

Abstract
The prevalence of abnormalities in androgen receptor gene (AR) among patients with ambiguous genitalia is unknown. Moreover, endocrinological data from prepubertal patients with AR mutation are very limited. Thus, the aim of this study was to examine the prevalence of abnormalities in AR among patients with both ambiguous genitalia, which was defined as a combination of two or more genital abnormalities (i.e. hypospadias, microphallus (penile length < 25 mm), hypoplastic scrotum, bifid scrotum, undescended testis) in this study, and normal to elevated T levels. We also compared the endocrinological data of prepubertal patients with AR mutation and ambiguous genitalia with that of those without the AR mutation. We screened 26 Japanese prepubertal 46,XY patients (five from three families were included) with both ambiguous genitalia and normal to elevated T levels. Mutations in AR were found in three (two of the three were related). Among the 23 patients without mutation in AR, the steroid 5-alpha-reductase 2 gene (SRD5A2) was also examined in eight patients with elevated T/dehydrotestosterone ratio after the hCG (>10) or with undervirilized family members. No mutation in SRD5A2 was found. Characteristics of the three patients with mutation in AR were compared with the 23 patients without mutation. In two patients, basal T levels (0.3, 0.2 ng/ml) and peak T levels after the hCG tests (8.3, 8.5 ng/ml) tended to be higher, and the peak LH/ peak FSH ratios after the GnRH tests (4.6, 4.0) were higher than in patients without mutation, at the ages of 1 yr and 9 mo and 3 yr and 8 mo, respectively. In conclusion, an abnormality in either AR or SRD5A2 was not common among patients with ambiguous genitalia and normal testosterone secretion. Elevated peak LH/peak FSH ratio (≥4) after the GnRH test in addition to detectable basal T levels and elevated peak T levels after the hCG test may infer AR abnormality in prepubertal patients with ambiguous genitalia at the age of one and over, although further study is needed, because our data were limited.
Keywords: androgen receptor; ambiguous genitalia; prevalence; 46,XY disorders of sex development
Background
Androgen receptor gene (AR) is a possible candidate gene for causes of 46,XY disorders of sex development (DSD) with normal T secretion. Androgen receptor (AR) dysfunction due to mutations in AR results in a wide spectrum of abnormalities of male sexual development ranging from phenotypic female to male with infertility (1). The expanded length of CAG repeats in the N-terminal region of AR may also cause AR dysfunction (2). However, abnormality in AR is an uncommon occurrence in isolated hypospadias, undescended testis or microphallus. The prevalence of abnormality in AR was reported to be from zero out of twenty-six to one out of nine among isolated hypospadias (3,4,5), and zero out of twenty-one among isolated undescended testis (6). However, the prevalence of mutation in AR among patients with ambiguous genitalia, which was defined as a combination of two or more genital abnormalities (i.e. hypospadias, microphallus (penile length <25 mm), hypoplastic scrotum, bifid scrotum, undescended testis) in this study, is unknown.
Steroid 5-alpha-reductase 2 gene (SRD5A2) is also a possible candidate gene in 46,XY DSD among patients with ambiguous genitalia and elevated T/dihydrotestosterone (DHT) ratio after the human CG (hCG) test. However, there is no report on the prevalence of mutation in SRD5A2 among such patients.
The endocrine characteristics of AR abnormality have been established in postpubertal patients and patients before the age of one year. In partially virilized postpubertal patients, major hormonal clues inferring AR abnormality are elevated T and LH levels (1, 7). In partially virilized patients before the age of one year, major hormonal clues inferring AR abnormality are normal to elevated T and LH levels (7,8,9). Endocrinological data from patients with AR dysfunction in prepubertal children after the age of one year are so limited that the criteria for establishing patients who should be screened for AR mutations remain unknown. Only one complete female patient with an AR mutation at this stage has been reported she had slightly elevated basal T and LH levels and markedly elevated peak LH levels after the GnRH test (basal T level, 1.05 ng/ml (normal, 0.03–0.12); basal LH level, 5.3 mIU/ml (normal, 0.1–4.7); peak LH and peak FSH levels after the GnRH test, 90.7 and 4.4 IU/L, respectively, at the age of 1 yr) (10). Endocrinological data of partially virilized children with AR dysfunction at the prepubertal stage are also limited. Normal basal levels of both T and LH (1, 11, 12) have been reported. Two patients in this study (patients 25 and 26) were reported with their endocrinological data, previously (13, 14), and these cases remind us that T after the hCG test and LH after the GnRH test are relatively elevated. Considering the quiescence of the GnRH-gonadal axis in the prepuberatal period and the elevated peak LH levels in the complete female patient, as described above (10), stimulated data are presumed to be of value in partially virilized cases for inferring a diagnosis of AR dysfunction.
In these circumstances, we hypothesized that mutation in AR is more frequent among patients with ambiguous genitalia and normal to elevated T levels than patients with isolated hypospadias, undescended testis or microphallus. Therefore, we screened 26 Japanese prepubertal 46,XY patients with both ambiguous genitalia and normal to elevated T levels (initial visit, 0–48 mo; hormonal examinations, 0–124 mo) to determine the prevalence of AR mutations. Three patients were found to have AR mutations. Subsequently, SRD5A2 was sequenced for eight patients with an elevated T/DHT ratio after the hCG test (>10) or family members with autosomal recessively inherited undervirilization among the 23 patients without mutation in AR. We also clarified the endocrinological data of the prepubertal patients with ambiguous genitalia and stimulated levels of T and LH, in addition to basal levels of T and LH, in two of the three patients and examined the differences between them and the patients without AR mutation.
Patients and Methods
Patients
From 1988 to 1999, 1,386 Japanese patients with hypospadia, undescended testis, or both were referred to our hospital. Out of these 1,386, gonadal function, secretion of testosterone, was further assessed endocrinologically using the stimulation tests (GnRH and hCG test) (n=76) or by a basal hormonal study (n=3) in patients with two or more genital abnormalities (i.e. hypospadias, microphallus (<25 mm), hypoplastic scrotum, bifid scrotum, undescended testis), which was defined as ambiguous genitalia in this study. Among these 79 patients, 29 patients with 46,XY karyotype (0–48 mo old at initial visits) and normal to elevated testosterone secretion were included in this study. Among these 29 patients, the 26 patients for whom written informed consent for genetic analysis was obtained are described in this study. Testosterone secretion was defined as normal to elevated if peak T level increased to 2.0 ng/ml and over after hCG provocation at least once (n=25). In addition, detectable basal T level (≥0.1 ng/ml) at an initial visit after the age of one year was defined as elevated testosterone secretion (n=1) in three patients for whom stimulation tests were not performed. Ovotesticular DSD and malformation syndrome such as Rubinstein Taybi syndrome were excluded.
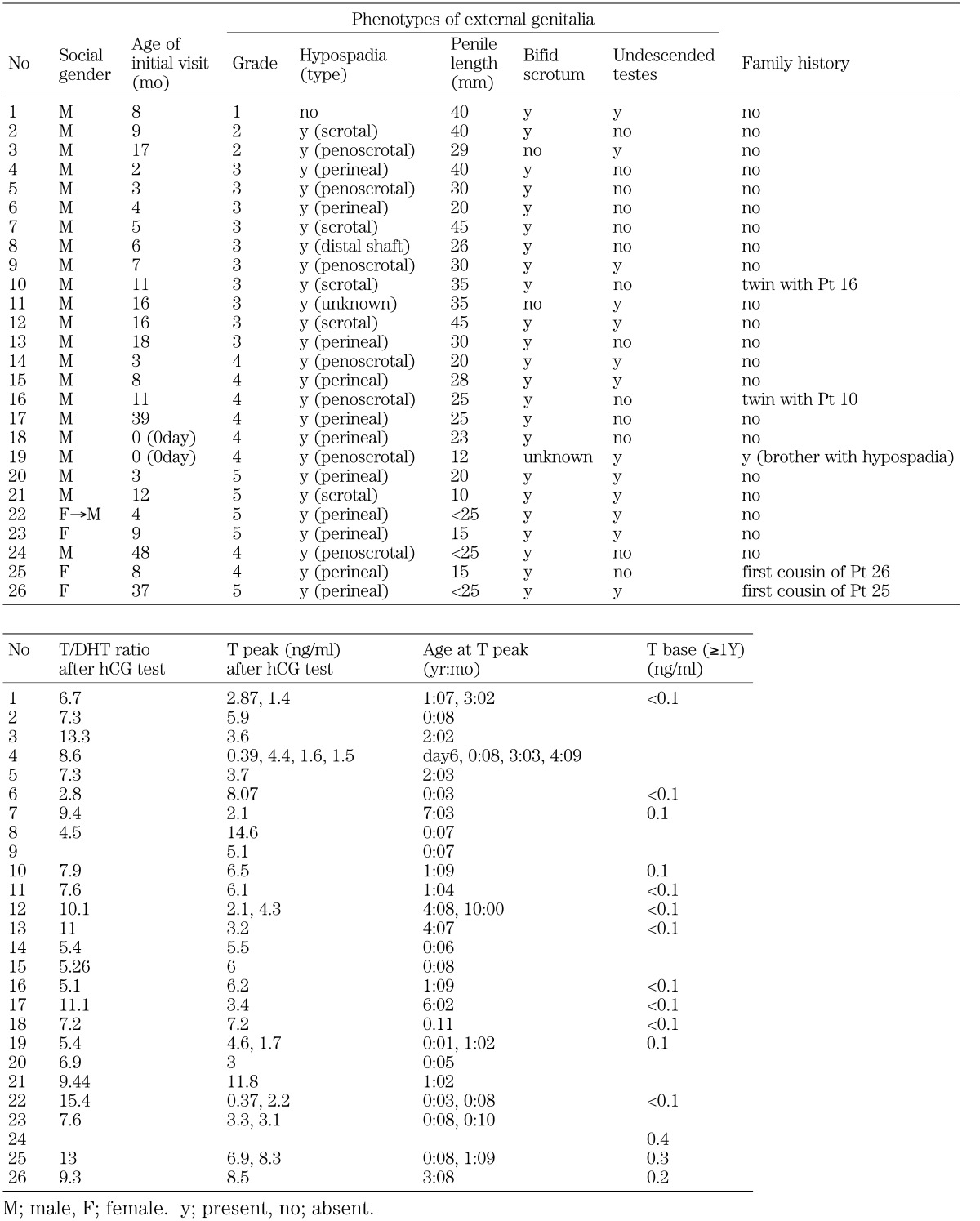
Features of the 26 patients are shown in Fig. 1 and are summarized in Table 1. Five patients were from three families. Patients 25 and 26 have been reported elsewhere (13, 14). The genital phenotypes of the patients were graded 2 to 5 in order of increasing severity of androgen resistance (1) (Fig. 1). Three were reared as social females, and 23 were reared as social males. Seven out of the 26 patients were tested with hCG, repeatedly. Out of the seven patients, four patients showed peak T levels of both above and below 2.0 ng/ml. Serum T levels and T/DHT after the hCG test were 0.39–14.6 ng/ml and 2.8–15.4, respectively (0–124 mo old at hormonal examinations).
Fig. 1.

Features of the patients. Genital phenotypes of our patients were graded 2 to 5 in order of increasing severity of androgen resistance. Grade 1: normal virilization in utero; Grade 6/7: complete female phenotype. *one patient had microphallus and undescended testis without hypospadias.
Table 1. Characteristics of patients.
This study was approved by a local ethics committee and written consents were obtained from the parents of all the participants.
Methods
Protocols of the provocation tests
GnRH test: Serum was obtained at 0, 30, 60, 90, and 120 min after intravenous administration of 100 μg/m2 GnRH (max 100 μg). Serum LH and FSH levels were measured by RIA. Intradaily and interdaily assay variations of LH and FSH measurements were less than 10% at three different concentrations. The lowest values of this assay for LH and FSH were less than 0.5 mIU/ml for both assays.
hCG test: For three consecutive days, a dose of 4000 unit hCG/m2/day (max 5000 unit/day) was administered intramuscularly each morning. T and DHT levels were measured by RIA before injection and at 48, 72, and 96 h after the last injection. Intradaily and interdaily assay variations of T and DHT measurements were less than 10% at three different concentrations. The lowest values of this assay for T and DHT were less than 0.1 ng/ml and 0.02 ng/ml, respectively. Normal basal T level is undetectable (<0.1 ng/ml) during the prepubertal period after the age of one year and an increase in the plasma T levels to 2.0 ng/ml or more is considered as normal (15,16,17) during the whole prepubertal period; a peak T/DHT ratio of less than 10 is considered normal (16).
Mutational analyses of AR and SRD5A2
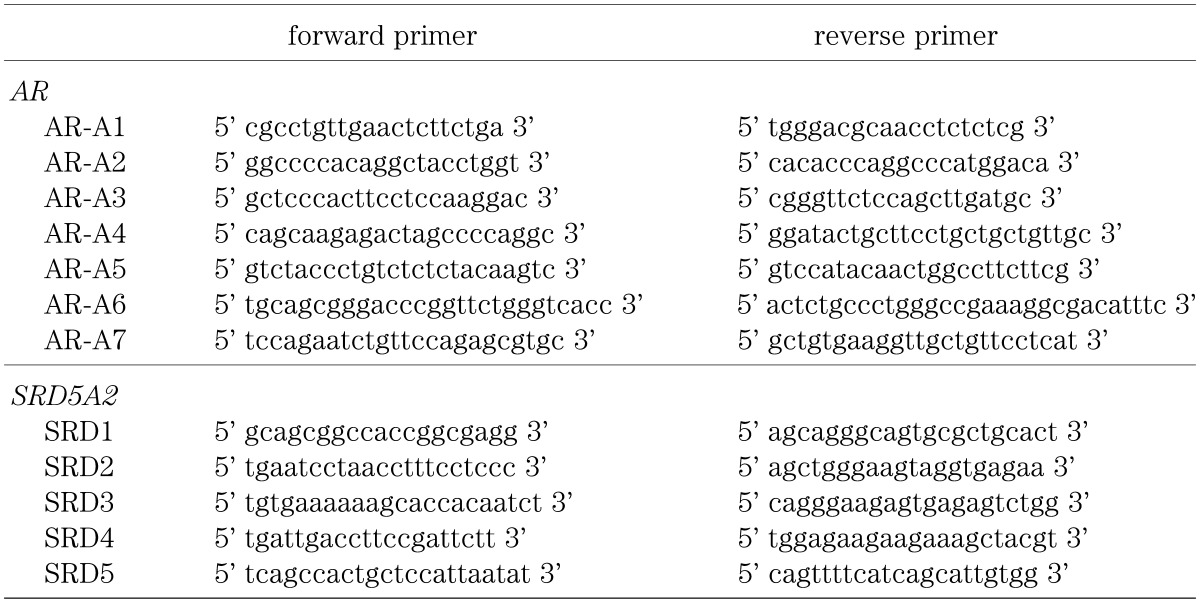
Genomic DNA was extracted from peripheral blood leukocytes of the patients. To examine the AR and SRD5A2 sequence, exons 1 to 8 of AR and exons 1 to 5 of SRD5A2 were amplified by PCR individually from genomic DNA using the oligonucleotide primer pairs. Primer pairs for exons 2 to 8 of AR were available upon reference (18). Primer pairs for exon 1 of AR are described in Table 2. Because at least 14 intronic mutations of AR have been reported (19), primers for AR were prepared long enough to include all the reported sites of mutation. Primer pairs for SRD5A2 are described in Table 2. The nucleotide sequence of the PCR products of the patients were directly determined by a sequence analyzer (ABI PRISM™ 310 Genetic analyzer; Applied Biosystems) in both directions. The number of the CAG repeats, a polymorphic site in exon 1 of AR, was also examined, because expansions of more than 40 repeats can result in androgen insensitivity (2, 20).
Table 2. Primer pairs for exon 1 of AR and exon 1 to 5 of SRD5A2.
AR, which is considered to be the most frequent genetic cause of 46,XY DSD, was sequenced for all the 26 patients. Subsequently, SRD5A2 was sequenced for eight patients with an elevated T/DHT ratio after the hCG test (>10) (n=5; T/DHT=10.1–15.4) or family members with an autosomal recessively inherited undervirilization (n=3 out of 2 families) among the 23 patients without mutation in AR.
Examination of characteristics of the patients with mutations in AR
Patients with mutations in AR were further examined for clinical features and laboratory data. All 26 patients were divided into two groups according to presence or absence of mutation (mutation (+) (n=3) and mutation (–) (n=23), respectively). As for clinical features, degree of external ambiguity according to Quigley’s grade (1) (n=26) and existence of vagina or prostatic utricle (n=11) were compared between the two groups.
As for laboratory data, basal levels of T, LH and FSH, peak levels of T after the hCG test, peak levels of LH and FSH, and peak LH / peak FSH ratio after the GnRH test were compared among the patient, with and without mutation, groups and a normal control group of 31 healthy prepubertal males aged from 4 mo to 10 yr and 4 mo. Multiple data from the same patients were included (multiple basal T levels, peak T levels after the hCG test (15), and LH peak levels after the GnRH test were available in 13, seven, and 19 patients, respectively). Data obtained after orchidopexy were not included.
Results
Mutational analysis of AR
Three (Patients 24, 25 and 26) of the 26 patients had two different mutations in AR (V746M, R840H). Mutation and functional analyses of patients 25 and 26 with R840H have been reported previously (13, 14). Mutations in AR were found in one out of 21 (4.8%) sporadic patients and one family out of three families. Mutations in AR were found in two out of 24 (8.3%) patients, if only probands were included.
The numbers of the CAG repeats were from 15 to 28 in all patients; the number of the CAG repeats in healthy populations ranges from 11-32 (20,21,22,23,24).
Mutational analysis of SRD5A2
No mutations in SRD5A2 were found among the eight patients examined.
Characteristics of the patients with mutations in the AR gene
Table 3. Characteristics of patients (Number of patients).

Fig. 2.

Basal and peak T levels after hCG test. Basal T levels (left) and peak testosterone levels (right) after the hCG test in patients with ambiguous genitalia (◆: without AR mutation, ■: with AR mutation) and normal control subject (△) in this study. A line shows previously reported normal mean basal T values(30, 31). Data of patients with AR mutation at ages of 8 mo (# 25a: basal and peak T level=0.41 and 6.94 ng/ml, respectively), 1 yr 9 mo (# 25b: basal and peak T level=0.3 and 8.3 ng/ml, respectively), 3 yr 8 mo (# 26: basal and peak T level=0.2 and 8.5 ng/ml, respectively), and 4 yr (# 24: basal T level=0.3 ng/ml) were obtained from patients 25, 25, 26 and 24, respectively. Patients 25 and 26 were cousins (R840H) and both were social females (Quigley’s grade 4 and 5, respectively). Patient 24 was a social male, Quigley’s grade 3.
Fig. 3.

Peak levels of LH and FSH after GnRH test. Peak LH (left) and peak FSH (right) levels after the GnRH test in patients with ambiguous genitalia (◆: without AR mutation, ■: with AR mutation) and normal control subject (△) in this study. Data of patients with mutation at the ages of 1 yr 9 mo (# 25b: peak LH level / peak FSH level=37.0 / 8.1 mIU/ml) and 3 yr 8 mo (# 26: peak LH level / peak FSH level=25.8 / 6.4 mIU/ml) were obtained from patients 25 and 26, respectively.
Fig. 4.

LH peak / FSH peak ratio after GnRH test. Peak LH / peak FSH ratio after the GnRH test in patients with ambiguous genitalia (◆: without AR mutation, ■: with AR mutation) and normal control subject (△) in this study. Data of patients with mutation at ages of 1 yr 9 mo (# 25b: peak LH / peak FSH ratio=4.6) and 3 yr 8 mo (# 26: peak LH / peak FSH ratio =4.0) were obtained from patients 25 and 26, respectively.
Clinical features of the patients with mutations in the AR gene
The clinical features of the patients are shown in Table 3. All three patients with mutation in AR showed hypospadias, microphallus and bifid scrotum. Patient 24, with external genitalia cllasified as grade 4, was a social male (V746M), and the other two (grades 4 and 5, patients 25 and 26, respectively) were social females who were the first cousins (R840H).
Of all the 26 patients, 11 patients (AR mutation (n=2), without AR mutation (n=9)) were examined for the existence of a vagina. A vagina or enlarged prostatic utricle was found in two patients with AR mutation and six without AR mutation.
Laboratory data of the patients with mutations in the AR gene
Because a decrease of T, LH and FSH levels with age is known during the first year of life, data were analyzed separately before and after the age of 1 yr. However only one T level (# 25a; see below) was available before one year of age. Data at the age of 8 mo (# 25a), 1 yr 9 mo (# 25b), 3 yr 8 mo (# 26), and 4 yr (# 24) were obtained from patients 25, 25, 26 and 24, respectively.
Basal and peak T levels after the hCG test are shown in Fig. 2. In the AR mutation group, basal T levels were 0.3, 0.2 and 0.4 ng/ml (# 25b, # 26 and #24), and peak T levels were 8.3 and 8.5 ng/ml (# 25b and # 26) at ages over one year. At over 1 yr of age, all basal T levels were undetectable in the group without AR except for the following three subjects: Patients 7, 10 and 19; T level 0.1 at 7 yr 3 mo, 1 yr 10 mo and 1 yr 2 mo, respectively. Peak T levels were 1.4–6.5 ng/ml, except for one at 11.8 ng/ml, in the group without AR mutation, and 2.2–8.0 ng/ml in the normal control group (15). Although both basal and peak T levels in the AR mutation group overlapped values in the group without AR mutation to some extent, they tended to be higher than values in the group without AR mutation and the normal control groups.
At the age of 8 mo, basal and peak T levels of patient 25 (# 25a, AR mutation) were 0.41 and 6.94 ng/ml, respectively, and overlapped data of the group without AR mutation (basal T levels, <0.1–3.7 ng/ml; peak T levels, 0.39–14.6 ng/ml) and the normal control group (basal T level, 0.9 ng/ml; peak T levels, 5.1, 10.1 ng/ml) at under one year of age.
Peak LH and FSH levels after the GnRH test are shown in Fig. 3. At ages of 1 yr and over, peak LH levels in the AR mutation group (37.0 and 25.8 mIU/ml, # 25b and # 26, respectively) were higher than those of the other two groups. Peak FSH levels (8.1 and 6.4 mIU/ml, # 25b and # 26, respectively) in the AR mutation group overlapped the other two groups. Peak LH / peak FSH ratios after the GnRH test were 4.6 (# 25b) and 4.0 (# 26) in the AR mutation group (Fig. 4). All the ratios in the group without mutation and normal control groups were below 4.0. Basal LH and FSH levels of the three groups overlapped (data not shown).
There was no data of LH and FSH levels in the patient with AR mutation before one year.
Discussion
Two types of mutations in AR were found in this study and they were thought to be the cause of ambiguous genitalia of three patients. Although the binding assay was not available for one patient with V746M, this mutation may be the cause of the ambiguous genitalia for following reasons. This mutation is located in the ligand binding domain and the amino acid is conserved among other species such as mus musculus, canis familiaris and rattus norvegicus. Two patients were reported to have the same mutation (19, 25). Patients 25 and 26 had the same mutation, R840H, which was the cause of ambiguous genitalia, because the two patients were related and the binding study revealed thermolability as reported previously (13, 14). In addition, at least 17 cases with mutations in the same amino acid (R840H, R840C, R840S) have been reported (19).
Among the patients with both ambiguous genitalia and normal to elevated T levels, the prevalence of AR was two out of 24 (8.3%), if only probands were included in this study. Mutation in AR was found in one out of 21 (4.8%) sporadic patients and one family out of three families. No mutation in SRD5A2 was found among the eight patients with elevated T/DHT ratio or autosomal recessively inherited family history of undervirilization out of the 23 patients without AR mutation. According to previous studies, abnormalities in either AR or SRD5A2 were not common among patients with isolated hypospadias, undescended testis or microphallus (3,4,5,6, 26, 27). In this study, mutations in AR and SRD5A2 were comparable in prevalence to those patients even among the more suspected patients in this study.
The causes of ambiguity in external genitalia in the 23 patients who had no mutations in AR or SRD5A2 are unknown. Three causes are possible. First, androgen insensitivity due to undetected mutations in introns or abnormalities in cofactors of AR is possible (28). All the intronic mutations reported so far were excluded in this study. However other intronic mutations, which result in AR abnormalities, are possible. Second, they may actually have decreased testosterone secretion. Peak T levels of over 2.0 ng/ml after the hCG test is not a clear cut-off, and lack of reproducibility of peak values was observed in this study. Lastly, SRD5A2 mutations may be responsible for the ambiguous external genitalia since SRD5A2 was not sequenced for 16 patients in this study.
On the basis of our hormonal studies, which were conducted for two of the three patients with AR mutation, patients with ambiguous genitalia between the ages of 1 and 10 yr showed elevated basal T levels, and elevated peak levels of T after the hCG test and LH after the GnRH test. These hormonal data are essentially similar to the data of adult patients with AR abnormality, that is, normal to elevated basal levels of T and LH (1). Endocrinological data for partially virilized patients with AR abnormality in prepubertal children after the age of one year are limited. Although basal levels of T and LH were reported to be normal in partially virilized patients with possible AR dysfunction during the prepubertal period after the age of one (1, 11, 12), our data showed that basal T levels were higher than the undetectable limit. Ascertainment bias could not be denied, because one of our inclusion criteria was detectable basal T level in one patient of this study.
Since the GnRH-gonadal axis is quiescent in the prepubertal period after the age of one, it is difficult to evaluate basal hormonal levels, and the hCG and GnRH tests may be useful for infering AR abnormality, particularly among patients with less severe phenotype as was the case in this study. Elevated levels of T after the hCG test and LH after the GnRH test were observed in this study. However, a previous report described normal peak T levels after the hCG test in partially virilized patients with possible AR dysfunction in the prepubertal period (29). A peak LH / peak FSH ratio over 4 is another possible marker for AR abnormality in patients with ambiguous genitalia during prepuberatal period after the age of one. However, the usefulness of the ratio is unknown due to limited data and further study is needed.
Conclusion
In conclusion, among the patients with both ambiguous genitalia and normal to elevated T levels, the prevalence of AR mutation was two out of 24 (8.3%), if only probands were included. Mutation in AR was found in one out of 21 (4.8%) sporadic patients and one family out of three families. Mutation in SRD5A2 was not found in eight patients with elevated T/DHT and no abnormality of AR. Elevated peak LH/peak FSH ratio (≥4) after the GnRH test in addition to elevated basal and peak T levels after the hCG test may infer AR abnormality in patients with ambiguous genitalia in the prepuberatal period after the age of one, although further study is needed.
References
- 1.Quigley CA, Bellins A, Marschke KB, El-Awady MK, Wilson EM, French FS. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr Rev 1995;16: 271–321 doi: 10.1210/edrv-16-3-271 [DOI] [PubMed] [Google Scholar]
- 2.Chamberlain NL, Driver ED, Miesfeld RL. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res 1994;22: 3181–6 doi: 10.1093/nar/22.15.3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutherland RW, Wiener JS, Hicks JP, Marcelli M, Gonzales ET, Jr, Lamb DJ, et al. Androgen receptor gene mutations are rarely associated with isolated penile hypospadias. J Urol 1996;156(2 Pt 2): 828–31 [DOI] [PubMed] [Google Scholar]
- 4.Allera A, Herbst MA, Griffin JE, Wilson JD, Schweikert HU, McPhaul MJ. Mutations of the androgen receptor coding sequence are infrequent in patients with isolated hypospadias. J Clin Endocrinol Metab 1995;80: 2697–9 [DOI] [PubMed] [Google Scholar]
- 5.Nordenskjold A, Friedman E, Tapper-Persson M, Soderhall C, Leviav A, Anvret M, et al. Screening for mutations in candidate genes for hypospadias. Urol Res 1999;27: 49–55 doi: 10.1007/s002400050088 [DOI] [PubMed] [Google Scholar]
- 6.Wiener JS, Marcelli M, Gonzales ET, Jr, Roth DR, Lamb DJ. Androgen receptor gene alterations are not associated with isolated cryptorchidism. J Urol 1998;160(3 Pt 1): 863–5 [DOI] [PubMed] [Google Scholar]
- 7.Grumbach MM, Hughes LA, Conte FA. Disorders of sex differentiation. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, editors. Williams textbook of endocrinology, 10th Edition. Philadelphia: Saunders; 2002. p.842–1002. [Google Scholar]
- 8.Nagel RA, Lippe BM, Griffin JE. Androgen resistance in the neonate: use of hormones of hypothalamic-pituitary-gonadal axis for diagnosis. J Pediatr 1986;109: 486–8 doi: 10.1016/S0022-3476(86)80123-8 [DOI] [PubMed] [Google Scholar]
- 9.Lee PA, Brown TR, LaTorre HA. Diagnosis of the partial androgen insensitivity syndrome during infancy. JAMA 1986;255: 2207–9 doi: 10.1001/jama.1986.03370160105034 [DOI] [PubMed] [Google Scholar]
- 10.Imasaki K, Okabe T, Murakami H, Tanaka Y, Takayanagi R, Nawata H, et al. Androgen insensitivity syndrome due to new mutations in the DNA-binding domain of the androgen receptor. Mol Cell Endocrinol 1996;120(1): 15–24 doi: 10.1016/0303-7207(96)03812-9 [DOI] [PubMed] [Google Scholar]
- 11.Tremblay RR, Foley Jr TP, Corvol P, Park IJ, Kowarski A, Migeon CJ, et al. Plasma concentration of testosterone, dihydrotestosterone, testosterone-oestradiol binding globulin, and pituitary gonadotrophins in the syndrome of male pseudo-hermaphroditism with testicular feminization. Acta Endocrinol (Copenh) 1972;70: 331–41 [DOI] [PubMed] [Google Scholar]
- 12.Cicognani A, Cacciari E, Tacconi M, Pascucci MG, Tonioli S, Balsamo A, et al. Effect of gonadectomy on growth hormone, IGF-1 and sex steroids in children with complete and incomplete androgen insensitivity. Acta Endocrinol (Copenh) 1989;121: 777–83 [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa T, Imasaki K, Haji M, Hasegawa Y, Aso T, Nawata H, et al. Incomplete androgen insensitivity associated with a thermolabile androgen receptor. Endocr J 1994;41: 31–5 doi: 10.1507/endocrj.41.31 [DOI] [PubMed] [Google Scholar]
- 14.Imasaki K, Hasegawa T, Okabe T, Sakai Y, Haji M, Nawata H, et al. Single amino acid substitution (840Arg→His) in the hormone-binding domain of the androgen receptor leads to incomplete androgen insensitivity syndrome associated with a thermolabile androgen receptor. Eur J Endocrinol 1994;130: 569–74 doi: 10.1530/eje.0.1300569 [DOI] [PubMed] [Google Scholar]
- 15.Tanaka N, Miyamoto J, Hasegawa Y. A response to human chorionic gonadotropin test in boys with normal gonadal function and with hypogonadism. Clin Pediatr Endocrinol 2001;10: 147–50. doi: 10.1297/cpe.10.147 [DOI] [Google Scholar]
- 16.Pang S, Levine LS, Chow D, Sagiani F, Saenger P, New MI. Dihydrotestosterone and its relationship to testosterone in infancy and childhood. J Clin Endocrinol Metab 1979;48: 821–6 doi: 10.1210/jcem-48-5-821 [DOI] [PubMed] [Google Scholar]
- 17.Walsh PC, Curry N, Mills RC, Siiteri PK. Plasma androgen responce to hCG stimulation in prepubertal boys with hypospadias and cryptorchidism. J Clin Endocrinol Metab 1976;42: 52–9 doi: 10.1210/jcem-42-1-52 [DOI] [PubMed] [Google Scholar]
- 18.Nakao R, Haji M, Yanase Y, Ogo A, Takayanagi R, Nawata H, et al. A single amino acid substitution (Met786→Val) in the steroid-binding domain of human androgen receptor leads to complete androgen insensitivity syndrome. J Clin Endocrinol Metab 1992;74: 1152–7 [DOI] [PubMed] [Google Scholar]
- 19.from AR database:http://www.androgendb.mcgill.ca/
- 20.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fishbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991;352: 77–9 doi: 10.1038/352077a0 [DOI] [PubMed] [Google Scholar]
- 21.Komori S, Kasumi H, Kanazawa R, Sakata K, Nakata Y, Koyama K, et al. CAG repeat length in the androgen receptor gene of infertile Japanese males with oligozoospermia. Mol Hum Reprod 1999;5: 14–6 doi: 10.1093/molehr/5.1.14 [DOI] [PubMed] [Google Scholar]
- 22.Lund A, Tapanainen JS, Lahdetie J, Savontaus ML, Aittomaki K. Long CAG repeats in the AR gene are not associated with infertility in Finnish males. Acta Obstet Gynecol Scand 2003;82: 162–6 doi: 10.1034/j.1600-0412.2003.00068.x [DOI] [PubMed] [Google Scholar]
- 23.Sasagawa I, Suzuki Y, Ashida J, Nakada T, Muroya K, Ogata T. CAG repeat length analysis and mutation screening of the androgen receptor gene in Japanese men with idiopathic axoospermia. J Androl 2001;22: 804–8 [PubMed] [Google Scholar]
- 24.Thangaraj K, Joshi MB, Reddy AG, Gupta NJ, Chakravarty B, Singh L. CAG repeat expansion in the androgen receptor gene is not associated with male infertility in Indian populations. J Androl 2002;23: 815–8 [PubMed] [Google Scholar]
- 25.Hiort O, Sinnecker GH, Holterhus PM, Nitsche EM, Kruse K. The clinical and molecular spectrum of androgen insensitivity syndromes. Am J Med Genet 1996;63: 218–22 doi: [DOI] [PubMed] [Google Scholar]
- 26.Silver RI, Russell DW. 5alpha-reductase type 2 mutations are present in some boys with isolated hypospadias. J Urol 1999;162(3 Pt 2): 1142–5 [DOI] [PubMed] [Google Scholar]
- 27.Gad YZ, Nasr H, Mazen I, Salah N, el-Ridi R. 5 alpha-reductase deficiency in patients with micropenis. J Inherit Metab Dis 1997;20: 95–101 doi: 10.1023/A:1005326027087 [DOI] [PubMed] [Google Scholar]
- 28.Adachi M, Takayanagi R, Tomura A, Imasaki K, Kato S, Nawata H, et al. Androgen-insensitivity syndrome as a possible coactivator disease. N Engl J Med 2000;343: 856–62 doi: 10.1056/NEJM200009213431205 [DOI] [PubMed] [Google Scholar]
- 29.Bertelloni S, Federico G, Baroncelli GI, Cavallo L, Corsello G, Saggese G, et al. Biochemical selection of prepubertal patients with androgen insensitivity syndrome by sex hormone-binding globulin response to the human chorionic gonadotropin test. Pediatr Res 1997;41: 266–71 doi: 10.1203/00006450-199702000-00018 [DOI] [PubMed] [Google Scholar]
- 30.Korth-Schutz S, Levine LS, New MI. Serum androgens in normal prepubertal and pubertal children and in children with precocious adrenarche. J Clin Endocrinol Metab 1976;42: 117–24 doi: 10.1210/jcem-42-1-117 [DOI] [PubMed] [Google Scholar]
- 31.Forest MG, de Peretti E, Bertrand J. Testicular and adrenal androgens and their binding to plasma proteins in the perinatal period: Developmental patterns of plasma testosterone, 4-androstenedione, dehydroepiandrosterone and its sulfate in premature and small for date infants as compared with that of full-term infants. J Steroid Biochem 1980;12: 25–36 doi: 10.1016/0022-4731(80)90247-2 [DOI] [PubMed] [Google Scholar]