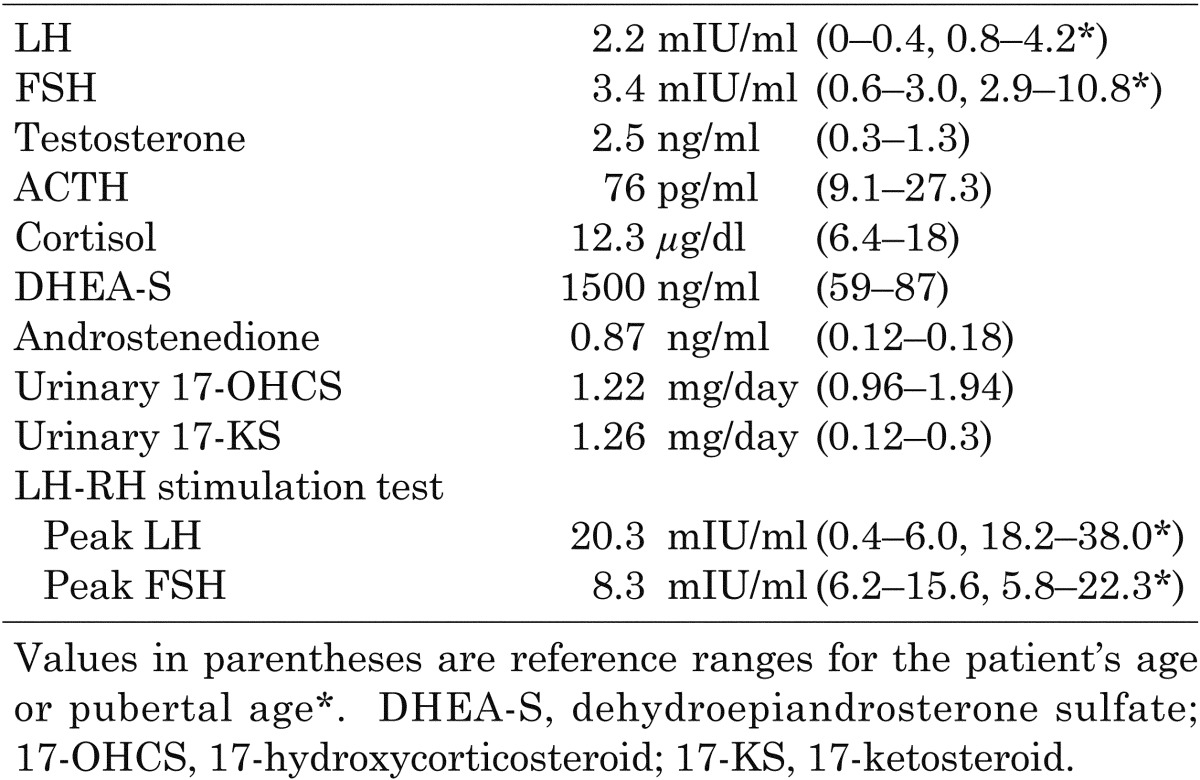
The patient was a 4-yr-old boy born to nonconsanguineous parents. He has a strong familial history of carcinoma on his mother’s side: mother, hemangioblastoma and breast cancer; grandmother, liver and breast cancer; great-grandfather, gastric cancer; and great-grandmother, renal cancer. He was admitted for evaluation of precocious puberty with acceleration of growth rate and maturation of external genitalia. His height was 116.9 cm (+3.62 SD), and his body weight was 23.6 kg (body mass index, 17.3 kg/m2). His Tanner stages were G2 and PH2, and each of his testicles was 8 ml in volume. No other abnormalities, such as cushingoid symptoms and hemihypertrophy, were found. The results of blood count and blood chemistry evaluation were all within normal ranges. Although his familial history reminded us of p53 gene mutation, genetic analysis was not performed because informed consent was not obtained. The endocrinological data are shown in Table 1. His LH, FSH and testosterone levels were elevated compared with his age’s reference. Although his dehydroepiandrosterone sulfate (DHEA-S), androstenedione and urinary 17-ketosteroid (17-KS) levels were increased, his cortisol and urinary 17-hydroxycorticosteroid (17-OHCS) levels were normal. The peaks of LH and FSH by LH-RH stimulation were at pubertal levels. His bone age was 11 years. Magnetic resonance imaging (MRI) showed a 2 × 3 cm mass in the left adrenal gland, and 131I-adosterol scintigraphy showed increased uptake there. No metastases were found based on radiological findings. An operation was performed based on the diagnosis of left androgen-secreting adrenocortical tumor. The tumor, which was 2 × 2.1 × 2.6 cm in size with focal necrosis and hemorrhage, was totally removed, and the pathological diagnosis was adrenocortical carcinoma.
Table 1. Endocrinological data.
Mitotane therapy was initiated as an adjuvant chemotherapy because the capsule and surgical margin were microscopically invaded by the carcinoma cells. We began with 0.5 g/day and increased the dose gradually by 0.5 g/1–2 weeks while monitoring the plasma level, up to 5.0 g/day (plasma level, 13.43 µg/ml, 3 mo after initiation). Leuprorelin acetate was also administered because an LH-RH test after the surgery still showed a response at the pubertal level (LH, 20.9 mIU/ml; FSH, 6.6 mIU/ml), suggesting secondary central precocious puberty. Hydrocortisone replacement was also initiated for treatment of adrenal insufficiency caused by mitotane.
Five months after initiation of mitotane, the patient became anorexic and his steps while walking became unsteady. These symptoms progressively worsened, and he began to have difficulty walking and speaking. He was apathetic to his surroundings and showed memory disturbance. His gait was unstable due to moderate spastic quadriplegia and ataxia. A head MRI revealed mild cerebral and cerebellar atrophy, and electroencephalograhy showed diffuse slowing of background activity. Thus, encephalopathy developed due to the increased level of mitotane (34.55 µg/ml, 6 mo after initiation). We discontinued mitotane and administered a high dose of hydrocortisone for adrenal crisis. The plasma mitotane level gradually decreased, and the neurological symptoms improved. It took 6 mo for the mitotane to disappear from the plasma and for clinical recovery from the encephalopathy.
No signs of tumor recurrence have been observed for 3 yr since discontinuation of mitotane. All neurological abnormalities found after initiation of mitotane, including memory disturbance and motor disability, are no longer present, thereby enabling him to preschool and play with similarly aged children without problems.
Discussion
Adrenocortical carcinoma is a rare disease with poor prognosis, especially in children. The efficacy of anti-cancer drugs cannot be expected, and the only effective treatment is complete resection (1,2,3). Although its efficacy is controversial, mitotane is widely used in adults for this disease, and its efficacy for prolonging recurrence-free survival has recently been reported (8). According to a report that recruited 254 pediatric patients with adrenocortical tumors, our patient is classified into stage III based on the possibility of a residual tumor identified by microscopic examination, which indicates a poor 5-yr event-free survival prognosis (2). Although he required adjuvant chemotherapy, anti-cancer drugs and genetic factors, such as p53 gene mutation, are presumed to be related to the occurrence of the second cancer (9). Therefore, we chose to use mitotane as the adjuvant chemotherapy.
There has been limited use of mitotane in children, and its usage remains to be established. Even in adults, a wide range of doses is used depending on the individual. Monitoring of the plasma level is emphasized to exploit its therapeutic effects and to avoid its toxic effects. Mitotane therapy at low plasma levels is known to be meaningless, and its level should be more than 14 µg/ml (4,5,6). A high dose of mitotane was reported to successfully treat a 14-yr-old patient with adrenocortical carcinoma and extensive pulmonary metastases (10). Therefore, we gradually increased the mitotane dose to achieve the therapeutic range with plasma level monitoring.
On the other hand, several side effects of mitotane are known, especially due to its narrow therapeutic range. A mitotane level of more than 20 µg/ml is associated with neurological toxicity (4, 6). In our patient, encephalopathy appeared after 5 mo despite monthly monitoring of the plasma level after determination of the dose. This might have been caused by gradual accumulation of lipid-soluble mitotane (11) after we determined the therapeutic dose. Therefore, for effective and safe treatment with mitotane, close monitoring of the plasma level is necessary even after the dose has been determined. However, the mitotane assay is not performed regularly for clinical use. In fact, it took 1–2 weeks to obtain the mitotane level data, and encephalopathy developed before we noticed the high level of mitotane. The mitotane assay should be more readily available so that the dose can be adjusted immediately according to the plasma level in order to obtain better effects with less toxicity.
The toxicity of mitotane has been reported to be reversible after discontinuation of the drug in adults (4, 7). Because its toxicity is reversible, a high dose of mitotane is recommended to control the disease. However, complete recovery in developing pediatric patients is questionable, and long-term follow-up is needed. De Leon et al. reported a 15-yr outcome in an infant with metastatic adrenocortical carcinoma (12). The patient showed long-term recovery from the toxic effects of mitotane, such as growth arrest and developmental delay. They concluded that long-term disease-free survival should justify use of mitotane therapy for adrenocortical carcinoma with an extremely poor prognosis. On the other hand, Nagasaki et al. reported a case of adrenocortical carcinoma in a 3-yr-old child with p53 gene mutation (13). They did not administer an adjuvant chemotherapy, and multiple lung metastases and a single liver metastasis were found 5 mo after surgery. Therefore, they suggested postoperative adjuvant chemotherapy for pediatric patients at high risk, even if macroscopic total surgical removal is performed. It took 6 mo for our patient to achieve clinical recovery from the encephalopathy, whereas he has had no signs of recurrence and metastasis for 3 yr since discontinuation of mitotane. The high concentration of mitotane may have prevented later recurrence and metastasis. We believe that our use of mitotane was also justified considering the poor prognosis and possibility of p53 gene mutation in our patient.
In conclusion, we report a case of adrenocortical carcinoma in a child. Although encephalopathy, which appeared reversible, developed and long-term follow-up of his growth, brain function and development is needed, adjuvant mitotane therapy was potentially effective for the cancer itself. Adrenocortical carcinoma requires further investigation in children, especially concerning effective and safe adjuvant chemotherapies.
References
- 1.Ciftci AO, Senocak ME, Tanyel FC, Buyukpamukcu N. Adrenocortical tumors in children. J Pediatr Surg 2001;36: 549–54 doi: 10.1053/jpsu.2001.22280 [DOI] [PubMed] [Google Scholar]
- 2.Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 2004;22: 838–45 doi: 10.1200/JCO.2004.08.085 [DOI] [PubMed] [Google Scholar]
- 3.Teinturier C, Pauchard MS, Brugieres L, Landais P, Chaussain JL, Bougneres PF. Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol 1999;32: 106–11 [DOI] [PubMed] [Google Scholar]
- 4.van Slooten H, Moolenaar AJ, van Seters AP, Smeenk D. The treatment of adrenocortical carcinoma with o,p'-DDD: prognostic implications of serum level monitoring. Eur J Cancer Clin Oncol 1984;20: 47–53 doi: 10.1016/0277-5379(84)90033-6 [DOI] [PubMed] [Google Scholar]
- 5.Haak HR, Hermans J, van de Velde CJ, Lentjes EG, Goslings BM, Fleuren GJ, et al. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer 1994;69: 947–51 doi: 10.1038/bjc.1994.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baudin E, Pellegriti G, Bonnay M, Penfornis A, Laplanche A, Vassal G, et al. Impact of monitoring plasma 1,1-dichlorodiphenildichloroethane (o,p'DDD) levels on the treatment of patients with adrenocortical carcinoma. Cancer 2001;92: 1385–92 doi: [DOI] [PubMed] [Google Scholar]
- 7.Bollen E, Lanser JB. Reversible mental deterioration and neurological disturbances with o,p'-DDD therapy. Clin Neurol Neurosurg 1992;94(Suppl): S49–51 doi: 10.1016/0303-8467(92)90020-4 [DOI] [PubMed] [Google Scholar]
- 8.Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007;356: 2372–80 doi: 10.1056/NEJMoa063360 [DOI] [PubMed] [Google Scholar]
- 9.Yonemoto T, Tatezaki S, Ishii T, Hagiwara Y, Inoue M. Multiple primary cancers in patients with osteosarcoma: influence of anticancer drugs and genetic factors. Am J Clin Oncol 2004;27: 220–4 doi: 10.1097/01.COC.0000054534.43117.76 [DOI] [PubMed] [Google Scholar]
- 10.Krzisnik C, Petric G, Jereb B. Complete response of metastatic adrenal cortical carcinoma to o,p'-DDD in a child. Pediatr Hematol Oncol 1988;5: 65–9 doi: 10.3109/08880018809031254 [DOI] [PubMed] [Google Scholar]
- 11.MOY RH Studies of the pharmacology of o,p'DDD in man. J Lab Clin Med 1961; 58: 296–304 [PubMed] [Google Scholar]
- 12.De Leon DD, Lange BJ, Walterhouse D, Moshang T. Long-term (15 years) outcome in an infant with metastatic adrenocortical carcinoma. J Clin Endocrinol Metab 2002; 87: 4452–6 doi: 10.1210/jc.2001-011978 [DOI] [PubMed] [Google Scholar]
- 13.Nagasaki K, Horikawa R, Nagaishi J, Honna T, Sekiguchi A, Tsunematsu Y, et al. Virilizing adrenocortical carcinoma invading the right atrium with histological high-grade malignancy and p53 mutation in a 3-year-old child: indication of post operative adjuvant chemotherapy. Clin Pediatr Endocrinol 2004; 13: 25–32. doi: 10.1297/cpe.13.25 [DOI] [PMC free article] [PubMed] [Google Scholar]