

Abstract
Nine previously-unreported farnesylphenols, involving eight neogrifolin derivatives (1–8) and one grifolin analogue (9), together with three known compounds, were isolated from the fruiting bodies of the mushroom Albatrellus caeruleoporus. Their structures were elucidated as (S)-17-hydroxy-18,20-ene-neogrifolin (1), (S)-18,19-dihydroxyneogrifolin (2), (S)-9-hydroxy-10,22-ene-neogrifolin (3), (9S,10R)-6,10-epoxy-9-hydroxyneo grifolin (4), (9S,10R)-6,9-epoxy-10-hydroxyneogrifolin (5), (−)-13,14-dihydroxyneogrifolin (6), albatrelin G (7), albatrelin H (8), and one grifolin analogue, (S)-10-hydroxygrifolin (9), grifolin (10), neogrifolin (11), and albatrellin (12) by extensive spectroscopic analyses and chemical methods. Compounds 7 and 8 showed weak cytotoxic activity to cell lines HL-60, SMMC-7721, A-549, and MCF-7, in vitro.
Electronic supplementary material
The online version of this article (doi:10.1007/s13659-014-0015-5) contains supplementary material, which is available to authorized users.
Keywords: Albatrellus caeruleoporus, Mushroom, Polyporaceae, Farnesylphenols
Introduction
Mushrooms of the Albatrellus genus are well known for producing farnesylphenols, such as grifolin, neogrifolin and their derivatives [1–6]. Farnesylphenols can be divided into two groups: monomers of grifolin and neogrifolin derivatives, and dimers of them. The monomers were reported to possess diverse biological activities, such as anti-oxidative activity [3], anti-microbial effect [7, 8], promotion of melanin synthesis [9], activity on human and rat vanilloid receptor 1 [10] inhibition of tumor-cell growth [11], and inhibition of nitric oxide production in RAW 264.7 cells [4]. And the dimers (fungal pigments) are regarded as the chemical base of the conspicuous fruiting bodies of these mushrooms [2, 4].
Albatrellus caeruleoporus is a nontoxic and inedible mushroom distributed in central and southwestern China. Its fruiting body is white with a light blue skin on the pileus [12]. Previous investigation on A. caeruleoporus led to three grifolin monomers, grifolin, neogrifolin, and grifolinone A, and one dimer, grifolinone B [4]. Their nitorite production inhibitory activities were reported [4]. In order to find more farnesylphenols with biological activities a systematic phytochemical investigation on the basidiomycete A. caeruleoporus was performed, it led to isolate eight new neogrifolin derivatives (1–8), a new grifolin analogue (9), grifolin (10) [10], neogrifolin (11) [10], and albatrellin (12) [2]. Their structures were identified by a combination of extensive spectroscopic analyses (NMR, MS, IR, UV, and [α]D) and chemical methods. Compounds 1–9 were oxygenated farnesylphenols, which have not previously been reported in the Albatrellus genus, and might be regarded as a chemotaxonomic evidence for identification of this mushroom. All new compounds were tested in a cytotoxicity assay in vitro against five human cancer cell lines.
Results and Discussion
The chloroform–methanol (1:1) extract of fruiting bodies of A. caeruleoporus was subjected to silica gel, RP-18, Sephadex LH-20 column chromatography (CC), and semipreparative HPLC purification steps to give compounds 1–12 (Fig. 1). Compounds 1–8, namely (S)-17-hydroxy-18,20-ene-neogrifolin (1), (S)-18,19-dihydroxyneogrifolin (2), (S)-9-hydroxy-10,22-ene-neogrifolin (3), (9S,10R)-6,10-epoxy-9-hydroxyneogrifolin (4), (9S,10R)-6,9-epoxy-10-hydroxyneogrifolin (5), (−)-13,14-dihydroxyneogrifolin (6), albatrelin G (7), albatrelin H (8), were neogrifolin derivatives, (S)-10-hydroxygrifolin (9) was a grifolin analogue, and compound 12 was a violet pigment named albatrellin.
Fig. 1.
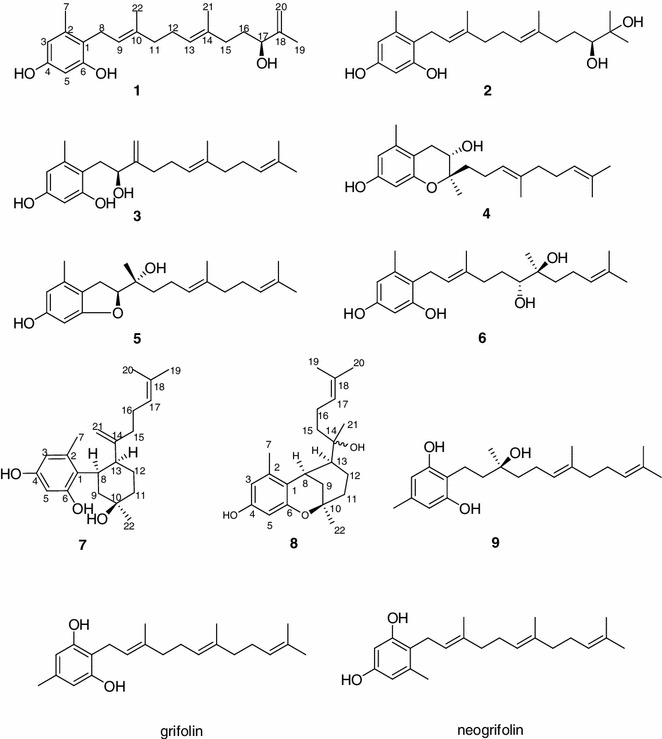
Chemical structures of compounds 1–9
Compound 1, colorless oil, displayed a [M]+ ion peak at m/z 344.2348 in positive HREIMS, corresponding to the molecular formula C22H32O3 and seven degrees of unsaturation. The IR spectrum showed absorption at 3421 cm−1 which indicated the presence of OH groups. The 1H NMR spectrum contained signals for two m-coupling aromatic protons at δH 6.25 (1H, d, J = 1.8 Hz) and 6.17 (1H, d, J = 1.8 Hz), four olefinic protons, and four singlet methyls. Combined with 13C NMR (DEPT) experiment, the existence of a 1,2,4,6-tetra-substituted phenyl ring, a terminal double bond, two tri-substituted double bonds, one oxygen-bearing methine, five methylenes, and four methyls were assigned. The 1H and 13C NMR spectroscopic data were close to those of neogrifolin, except that a methylene and a methyl group in neogrifolin were replaced by a hydroxyl methine (δC 75.3) and a terminal double bond (δC 110.3 and 149.3), respectively. According to the observed HMBC correlations from δH 6.25 and 6.17 (H-20) to δC 17.8 (C-19) and 75.3 (C-17), the terminal double bond was located at the end of the farnesyl side chain, and the hydroxyl group was at C-17 (δC 75.3). This conclusion was supported by cross peaks from δH 3.73 (OH-17) to δC 34.5 (C-16), 75.3 (C-17) and 149.3 (C-18), and from 3.97 (H-17) to δC 36.4 (C-15) in HMBC spectrum (Fig. 2). The absolute configuration of the only chiral center (C-17) of 1 was deduced to be S by comparing the optical rotation value of 1 ([α]21D −9.0, MeOH) with that of (R)-(+)-3-methyl-3-buten-2-ol ([α]D +7.6, CHCl3) [13]. Therefore, compound 1 was elucidated and named as (S)-17-hydroxy-18,20-ene-neogrifolin.
Fig. 2.
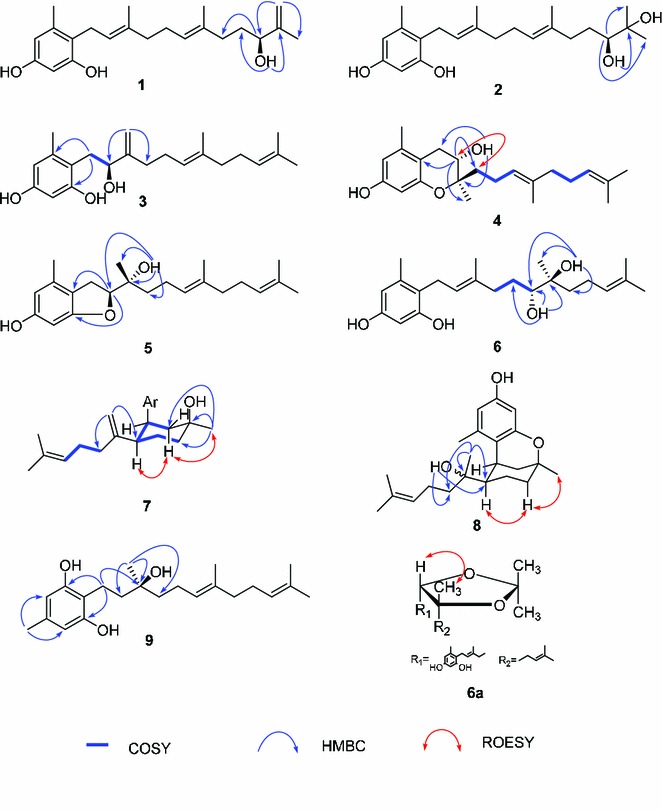
Selective 2D NMR correlations for compounds 1–9
Compound 2 possessed a molecular formula of C22H34O4 according to HREIMS which showed a molecular ion peak at m/z 362.2452, requiring six degrees of unsaturation. Inspection of the 1H and 13C NMR (DEPT) spectra indicated five methyls, five methylenes, four sp2 methines, one oxygen-bearing methine, and seven quaternary carbons. The 1D NMR spectroscopic data were similar to those of 1, except for the terminal double bond being replaced by a methyl and an oxygen-bearing quaternary carbon, which was confirmed by HMBC correlations from δH 3.54 (OH-17) to δC 72.9 (C-18), from δH 3.24 (H-17) to δC 25.8 (C-19 and C-20), from δH 1.11 (H-20) to δC 25.8 (C-19), 72.9 (C-18) and 78.5 (C-17). The absolute configuration of C-17 in 2 was assigned to be S, the same as 1, on a biogenetic point of view. And this supposition was further confirmed by a comparison of the optical rotation values between 2 ([α]21D −9.2, MeOH) and (R)-2-methylpentane-2,3-diol ([α]18.5D +27.3, ether) [14]. Therefore, compound 2 was identified as (S)-18,19-dihydroxyneogrifolin.
Compound 3 was determined to have the molecular formula of C22H32O3 from HREIMS at m/z 344.2364 ([M]+). The 13C NMR (DEPT) spectra showed signals of a tetra-substituted phenyl moiety, a terminal double bond, two tri-substituted double bonds, a hydroxyl methine, four methyls, and five methylenes, which resembled those of compound 1. Extensive 2D NMR (COSY, ROESY and HMBC) analyses revealed that the locations of the double bond and the oxygen-bearing methine were different with those of 1. COSY correlations from δH 2.82 and 2.68 (H-8) to δH 4.28 (δC = 77.0) suggested that C-9 was the oxygenated carbon. Moreover, δH 5.11 and 4.84 (δC = 108.9, t) gave HMBC correlations to δC 32.5 (C-11) and 77.0 (C-9), revealing that the Me-22 in compound 1 converted to be a double bond in 3. Therefore, the structure of compound 3 was identified as 9-hydroxy-10,22-ene-neogrifolin. The absolute stereochemistry of the chiral center of C-9 was determined to be S by comparing the optical rotation value of 3 ([α]D −8.8, MeOH) with (S)-3-methyl-1-phenylbutan-2-ol ([α]D −29.5, CHCl3) [15].
The HREIMS of compound 4 showed a [M]+ ion peak at m/z 344.2348, indicating a molecular formula of C22H32O3 and seven degrees of unsaturation. A comparison of the MS and 1D NMR data of 4 with those of 3 revealed that 4 was another neogrifolin analogue resembled 3 except for the double bond between C-10 and C-22 in 3 being replaced by a methyl (Me-22) and an oxygenated quaternary carbon (C-10) in 4. This structure requires six degrees of unsaturation, and an additional ring was needed to complete the unsaturation. There were two plausible proposals: an epoxy ring between C-6 and C-9 or between C-6 and C-10. HMBC correlations from H-9 (δH 3.85) to C-1 (δC 111.0), C-10 (δC 78.6), C-11 (δC 38.7), and C-22 (δC 17.9), and from the OH at δH 4.14 to C-8 (δC 29.7), C-9 (δC 68.4) and C-10 (δC 78.6) revealed the location of the free OH at C-9, placing the epoxy ring between C-6 and C-10. Therefore, the planar structure of 4 was elucidated as 6,10-epoxy-9-hydroxyneogrifolin. The ROESY spectrum displayed cross peaks of H-9/H-11 and H-9/H-12, suggesting the same orientation of H-9 and the geranyl group. From a biogenetic point of view, compounds 4 and 3 should share the same absolute configuration on C-9. So the absolute stereochemistry of compound 4 was deduced to be 9S, 10R.
Compound 5 was proposed to be a neogrifolin derivative on basis of HREIMS which displayed the molecular ion peak at m/z 344.2358. A comparison of the 13C NMR (DEPT) spectra of 5 with those of 4 revealed the resemblance of the two structures, for example, the presence of the 1-(2-methyl-4,6-dihydroxyl)-phenyl group and the geranyl moiety. The structural difference between compounds 4 and 5 was the fragment from C-8 to C-10, according to the different chemical shifts of the corresponding carbons and protons (Tables 1 and 2). In order to establish the structure of 5, extensive 2D NMR experiments were employed. The HMBC correlations of H-9/C-1 and H-9/C-6 indicated the existence of an oxygen bridge between C-6 and C-9. The free hydroxyl group was determined to be located at C-10 by HMBC correlations from OH-10 to C-9, C-10, C-11 and C-22. Biogenetically speaking, compound 5 would show the same stereochemistry at C-9 and C-10 as compound 4. Therefore, compound 5 was elucidated as (9S,10R)-6,9-epoxy-10-hydroxyneogrifolin.
Table 1.
1H NMR spectroscopic data for compounds 1–6 in acetone-d6 (δ in ppm, J in Hz)
| No | 1 a | 2 c | 3 a | 4 b | 5 a | 6 c |
|---|---|---|---|---|---|---|
| 3 | 6.17, d (1.8) | 6.17, d (2.3) | 6.21, s | 6.25, d (2.1) | 6.12, br. s | 6.17, s |
| 5 | 6.25, d (1.8) | 6.25, d (2.3) | 6.22, s | 6.12, d (2.1) | 6.03, br. s | 6.25, s |
| 7 | 2.15, s | 2.15, s | 2.20, s | 2.11, s | 2.11, s | 2.15, s |
| 8 | 3.26, d (6.7) | 3.26, d (6.7) | 2.82, dd (14.4, 2.2) | 2.79, dd (16.1, 5.9) | 3.06, dd (15.3, 7.9) | 3.27* |
| 2.68, dd (14.4, 9.3) | 2.45, dd (16.1, 8.5) | 2.93, dd (15.3, 9.5) | ||||
| 9 | 5.09, t (6.7) | 5.09, t (6.7) | 4.28, dd (9.3, 2.2) | 3.85, td (8.5, 5.8) | 4.62, dd (9.5, 7.9) | 5.11* |
| 11 | 1.90–2.00, m | 1.97–1.99, m | 2.22–2.25* | 1.66–1.75, m | 1.50–1.56, m | 2.25, t (9.9) |
| 2.14–2.17, m | ||||||
| 1.96–2.03* | ||||||
| 12 | 2.03–2.11, m | 2.07–2.10, m | 2.22–2.25* | 2.14–2.24, m | 2.09–2.20, m | 1.69–1.74, m |
| 1.33–1.40, m | ||||||
| 13 | 5.12, t (6.6) | 5.14, t (7.0) | 5.20, br. s | 5.17, t (7.2) | 5.18, t (6.9) | 3.29* |
| 15 | 1.90–2.00, m | 2.21–2.26, m | 1.98, t (7.5) | 1.97, t (7.6) | 1.98, t (7.5) | 1.54–1.58, m |
| 1.94–1.97, m | 1.33–1.40, m | |||||
| 16 | 1.54–1.58, m | 1.62–1.68, m | 2.07–2.09* | 2.06–2.08* | 2.06–2.09* | 2.09–2.13, m |
| 1.28–1.35, m | 2.03–2.05, m | |||||
| 17 | 3.97, m | 3.24, td (5.3, 1.8) | 5.10* | 5.10, t (6.9) | 5.10, t (7.0) | 5.11*1 |
| 19 | 1.68, s | 1.11, s | 1.59, s | 1.58, s | 1.59, s | 1.58, s |
| 20 | 4.89, br. s | 1.11, s | 1.65, s | 1.65, s | 1.65, s | 1.64, s |
| 4.74, br. s | ||||||
| 21 | 1.58, s | 1.58, s | 1.63, s | 1.61, s | 1.63, s | 1.07, s |
| 22 | 1.75, s | 1.75, s | 5.11, br. s | 1.16, s | 1.20, s | 1.76, s |
| 4.84, br. s | ||||||
| 4-OH | 7.86, s | 7.88, s | 8.01, s | 7.89, s | 8.05, s | 7.89, s |
| 6-OH | 8.02, s | 8.06, s | 8.56, s | 8.05, s | ||
| 9-OH | 4.88, d (2.8) | 4.14, d (5.6) | ||||
| 10-OH | 3.54, s | |||||
| 13-OH | 3.56, d (5.8) | |||||
| 14-OH | 3.20, s | |||||
| 17-OH | 3.73, d (4.2) | 3.54, d (5.3) | ||||
| 18-OH | 3.40, s |
aMeasured at 400 MHz
bMeasured at 500 MHz
cMeasured at 600 MHz
* Signals were overlapped
Table 2.
13C NMR spectroscopic data for compounds 1–6 in acetone-d6 (δ in ppm)
| No | 1 a | 2 c | 3 a | 4 b | 5 a | 6 c |
|---|---|---|---|---|---|---|
| 1 | 118.3, C | 118.3, C | 117.1, C | 111.0, C | 117.8, C | 118.3, C |
| 2 | 138.9, C | 138.9, C | 139.0, C | 138.6, C | 135.2, C | 138.8, C |
| 3 | 109.4, CH | 109.3, CH | 109.8, CH | 110.1, CH | 108.5, CH | 109.3, CH |
| 4 | 156.5, C | 156.5, C | 157.1, C | 157.1, C | 158.4, C | 156.5, C |
| 5 | 101.0, CH | 101.0, CH | 102.2, CH | 102.0, CH | 95.2, CH | 101.0, CH |
| 6 | 156.4, C | 156.4, C | 157.9, C | 154.7, C | 161.6, C | 156.4, C |
| 7 | 19.9, CH3 | 19.9, CH3 | 20.5, CH3 | 19.3, CH3 | 19.0, CH3 | 19.9, CH3 |
| 8 | 25.1, CH2 | 25.1, CH2 | 34.4, CH2 | 29.7, CH2 | 29.1, CH2 | 25.1, CH2 |
| 9 | 124.8, CH | 124.8, CH | 77.0, CH | 68.4, CH | 89.8, CH | 124.4, CH |
| 10 | 134.1, C | 134.1, C | 153.4, C | 78.6, C | 73.3, C | 134.6, C |
| 11 | 40.4, CH2 | 40.4, CH2 | 32.5, CH2 | 38.7, CH2 | 39.3, CH2 | 37.8, CH2 |
| 12 | 27.2, CH2 | 27.2, CH2 | 27.3, CH2 | 22.1, CH2 | 22.5, CH2 | 30.2, CH2 |
| 13 | 124.8, CH | 124.8, CH | 125.1, CH | 125.4, CH | 125.6, CH | 77.8, CH |
| 14 | 135.5, C | 135.8, C | 135.7, C | 135.4, C | 135.3, C | 74.2, C |
| 15 | 36.4, CH2 | 37.6, CH2 | 40.4, CH2 | 40.4, CH2 | 40.4, CH2 | 38.5, CH2 |
| 16 | 34.5, CH2 | 30.7, CH2 | 27.3, CH2 | 27.4, CH2 | 27.3, CH2 | 22.6, CH2 |
| 17 | 75.3, CH | 78.5, CH | 125.1, CH | 125.1, CH | 125.1, CH | 126.2, CH |
| 18 | 149.3, C | 72.9, C | 131.6, C | 131.6, C | 131.6, C | 131.1, C |
| 19 | 17.8, CH3 | 25.8, CH3 | 17.7, CH3 | 17.7, CH3 | 17.7, CH3 | 17.6, CH3 |
| 20 | 110.3, CH2 | 25.8, CH3 | 25.8, CH3 | 25.8, CH3 | 25.8, CH3 | 25.8, CH3 |
| 21 | 16.1, CH3 | 16.1, CH3 | 16.1, CH3 | 16.0, CH3 | 16.0, CH3 | 22.8, CH3 |
| 22 | 16.1, CH3 | 16.1, CH3 | 108.9, CH2 | 17.9, CH3 | 22.3, CH3 | 16.3, CH3 |
aMeasured at 100 MHz
bMeasured at 125 MHz
cMeasured at 150 MHz
Compound 6 was obtained as a colorless oil, with a molecular formula of C22H34O4 according to the HREIMS at m/z 362.2432 ([M]+). Inspection of the 1H, 13C (DEPT) and HSQC NMR spectra allowed the assignment of five methyls, five methylenes, five methines, seven quaternary carbons, and four active protons. Comparing the 1H and 13C NMR spectroscopic data of 6 with those of neogrifolin indicated that compound 6 shared the “1-(2-methyl-4,6-dihydroxyl)-phenyl” partial structure with neogrifolin, but had a different side chain, in which one double bond in the farnesyl group was replaced by two oxygen-bearing sp3 carbons. The COSY cross peaks of H-11/H-12/H-13, and HMBC correlations from OH at δH 3.56 to C-12, C-13 and C-14, and from δH 3.20 to C-21, C-13, C-14, and C-15 (Fig. 2) suggested the oxygenated carbons being located at C-13 (δC 77.3, CH) and C-14 (δC 74.2, C). In order to identify the relative configuration of the two chiral centers C-13 and C-14, compound 6 was reacted with 2,2-dimethoxypropane in DMF for 30 min at room temperature to yield its di-O-isopropylidene derivative 6a. The observed ROESY correlations of Me-21/H-13 (Fig. 2) indicated that the 13,14-diol existed as the erythro form. So, the absolute configuration of 6 should be 13S, 14S or 13R, 14R.
Compound 7 possessed a molecular formula of C22H32O3 from its HREIMS, which displayed a molecular ion peak at m/z 344.2348. A comparison of the 1H and 13C NMR data of 7 with those of 1 revealed the presence of the 1-(2-methyl-4,6-dihydroxyl)-phenyl group. Combined with MS spectral data, compound 7 was determined to be a neogrifolin derivative unambiguously. Unlike the other neogrifolin analogues (1–6) which had straight-chains as “tails”, compound 7 had a cyclohexane moiety—by C-8 connecting to C-13—in its tail. It was supported by COSY correlations of H-9/H-8/H-13/H-12/H-11, and HMBC cross peaks from Me-22 to C-9, C-10 and C-11 (Fig. 2). The remaining part of the “tail” was a 2-(6-methyl)-1,5-heptadiene residue. This residue was connected with C-13, because COSY correlations of H-15/H-16/H-17, and HMBC correlations from H-21 to C-13 and C-15, and from Me-19 and -20 to C-18 and C-17 (Fig. 2) were observed. In order to determine the relative stereochemistry of C-8, C-10 and C-13, a ROESY experiment was performed, combined with further analysis of the coupling constants of several signals in 1H NMR spectrum. The observed broad singlet (br. s) signal of H-8 (δH = 3.22) in its 1H NMR spectrum indicated that H-8 existed as an equatorial bond in the stable boat conformation of the cyclohexane moiety, as shown in Fig. 2. Likewise, H-13 was proposed to be in an axial position because of the doublet of triplets at δH 3.22 with coupling constants of 12.5 and 2.7 Hz, respectively. Furthermore, The ROESY correlations of H-13/Hax-9 and Me-22/Hax-9 revealed the same orientation of H-13 and Me-22. Therefore, H-8, H-13 and Me-22 were deduced to be α-, α-, α- orientated. In compound 7, a ring was formed by new C–C bond connection between C-8 and C-13 in side chain.
Compound 8 exhibited a molecular ion peak at m/z 344.2345 in HREIMS, indicating the molecular formula of C22H32O3 which required seven degrees of unsaturation. According to the 1H and 13C NMR (DEPT) spectra, 22 carbon signals were recognized as five methyls, four methylenes, five methines, and seven quaternary carbons. Extensive NMR analyses suggested that the structure of 8 resembled that of 7, except for the terminal double bond C-14=C-21 in 7 being saturated to be a methyl and an oxygen-bearing quaternary carbon, which was confirmed by HMBC correlations from δH 1.13 (Me-21) to δC 36.8 (C-15), 55.9 (C-13), and 74.0 (C-14), and from δH 3.30 (OH-14) to δC 36.8 (C-15), 55.9 (C-13), and 74.0 (C-14). So far, six degrees of unsaturation was assigned, and one more ring should be constructed to complete the structure of 8. The only possible ring to be formed was the oxygen bridge between C-6 and C-10. The stereochemistry of C-13 was identified by analysis of 1H NMR spectrum, in which H-13 showed a doublet-of-triplets peak with the coupling constants of 12.8 and 2.0 Hz, respectively, suggesting the axial bond of H-13. Me-22 had the same orientation as H-13 by the observed ROESY correlations of H-13/Hax-11/Me-22, and because of the planar structure of the phenyl group, H-8 and Me-22 should be on the same orientation. Thus, H-8, H-13 and Me-22 were determined to be α-, α-, α- orientated, the same as for compound 7.
Compound 9 was proposed to possess a molecular formula of C22H34O3 on basis of HREIMS at m/z 346.2505 ([M]+). Its 13C NMR (DEPT) spectrum showed 20 carbon signals, including two signals at δC 108.4 (CH) and 156.7 (C) which represented two carbons respectively. The overlapped carbon signals indicated that 9 was a grifolin derivative possessing a symmetric aromatic ring, which was confirmed by HMBC cross peaks from δH 2.10 (Me-7) to δC 108.4 (C-3 and -5) and 136.5 (C-4), from δH 2.67 (H-8) to δC 113.9 (C-1) and 156.7 (C-2 and -6), and from δH 8.05 (OH-2 and -6) to δC 108.4 (C-3 and -5), 113.9 (C-1) and 156.7 (C-2 and -6). Besides the aromatic ring, the remaining signals represented an oxygenated farnesyl group with four methyls, six methylenes, two pairs of tri-substituted double bonds, and one oxygenated quaternary carbon. The next problem to be resolved was the position of oxygenation, which was addressed by 2D NMR (HMBC and COSY) experiments. The HMBC correlations from δH 2.67 (H-8) to δC 72.6 (C-10), and from δH 1.20 (Me-22) to δC 41.2 (C-9), 72.6 (C-10) and 42.6 (C-11) revealed the hydroxylation of C-10. Hence, the planar structure of 9 was established as 10-hydroxygrifolin. The absolute stereochemistry of C-10 was deduced to be S by a comparison of the optical rotation value of 9 ([α]D −8.7, MeOH) with that of (S)-3-methyl-1-phenyl-3-pentanol ([α]D −1.6, CHCl3) [16].
All the new compounds were assayed for their cytotoxicity against five human cancer cell lines (HL-60, SMMC-7712, A-549, MCF-7, and SW480) by the MTT method in vitro, with DDP and taxol as positive controls. Compound 7 showed cytotoxic activities to cell lines HL-60, SMMC-7721, A-549, and MCF-7, with IC50 of 12.8, 33.8, 33.0, and 33.2 μM, respectively, and 8 exhibited weak growth inhibition activity to human tumor cell lines HL-60 and A-549, with IC50 of 21.8 and 30.3 μM, respectively.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Jasco model 1020 polarimeter (Jasco International Co. Ltd, Tokyo, Japan). UV spectra were recorded on a Shimadzu double-beam 2401A spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were obtained on a Bruker Tensor 27 FT-IR spectrometer (Bruker, Ettlingen, Germany) using KBr pellets. 1D and 2D NMR spectra were acquired on Bruker AV-600, DRX-500 and AM-400 instruments at room temperature with TMS as internal standard (Bruker, Rheinstetten, Germany). Chemical shifts (δ) were expressed in ppm with reference to the solvent signals. Mass spectra (MS) were recorded on a VG Autospec-3000 spectrometer (VG, Manchester, England). Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), Sephadex LH-20 (Amersham Biosciences, Sweden), and RP-18 gel (40–75 μm, Fuji Silysia Chemical Ltd. Japan) were used for CC. HPLC analysis (Zorbax SB-C18, 5 μm, 4.6 × 150 mm) was performed on an Agilent 1100 liquid chromatography system, and semi-preparative HPLC was performed on an Agilent 1200 liquid chromatography system equipped with a Zorbax SB-C18 column (9.4 mm × 150 mm). Pre-coated silica gel GF254 plates (Qingdao Marine Chemical Inc., Qingdao, China) were used for TLC. Fractions were monitored by TLC, and spots were visualized by heating silica gel plates sprayed with 10 % H2SO4 in ethanol.
Fungal Material
The fungus A. caeruleoporus was collected in Anhui province, China, in October, 2011. The voucher specimen (GDGM 29146) has been deposited in the Herbarium of Microbiology Institute of Guangdong.
Extraction and Isolation
The dried fruiting bodies of A. caeruleoporus (about 200 g) were extracted with chloroform/methanol (1/1) for three times (5 L × 3). Evaporation of the solvent under reduced pressure gave the crude extract (20 g), which was subjected to silica gel CC using a petroleum ether–acetone gradient (1:0 → 0:1) to afford fractions A–E. Fraction B was purified by CC over silica gel with a petroleum ether–acetone system (20:1 → 10:1) to yield two fractions B1 and B2. Fraction B1 was purified by semi-preparative HPLC (CH3CN/H2O, 6:4) to gave 1 (5.2 mg) and 10 (12.0 mg), while B2 was applied on a Sephadex LH-20 (CHCl3/MeOH 1/1) column and then on semi-preparative HPLC eluting with MeCN-H2O (6:4) to yield compounds 2 (6.0 mg) and 11 (21.0 mg). Fraction C was subjected to CC with RP-18 silica gel eluting with 90 % methanol, and then purified by semi-preparative HPLC (CH3CN/H2O, 55:45) to get compound 6 (5.1 mg). Fraction D was submitted to silica gel CC eluting with petroleum ether-acetone gradient (15:1) to gave two fractions D1 and D2, which were purified first by PR-18 and then by Sephadex LH-20 CC to yield fractions D1′ and D2′, respectively. Fraction D1′ was loaded on a semi-preparative HPLC eluting with MeCN-H2O (60:40 → 65:35) to afford compounds 9 (4.8 mg) and 3 (4.5 mg), while fraction D2′ was passed through a semi-preparative HPLC (MeCN/H2O, 60:40 → 65:35) to yield compounds 8 (6.4 mg) and 5 (4.0 mg). Fraction E was applied on CC over RP-18 to give fractions E1 and E2. Compound 4 (6.5 mg) was obtained from fraction E1 which was passed through silica gel column (petroleum ether-acetone, 10:1), Sephadex LH-20 column (chloroform–methanol, 1:1), and semi-preparative HPLC (MeCN/H2O 7:3), successively. Fraction E2 was passed through Sephadex LH-20, and then loaded on semi-preparative HPLC to yield compound 7 (4.5 mg).
(S)-17-hydroxy-18,20-ene-neogrifolin (1)
Colorless oil; [α]21D −9.0 (c 0.20, MeOH); UV λmax (MeOH) (log ε) 283 (2.67) nm; IR (KBr) νmax 3421, 3075, 2970, 2922, 2855, 1611, 1447, 1140 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; EI-MS m/z: 344 [M]+, 326 [M–H2O]+, 191, 175, 137; HR-EI-MS m/z: 344.2348 [M]+ (calcd for C22H32O3, 344.2351).
(S)-18,19-dihydroxyneogrifolin (2)
Colorless oil; [α]21D −9.2 (c 0.18, MeOH); UV λmax (MeOH) (log ε) 282 (2.67) nm; IR (KBr) νmax 3423, 2974, 2925, 2855, 1613, 1467, 1141 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; EI-MS m/z: 363 [M+H]+, 362 [M]+, 344 [M–H2O]+, 191, 175, 137; HR-EI-MS m/z: 362.2452 [M]+ (calcd for C22H34O4, 362.2457).
(S)-9-hydroxy-10,22-ene-neogrifolin (3)
Colorless oil; [α]21D −8.8 (c 0.24, MeOH); UV λmax (MeOH) (log ε) 283 (2.90) nm; IR (KBr) νmax 3420, 2966, 2923, 2855, 1614, 1447, 1143 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; EI-MS m/z: 344 [M]+, 326 [M–H2O]+, 137; HR-EI-MS m/z: 344.2364 [M]+ (calcd for C22H32O3, 344.2351).
(9S,10R)-6,10-epoxy-9-hydroxyneogrifolin (4)
Colorless oil; [α]22D −6.7 (c 0.28, MeOH); UV λmax (MeOH) (log ε) 282 (2.74) nm; IR (KBr) νmax 3422, 3038, 2967, 2923, 2854, 1618, 1601, 1460, 1138 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; EI-MS m/z: 344 [M]+, 191, 137; HR-EI-MS m/z: 344.2348 [M]+ (calcd for C22H32O3, 344.2351).
(9S,10R)-6,9-epoxy-10-hydroxyneogrifolin (5)
Colorless oil; [α]22D −8.6 (c 0.19, MeOH); UV λmax (MeOH) (log ε) 282 (2.82) nm; IR (KBr) νmax 3513, 3405, 2969, 2922, 2856, 1629, 1602, 1495, 1449, 1128 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; EI-MS m/z: 344 [M]+, 191, 175, 150; HR-EI-MS m/z: 344.2358 [M]+ (calcd for C22H32O3, 344.2351).
(−)-13,14-Dihydroxyneogrifolin (6)
Colorless oil; [α]22D −9.2 (c 0.20, MeOH); UV λmax (MeOH) (log ε) 282 (2.65) nm; IR (KBr) νmax 3440, 2969, 2924, 2856, 1628, 1452, 1141 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; EI-MS m/z: 362 [M]+, 344 [M–H2O]+, 326 [M–2 × H2O]+, 191, 175, 137; HR-EI-MS m/z: 362.2432 [M]+ (calcd for C22H34O4, 362.2457).
Albatrelin G (7)
Colorless oil; [α]22D −15.2 (c 0.19, MeOH); UV λmax (MeOH) (log ε) 283 (2.93) nm; IR (KBr) νmax 3441, 2969, 2929, 2855, 1640, 1615, 1495, 1452, 1141 cm−1; 1H and 13C NMR spectroscopic data, see Table 3; EI-MS m/z: 344 [M]+, 326 [M–H2O]+, 175; HR-EI-MS m/z: 344.2348 [M]+ (calcd for C22H32O3, 344.2351).
Table 3.
1H and 13C NMR spectroscopic data for compounds 7–9 in acetone-d6 (δ in ppm, J in Hz)
| No | 7 b | 8 a | 9 a | |||
|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| 1 | 113.7, C | 115.6, C | 113.9, C | |||
| 2 | 139.1, C | 139.1, C | 156.7, C | |||
| 3 | 109.3, CH | 6.11, d (2.3) | 109.6, CH | 6.13, d (2.2) | 108.4, CH | 6.19, s |
| 4 | 157.0, C | 157.0, C | 136.5, C | |||
| 5 | 100.6, CH | 6.09, d (2.3) | 100.7, CH | 6.07, d (2.2) | 108.4, CH | 6.19, s |
| 6 | 158.6, C | 158.6, C | 156.7, C | |||
| 7 | 20.0, CH3 | 2.01, s | 20.9, CH3 | 2.36, s | 21.2, CH3 | 2.10, s |
| 8 | 33.9, CH | 3.22, br. s | 30.3, CH | 3.52, br. s | 18.2, CH2 | 2.67, m |
| 9 | 38.9, CH2 | 1.97–2.00, dd (12.8, 3.0) | 40.4, CH2 | 1.87, dd (12.8, 3.2) | 41.2, CH2 | 1.66–1.70, m |
| 1.69–1.73, dd (12.8, 3.0) | 1.62, dd (12.8, 8.0) | |||||
| 10 | 74.2, C | 74.5, C | 72.6, C | |||
| 11 | 40.8, CH2 | 1.89, br. d (11.0) | 41.5, CH2 | 1.89–1.92, m | 42.6, CH2 | 1.50–1.54, m |
| 1.62–1.64, m | 1.53–1.56* | |||||
| 12 | 24.6, CH2 | 1.49–1.55, m | 21.6, CH2 | 1.53–1.56* | 23.3, CH2 | 2.10–2.15, m |
| 1.20–1.34, m | 1.31–1.35, m | |||||
| 13 | 48.7, CH | 2.34, dt (12.5, 2.7) | 55.9, CH | 1.76, dt (12.8, 2.0) | 125.9, CH | 5.15, t (6.9) |
| 14 | 152.5, C | 74.0, C | 135.0, C | |||
| 15 | 37.4, CH2 | 2.09–2.25* | 36.8, CH2 | 1.35–1.42,m | 40.4, CH2 | 1.96, t (7.5) |
| 1.00, m | ||||||
| 16 | 27.7, CH2 | 2.09–2.25* | 23.0, CH2 | 2.09–2.14, m | 27.4, CH2 | 2.07* |
| 1.96–2.01, m | ||||||
| 17 | 125.2, CH | 5.16, t (6.6) | 126.1, CH | 4.98, t (7.0) | 125.1, CH | 5.10, t (7.1) |
| 18 | 131.9, C | 130.9, C | 131.6, C | |||
| 19 | 17.7, CH3 | 1.60, s | 17.6, CH3 | 1.56, s | 17.7, CH3 | 1.58, s |
| 20 | 25.8, CH3 | 1.66, br. s | 25.8, CH3 | 1.59, s | 25.8, CH3 | 1.65, s |
| 21 | 109.7, CH2 | 4.64, br. s | 27.3, CH3 | 1.13, s | 15.9, CH3 | 1.62, s |
| 4.28, br. s | ||||||
| 22 | 29.1, CH3 | 1.28, s | 29.0, CH3 | 1.26, s | 27.5, CH3 | 1.20. s |
| 4-O H | 7.96, s | 7.91, s | 8.05, s | |||
| 6-O H | 8.05, s | |||||
| 10-O H | 3.54, s | |||||
| 14-O H | 3.30, s | |||||
a1H NMR spectra were measured at 400 MHz, and 13C NMR spectra at 100 MHz
b1H NMR spectra was measured at 500 MHz, and 13C NMR spectra at 125 MHz
* Signals were overlapped
Albatrelin H (8)
Colorless oil; [α]22D −9.1 (c 0.21, MeOH); UV λmax (MeOH) (log ε) 284 (2.63) nm; IR (KBr) νmax 3441, 2968, 2929, 2872, 2855, 1615, 1595, 1452, 1145 cm−1; 1H and 13C NMR spectroscopic data, see Table 3; EI-MS m/z: 344 [M]+, 175, 137; HR-EI-MS m/z: 344.2345 [M]+ (calcd for C22H32O3, 344.2351).
(S)-10-hydroxygrifolin (9)
Colorless oil; [α]22D −8.7 (c 0.20, MeOH); UV λmax (MeOH) (log ε) 276 (2.50) nm; IR (KBr) νmax 3441, 2967, 2925, 2856, 1628, 1598, 1452, 1381, 1050 cm−1; 1H and 13C NMR spectroscopic data, see Table 3; EI-MS m/z: 346 [M]+, 328 [M–H2O]+, 175, 137; HR-EI-MS m/z: 346.2505 [M]+ (calcd for C22H34O3, 346.2508).
Preparation of 6a
To a solution of compound 6 (2.3 mg, 6.35 μmol) in DMF (2 mL) were added 2,2-dimethoxypropane (1.3 mg, 12.7 μmol) and p-toluenesulfonic acid monohydrate (0.6 mg, 3.18 μmol), and the mixture was stirred for 30 min at room temperature. The reaction mixture was added into water, and then extracted by EtOAc for three times. The organic layer was evaporated and the residue was chromatographed on a column of silica gel eluting with petroleum ether-acetone 40:1 to yield 6a (2.1 mg).
Cytotoxic assay
The following human tumor cell lines were used: HL-60, SMMC-7712, A-549, MCF-7, and SW480. All the cells were cultured in RMPI-1640 or DMEM medium (Hyclone, Logan, UT), supplemented with 10 % fetal bovine serum (Hyclone) at 37 °C in a humidified atmosphere with 5 % CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan formed in living cells based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO). Briefly, 100 μL of adherent cells were seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1 × 105 cells/mL in 100 μL of medium. Each tumor cell line was exposed to the test compounds at various concentrations in triplicate for 48 h, with DDP and toxal as positive controls. After the incubation, MTT (100 μg) was added to each well, and the incubation continued for 4 h at 37 °C. The cells lysed with 200 μL SDS after removal of 100 μL of medium. The optical density of lysate was measured at 595 nm in a 96-well microtiter plate reader (Bio-Rad 680). The IC50 value of each compound was calculated by Reed and Muench’s method [17].
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgment
This project was supported by the National Natural Sciences Foundation of China (U1132607).
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.Ding ZH, Dong ZJ, Liu JK. Helv. Chim. Acta. 2001;84:259–262. doi: 10.1002/1522-2675(20010131)84:1<259::AID-HLCA259>3.0.CO;2-O. [DOI] [Google Scholar]
- 2.Koch B, Steglich W. Eur. J. Org. Chem. 2007;2007:1631–1635. doi: 10.1002/ejoc.200601022. [DOI] [Google Scholar]
- 3.Nukata M, Hashimoto T, Yamamoto I, Iwasaki N, Tanaka M, Asakawa Y. Phytochemistry. 2002;59:731–737. doi: 10.1016/S0031-9422(02)00050-X. [DOI] [PubMed] [Google Scholar]
- 4.Quang DN, Hashimoto T, Arakawa Y, Kohchi C, Nishizawa T, Soma GI, Asakawa Y. Bioorg. Med. Chem. 2006;14:164–168. doi: 10.1016/j.bmc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Yang XL, Qin C, Wang F, Dong ZJ, Liu JK. Chem. Biodivers. 2008;5:484–489. doi: 10.1002/cbdv.200890047. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Dong ZJ, Liu JK. Acta Bot. Yunn. 2009;31:187–189. doi: 10.3724/SP.J.1143.2009.08234. [DOI] [Google Scholar]
- 7.Hashimoto T, Quan DN, Nukada M, Nukada M, Asakawa Y. Heterocycles. 2005;65:2431–2439. doi: 10.3987/COM-05-10501. [DOI] [Google Scholar]
- 8.Luo DQ, Shao HJ, Zhu HJ, Liu JK. Z. Naturforschung. 2005;C60:50–56. doi: 10.1515/znc-2005-1-210. [DOI] [PubMed] [Google Scholar]
- 9.Kawagishi H, Tanaka A, Sugiyama K, Mori H, Sakamoto H, Ishoguro Y, Kobayashi K, Uramoto M. Phytochemistry. 1996;42:547–548. doi: 10.1016/0031-9422(95)00881-0. [DOI] [PubMed] [Google Scholar]
- 10.Hellwig V, Nopper R, Mauler F, Freitag J, Liu JK, Ding ZH, Stadler M. Arch. Pharm. 2003;336:119–126. doi: 10.1002/ardp.200390008. [DOI] [PubMed] [Google Scholar]
- 11.Ye M, Liu JK, Lu ZX, Zhao Y, Liu SF, Li LL, Tan M, Weng XX, Li W, Cao Y. FEBS Lett. 2005;579:3437–3443. doi: 10.1016/j.febslet.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Zheng HD, Liu PG. Microbiol. China. 2006;33:104–107. [Google Scholar]
- 13.Jones S, Valette D. Org. Lett. 2009;11:5358–5361. doi: 10.1021/ol902280d. [DOI] [PubMed] [Google Scholar]
- 14.Nakata M, Arai M, Tomooka K, Ohsawa N, Kinoshita M. Bull. Chem. Soc. Jpn. 1989;62:2618–2635. doi: 10.1246/bcsj.62.2618. [DOI] [Google Scholar]
- 15.Ziffer H, Kawai KI, Kasai M, Imuta M, Froussios C. J. Org. Chem. 1983;48:3017–3021. doi: 10.1021/jo00166a016. [DOI] [Google Scholar]
- 16.Mukaiyama T, Shintou T, Fukumoto K. J. Am. Chem. Soc. 2003;125:10538–10539. doi: 10.1021/ja0303844. [DOI] [PubMed] [Google Scholar]
- 17.Reed LJ, Muench H. Am. J. Hyg. 1938;27:493–497. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.