

Abstract
Background
Longitudinal, genetically informed, prospective data collected on a large population of male twins (n = 1037) were used to examine developmental differences in the etiology of antisocial behavior.
Method
Analyses were carried out on both mother- and child-reported symptoms of conduct disorder (CD) in 10- to 17-year-old twins from the Virginia Twin Study of Adolescent Behavioral Development (VTSABD) and self-reported antisocial behavior by the twins as young adults from the Young Adult Follow-Up (YAFU) study.
Results
The following trends were identified: (1) a single genetic factor influencing antisocial behavior beginning at age 10 through young adulthood (‘life-course persistent’); (2) a shared-environmental effect beginning in adolescence (‘adolescent-onset’); (3) a transient genetic effect at puberty; and (4) a genetic influence specific to adult antisocial behavior.
Conclusions
Overall, these etiological findings are consistent with predictions from Moffitt’s developmental theory of antisocial behavior. The genetic effect at puberty at ages 12–15 is also consistent with a genetically mediated influence on the timing of puberty affecting the expression of genetic differences in antisocial outcomes.
INTRODUCTION
Antisocial behavior is markedly heterogeneous. Because the broad class of antisocial behavior probably conceals varied forms, studying the manifestation, correlates and underlying etiology of these different subtypes has important implications for prevention and treatment. A meaningful distinction involves antisocial behaviors that are, and those that are not, associated with early temperamental difficulties (i.e. aggression and impulse-control problems), attention deficit hyperactivity disorder (ADHD), poor peer relations, and cognitive impairment (Rutter et al. 1998). Moffitt (1993) delineated two forms of antisocial behavior in her taxonomy: (1) lifelong persistent antisocial behavior that begins in early childhood and is characterized by major impairments continuing into adult life, and (2) adolescence-limited conduct disorder, showing later onset and less adult impairment.
Although they cannot be differentiated by their overt behavioral manifestations, the two forms have been shown to be etiologically heterogeneous, with persistent antisocial problems representing a more genetically influenced form of the condition (DiLalla & Gottesman, 1989; Hinshaw, 1994; Lyons et al. 1995; Miles & Carey, 1997; Rutter et al. 1997; Taylor et al. 2000). Life-course-persistent antisocial behavior is also associated with a different set of childhood variables (Moffitt, 1990; Moffitt et al. 1996; Moffitt & Caspi, 2001), and antisocial behavior co-morbid with ADHD shows a different profile of family risk factors (Lahey et al. 1988; Moffitt, 1990; Barkley et al. 1991; Hinshaw, 1994; Faraone et al. 1995) and a more severe prognosis (Moffitt, 1990). The co-morbid type that begins earlier may also be more strongly genetically influenced (Silberg et al. 1996).
Moffitt’s taxonomy has sometimes been misinterpreted as specifying a categorical differentiation but that is no part of the claim. Rather, she pointed to the evidence that some forms of antisocial behavior began unusually early and that these varieties were particularly likely to persist into adult life. Strikingly, the early onset was associated with a high rate of both individual risk factors (such as hyperactivity and neurodevelopmental impairment) and family adversity, which might reflect either genetic or environmental mediation. Both sets of risk factors are much less frequent in the case of antisocial behavior that begins in adolescence. The only risk factor with a substantial effect in both varieties is being part of a deviant peer group. Moffitt (2003) noted that the implication that genetic influences were likely to be stronger in the case of life-course-persistent antisocial behavior had a modest amount of support from the few studies that had relevant data.
By contrast, the adolescence-limited variety of antisocial behavior is much less likely to be associated with either the individual or family risk factors. It is not that the risk factors are different in type from those associated with life-course-persistent antisocial behavior, but rather that they are much more weakly associated. The one exception concerns membership of a deviant peer group (DiLalla & Gottesman, 1989).
Behavioral genetic studies have provided some empirical support for these etiological distinctions. The study by Lyons et al. (1995) is illustrative in showing a sixfold increase in shared-environmental factors on juvenile conduct disorder (recalled retrospectively) compared to antisocial behavior reported in adulthood. Whereas only 7% of individual differences in the liability to adolescent antisocial behavior could be explained by differences in genetic liability, additive genetic factors accounted for 43% of the variance in antisocial behavior in adulthood. Other studies have found similar results, with greater genetic influence demonstrated for early-onset versus later-onset (i.e. adolescent) delinquency (Taylor et al. 2000; Jacobson et al. 2006).
Previous studies have been limited in their reliance on retrospective assessments of adolescent antisocial behavior. Retrospective reports may be subject to recall bias, which may lead to bias in the estimates of heritability in adolescent versus adult antisocial behavior. No study published to date has directly compared retrospective and concurrent assessments of antisocial behavior. More importantly, there have yet to be sufficient data to examine the trajectory of antisocial behavior across specific ages from childhood through young adulthood within a genetically informed framework.
The evidence on adult-onset antisocial behavior is very limited, although work by Elander et al. (2000a, b) noted that it tended to be associated with either substance use/misuse or the development of serious mental disorder. Possibly, both of these might bring different genetic influences into play.
The present report describes the analysis of prospective and retrospective assessments of antisocial behavior in a large, population-based, longitudinal study of male twins from age 10 through young adulthood. The juvenile twins and their parents were part of the Virginia Twin Study of Adolescent Behavioral Development (VTSABD), a longitudinal, genetically informed study of childhood and adolescent psychopathology. The twins were contacted for follow-up as part of a Young Adult Follow-Up (YAFU) study after the age of 18.
METHOD
Study design and assessment
Details regarding the original ascertainment of juvenile twins in the VTSABD are included in previous publications (Eaves et al. 1997; Silberg et al. 2001). In brief, the sample was ascertained by statewide recruitment through Virginia schools when the twins were between ages 8 and 17; the families were then followed across three waves of study if the twins were younger than age 18 and still enrolled in high school. Of the 1894 Virginia families that were initially eligible for study, 74·5% participated in the first wave of data collection (n = 1412); 1047 out of the 1302 families that continued to meet the age and Virginia-residence requirements of the study completed a second home interview (80% participation rate), and 628 of the 777 eligible families (81%) participated in a third wave of assessment. The twins were evaluated using an extensive psychiatric battery that included the Child and Adolescent Psychiatric Assessment (CAPA; Angold et al. 1995) administered to both twins and at least one parent. For the present analysis, the nine CAPA symptoms comprising a DSM-III-R diagnosis of conduct disorder (CD) as well as the diagnosis itself were used. Each subject was assessed for CD between one and three times during the course of the study. Because of the likelihood of both overt- and covert-conduct symptoms, a CD symptom was considered to be present if either the child or the parent indicated that it had occurred in the previous 3 months.
Ascertainment of young adult twins
The YAFU is the outcome study of all the twins who participated in the first wave of the VTSABD. At 18 years or older, these twins were recontacted and evaluated as young adults using a telephone interview to generate DSM-III-R diagnoses of antisocial personality disorder (ASPD), major depression, generalized anxiety, eating disorders, and substance use. A semi-structured interview format was used for the evaluation of adult ASPD, identical to that administered to the parents of the VTSABD twins. As part of the assessment of adult ASPD, the young adult twins were asked about CD symptoms before age 16, using a protocol identical to that used in the adolescent study. However, as the goal of this analysis was to assess the relative role of genetic and environmental influences on antisocial behavior during and across different developmental time periods, juvenile CD was not included in the evaluation of adult ASPD symptoms, but was entered into the analysis separately, along with the prospective assessment of CD behaviors from the VTSABD.
Young adult sample
Of the 2824 subjects who were considered eligible for participation in the YAFU, data have thus far been collected on 1037 individual male and 1245 female twins (total n = 2282). The study is ongoing. For this present analysis, data on only male twins were analyzed, comprising 243 monozygotic male (MZM) pairs and 138 dizygotic male (DZM) pairs. Individual male twins from 222 opposite sex pairs and the 53 twins from incomplete pairs were also included. Ages of the total sample of 1037 individual male twins during participation in the YAFU ranged from 18 to 27 with a mean age of 21·4. Table 1 shows the number of male twins from the YAFU and their participation in the adolescent study. The majority of twins participated in at least two waves of the three-wave study (76%); 45% provided data at all three time points.
Table 1.
Number of YAFU male twins participating in the waves of the VTSABD study
| Wave I | Wave 1–Wave II only | Wave 1–Wave III only | Waves I–II–III | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of male individuals (n=1037)a | 216 | 317 | 35 | 469 | ||||||||
| No. of complete twin pairs (n=603) | MZM 42 | DZM 33 | DZO 49 | MZM 83 | DZM 34 | DZO 71 | MZM 9 | DZM 6 | DZO 4 | MZM 109 | DZM 65 | DZO 98 |
YAFU, Young Adult Follow-Up; VTSABD, Virginia Twin Study of Adolescent Behavioral Development; MZM, monozygotic male twin pairs; DZM, dizygotic male twin pairs; DZO, male twins from opposite sex pairs.
Includes 53 twins from incomplete pairs.
Six sequential age bands organized the data, representing twins assessed at ages 10–11, 12–13, 14–15, 16, 17, and 18 years or older. The 16- and 17-year-olds were separated, given the putative differences at these two ages (e.g. driving behavior). Available data on 8- to 9-year-olds were not included because of low confidence in children’s measures of their own behavior. The age structure of the male sample is illustrated in Table 2 by presenting the number of individual twins in each of the six age categories.
Table 2.
Number of male twins within each age category of the VTSABD and the YAFU (ages 18–28)
| Ages 10–11 | Ages 12–13 | Ages 14–15 | Age 16 | Age 17 | YAFU | |
|---|---|---|---|---|---|---|
| Ages 10–11 | 367 | |||||
| Ages 12–13 | 150 | 404 | ||||
| Ages 14–15 | 150 | 147 | 517 | |||
| Age 16 | 38 | 67 | 94 | 294 | ||
| Age 17 | 28 | 87 | 90 | 64 | 213 | |
| YAFU | 347 | 386 | 498 | 283 | 206 | 1037 |
YAFU, Young Adult Follow-Up; VTSABD, Virginia Twin Study of Adolescent Behavioral Development.
Subjects may be represented in multiple cells if they participated in more than one wave of the adolescent study.
Descriptive statistics
Data from the three waves of assessment were used to calculate the rate of CD symptoms by age. We tested for differences in the rates of CD diagnosis as a function of age at the date of assessment, adjusting for the non-independence of measures across waves and observations within twin pairs using the SAS genmod procedure. Pearson correlations between antisocial behavior before the onset of puberty (ages 10–13), during adolescence (ages 14–17) and in young adulthood were also estimated using SAS version 8 (SAS Institute, 2000).
Twin similarity in antisocial behavior
The partitioning of the total variation of a single trait into its genetic, shared and individual-specific environmental components was made by comparing the within-pair association between MZ and DZ twins within a single time point (e.g. twin 1’s and twin 2’s antisocial behavior in young adulthood). The relative influence of genes and environment on antisocial behavior was estimated within single phases of development by examining the twin correlations within the seven sequential age groups (which also included retrospective reports of CD).
The information for determining the extent to which a common set of genetic and environmental factors influence antisocial behavior across development derives from the covariance between antisocial behavior in one twin at one age (e.g. at age 12) and antisocial behavior in their co-twin at a subsequent age (e.g. young adulthood). For example, if the developmental association between symptoms of pre-pubertal CD and young adult ASPD can be attributed to a common set of genes acting additively, then the correlation between CD in one twin before age 12 (for example) and ASPD in the young adult co-twin, will be (on average) two times higher in MZ than DZ twins. The effect of genetic non-additivity or epistasis will be reflected in a DZ correlation less than one half the MZ correlation. If a common shared-environmental factor accounts for the covariance over time, then the correlation should be approximately the same in the two zygosity groups. Individually specific environmental factors (those influences that make one MZ twin different from their co-twin) will be reflected in a correlation within individuals across time but not within MZ twins.
Modeling of antisocial behavior
To test for differences in the etiology of antisocial behavior across multiple time points, a series of multivariate structural equation genetic models were fitted to the twin data using the statistical program Mx (Neale et al. 2003). The Cholesky decomposition model was selected as the basis for analyzing the underlying genetic and environmental structure of these longitudinal data. The full Cholesky model does not make any assumptions about the underlying genetic and environmental structure in terms of the number of factors or their rotation. In general, the Cholesky decomposition, like principal components, is merely a convenient saturated-factor model for the covariance matrices.
As outlined above for a two-variable case, the phenotypic covariance between a series of time points can be decomposed into latent additive-genetic covariance matrices, shared-environmental covariance matrices, and individual-specific covariance matrices. A triangular Cholesky factorization decomposes the covariance matrix into the product of the matrix and its transpose, yielding positive, definite maximum-likelihood (ML) estimates of the latent genetic and environmental covariance matrices. The three-component covariance matrices are required to be positive definite. This constraint is imposed during ML estimation by recognizing that the covariance matrices for the three latent sources of variation may each be expressed by a factor model in which the loadings form a lower triangular matrix of the same dimensions as the original covariance matrices.
Thus, for data from three time points, the Cholesky factors of A, when A is a 3 × 3 additive-genetic matrix (of occasions 1 through 3), are the product of H (a lower diagonal additive-genetic covariance matrix) post-multiplied by its transpose H′, giving rise to the following form (Neale & Cardon, 1992):
where the first Cholesky genetic factor influences time 1 through time 3 (a11–a31); the second factor is specific to time 2 and 3 (a22, a32); and the third factor unique to time 3. If there are common genetic effects influencing more than a single time point, the off-diagonal elements or factor loadings (e.g. a21, a31, a32) would be significantly different from zero. The paths or loadings are squared to estimate the proportion of variance in the outcome that is accounted for by each factor. This model is depicted as a path diagram in Fig. 1.
Fig. 1.
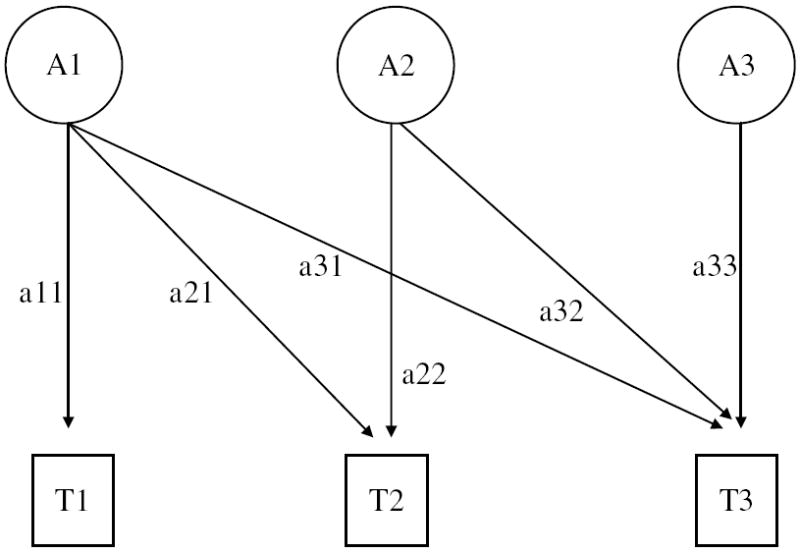
Path diagram of a three-variable case Cholesky decomposition model (additive genetic factors). Paths, which are squared to estimate the proportion of variance accounted for, are represented by lowercase letters. The first genetic factor represents the contributions of the first occasion (T1) to all subsequent occasions (T2, T3), the second gives the contributions of every new source of variation at the second occasion (T2, T3) to subsequent occasions, and the last factor is the contribution of effects specific to the last occasion (T3).
For present purposes, the Cholesky model that was fitted to the twin data included seven additive genetic factors, seven shared-environmental factors, and seven individual-specific environmental factors. The first genetic and environmental factors influence all seven occasions (i.e. CD symptoms at ages 10–11, 12–13, 14–15, 16, 17, adult ASPD symptoms, and retrospective CD) with each subsequent factor influencing one less variable than the preceding one. The final path in the model is variable specific. For repeated measures such as those obtained in this study, the Cholesky factors of the covariance matrices, in which the variables are ordered in time, have an intuitively simple and natural developmental interpretation.
Individual parameters or ‘paths’ were dropped from the model, beginning with the smallest parameters first. Initially, we sought a simplification of the shared-environmental structure and then the additive-genetic structure, with the goal of obtaining the most parsimonious account of the data with the least number of parameters. By eliminating single parameters at a time, the potential problem of invalid fit functions arising from incorrect degrees of freedom (Carey, 2005) in fitting Cholesky models was circumvented.
Each reduced model’s fit was evaluated using likelihood ratio χ2 tests in which the difference between −2(log-likelihood) of two alternative models is distributed as a χ2, with degrees of freedom (df) equal to the difference in the number of parameters estimated. To minimize skewness in the scores for model fitting, the data were first square-root transformed and the model fitted to the transformed raw data from the seven groups by ML.
Given the potentially large number of missing values, models were fitted to the raw-transformed data by ML pedigree analysis (Lange et al. 1976), which yields the correct likelihood provided that missing values are missing at random (MAR) or missing completely at random (MCAR) (Little & Rubin, 1987). This is a standard approach in the analysis of pedigree data in which family structures are highly irregular.
RESULTS
Prevalence rates
Approximately 6% of this young adult male sample met a diagnosis of ASPD. Figure 2 shows the rates of CD by age, and by age and adult ASPD diagnosis. The rates of CD increased from 4·7% at ages 10 to 11 to 9·6% at ages 16 to 17. The formal test of the overall increase in CD was significant (p < 0·002). The best-fitting cubic regression of CD on age showed the most rapid change at age 13·3 years. As illustrated in the figure, the rates of CD in those individuals with adult ASPD are at least twice as high as those without a diagnosis of ASPD across all age bands.
Fig. 2.
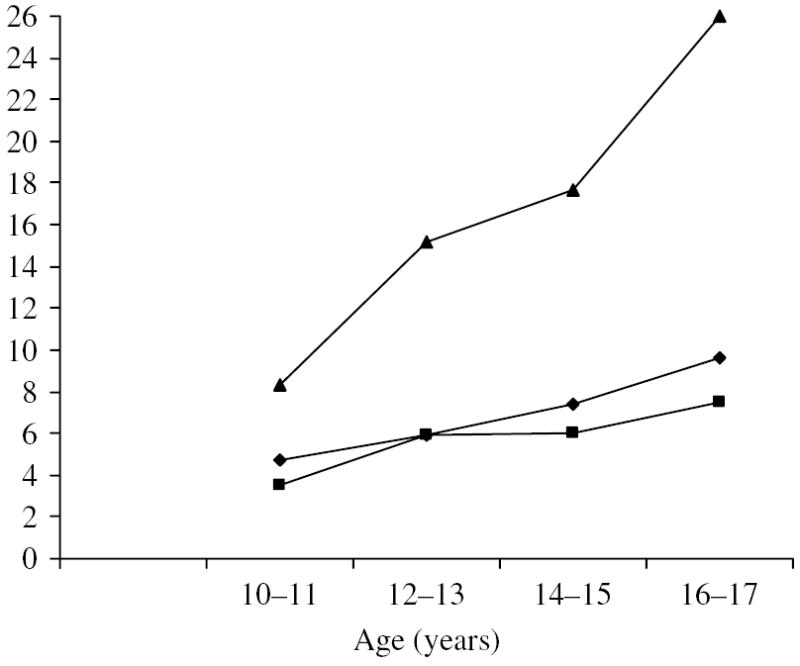
Rates of conduct disorder in Young Adult Follow-Up (YAFU) subjects and in subjects diagnosed with and without adult antisocial personality disorder (ASPD). –◆–, All subjects (n=2287); –■–, ASPD=0 (n=2138); –▲–, ASPD=1 (n=149).
Phenotypic associations
The phenotypic associations between CD symptoms before and during adolescence, recall of juvenile-conduct symptoms, and adult antisociality are presented in Table 3. These correlations showed a moderate degree of persistence, particularly from childhood through adolescence (r ≈ 0·3). Although somewhat attenuated, the relationship between childhood and adolescent CD and adult ASPD remained significant. We also note a strong correlation (0·46) between antisocial behavior assessed within the same point in time (retrospective CD and concurrent adult ASPD).
Table 3.
Correlations between symptoms of conduct disorder (CD) and antisocial behavior: middle childhood through young adulthood (number of twin pairs)
| CD ages 10–11 | CD ages 12–17 | Adult ASPD | CDr | |
|---|---|---|---|---|
| CD ages 10–11 | 1·00 (309) | |||
| CD ages 12–17 | 0·33 (190) | 1·00 (937) | ||
| Adult ASPD | 0·20 (232) | 0·24 (785) | 1·00 (1037) | |
| CDr | 0·19 (232) | 0·38 (785) | 0·46 (1037) | 1·00 (1037) |
ASPD, Antisocial personality disorder; CDr, retrospective CD.
Twin correlations for antisocial behavior
The MZ and DZ twin correlations for the different indices of antisocial behavior are presented in Table 4. The pattern of twin correlations suggest a shift from genetic influences before puberty (ages 10 and 11) to genetic- and shared-environmental influences in adolescence, and mostly genetic effects at age 17 through young adulthood. The twin correlations for retrospective recall of juvenile conduct symptoms (MZ=0·6, DZ=0·35) are consistent with the adolescent pattern, the MZ correlation being less than twice the DZ correlation. By contrast, the correlation for antisocial behavior in 17-years-olds and the young-adult sample strongly implicate genetic effects (possibly epistatic), given an MZ correlation more than twice the DZ correlation.
Table 4.
Twin correlations for symptoms of conduct disorder (CD) and antisocial behavior from age 10 through young adulthood (number of twin pairs)
| Age 10–11
|
Age 12–13
|
Age 14–15
|
Age 16
|
Age 17
|
Adult ASPD
|
CDr
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MZ | DZ | MZ | DZ | MZ | DZ | MZ | DZ | MZ | DZ | MZ | DZ | MZ | DZ |
| 0·65 (121) | 0·31 (87) | 0·56 (144) | 0·31 (67) | 0·54 (180) | 0·36 (114) | 0·63 (93) | 0·45 (45) | 0·46 (62) | 0·06 (33) | 0·52 (297) | 0·24 (169) | 0·59 (297) | 0·35 (169) |
ASPD, Antisocial personality disorder; MZ, monozygotic; DZ, dizygotic; CDr, retrospective CD.
Model fitting results
The twin correlations suggest that there may be important differences underlying the expression of antisocial behavior across different points in development. A systematic test of these differences is shown by the results of model fitting. The baseline index of fit of the full Cholesky model minus twice the log-likelihood (−2 ln L) was 7440·021 (df = 3844). The likelihood ratio test of the comparison in goodness of fit between this full model and a restricted model (7453·022, df = 3883) was not significantly different (χ2diff =13·001, p > 0·99). Table 5 presents the parameter estimates and confidence intervals under this best-fitting reduced-multivariate genetic model. A path diagram of the four additive-genetic and single shared-environmental factors model is also shown in Fig. 3. As the majority of the off-diagonal elements underlying the non-shared-environmental structure were primarily variable specific, these estimates are not shown. The model reveals the following pattern: (1) a genetic factor influencing antisocial behavior through all phases of development; (2) a transient genetic effect at puberty; (3) a shared-environmental effect beginning in adolescence; (4) a common genetic factor influencing both current ASPD and retrospective CD; and (5) specific genetic effects on young adult antisociality.
Table 5.
Parameter estimates and confidence intervals under the best-fitting multivariate genetic model for antisocial behavior
| Additive genetic factors
|
Shared environment
|
||||
|---|---|---|---|---|---|
| I Life-course persistent | II Pubertal specific | III Measurement specific | IV Adult specific | I Adolescent onset | |
| CD age 10–11 | 0·43 (0·49–0·56) | ||||
| CD age 12–13 | 0·29 (0·39–0·49) | 0·11 (0·29–0·40) | |||
| CD age 14–15 | 0·05 (0·17–0·30) | 0·18 (0·32–0·41) | 0·15 (0·29 0·41) | ||
| CD age 16 | 0·17 (0·42–0·56) | 0·14 (0·35–0·53) | |||
| CD age 17 | 0·16 (0·35–0·50) | 0·06 (0·27–0·43) | |||
| CD retrospective | 0·20 (0·29–0·39) | 0·26 (0·38–0·47) | 0·23 (0·34–0·45) | ||
| Young adult ASPD | 0·13 (0·22–0·31) | 0·21 (0·33–0·44) | 0·08 (0·29–0·37) | 0·11 (0·23–0·34) | |
CD, Conduct disorder; ASPD, antisocial personality disorder.
Fig. 3.
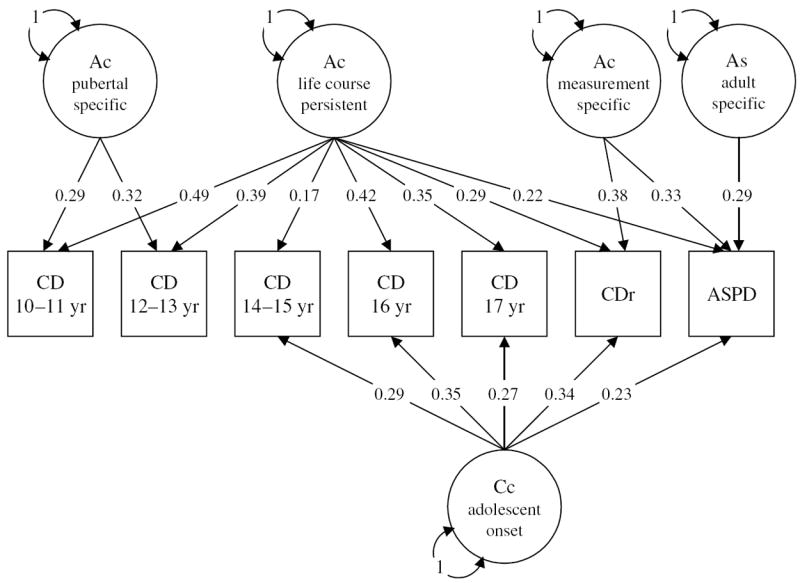
Parameter estimates under the best-fitting multivariate genetic model for antisocial behavior. Ac, Common additive genetic factors; As, specific additive genetic factor; Cc, common shared environmental factor.
DISCUSSION
The results of this study demonstrate important etiological differences in the development of antisocial behavior. Although the ‘best model’ to emerge from our analysis is consistent with Moffitt’s view of antisocial behavior, there are methodological limitations that need to be considered. Most importantly, the problem of relatively small sample sizes is a significant impediment to developing reliable models for longitudinal twin data. Clearly, these findings require replication in other populations. Nevertheless, the overall pattern of results appears remarkably consistent with Moffitt’s theory in demonstrating a genetically mediated effect that persists from childhood through adulthood and a shared-environmental effect beginning at adolescence (ages 14 to15). Our findings suggest that there are etiological differences, as well as epidemiological contrasts, between the two, in keeping with a broad range of both categorical and dimensional findings (Moffitt, 2003).
Our model also reveals specific genetic effects expressed at puberty (ages 12 to 15) that do not feature in Moffitt’s conception but may nevertheless have important implications for how we conceive of the development of conduct disorder. A significant genetic effect specific to puberty, in a genetic time series such as ours, is consistent with a mechanism in which the genetic control of the timing of puberty interacts with the expression of genetic differences in CD (Eaves & Silberg, 2003). Recent work has shown the timing of puberty to be largely genetically mediated (Eaves et al. 2004), so that the expression of genetic differences in antisocial behavior may depend upon those genes that affect the timing of puberty in boys.
The rates of CD are also consistent with other population samples in showing a rise in the mid-teens (Rutter et al. 1998; Maughan et al. 2006). The estimate of age of greatest change was 13·3 years for the diagnostic outcome and 14·4 years for total CD-symptom count. These phenotypic results coincide with our findings from model fitting showing a significant effect of the shared environment at age 14.
The nature of the shared-environmental effects is not altogether clear. It has generally been conceived to reflect the influence of conduct-disordered peers. However, there is increasing evidence that the selection of peers by twins is strongly influenced by an individual’s genotype (Cleveland et al. 2005). Such effects would not be expressed as a shared-environmental effect but included in the genetic estimates. The most likely explanation is that the shared environment is a combination of peers’ effects as well as other shared aspects of the family environment (e.g. lack of parental supervision and discord, lack of family cohesiveness, and general family stress). However, the degree to which such family variables represent a consequence of a difficult child’s genotype (i.e. evocative-genotype-environment correlation) also needs to be considered in these general models of risk.
The particular genes that are associated with the life-course-persistent subtype are not known. Persistent antisocial behavior has been associated with a large number of risk factors including early-onset CD and co-morbid hyperactivity. Given the strong genetic commonality between childhood CD and adult antisociality shown here, and the association between hyperactivity and persistent antisocial behavior, it may be the genes for early-onset aggression that differentiate the life-course-persistent versus adolescent-limited subtype, or alternatively, the genes associated with early ADHD. Using a trajectory analysis to contrast subgroups, Odgers et al. (in press) found a substantial subgroup with antisocial behavior that begins early but is largely limited to childhood. One possible implication is that the distinctive feature of life-course-persistent antisocial behavior is not so much that the onset is early but rather that the early onset is associated with hyperactivity and neurodevelopmental impairment. If so, the genetic liability might particularly apply to this constellation of features. Clearly, the search for the genes influencing the life-course-persistent form of antisocial behavior should necessarily take into account the co-morbid aspects of the disorder.
The fact that we did not find an environmental effect on persistent antisocial behavior does not preclude the importance of shared-environmental factors in its etiology. If the genetic susceptibility to antisocial behavior is expressed only in certain environments (a genotype × shared environment interaction), as shown by Caspi et al. (2002), any shared-environmental effect on life-course-persistent behavior would not be detected in these types of models but would be confounded in the overall estimate of the genetic effect (Jinks & Fulker, 1970; Eaves et al. 1977).
Although of secondary importance, the inclusion of both retrospective and prospective assessments of juvenile CD is informative. Retrospective CD loads on the general genetic factor and the later shared-environmental factor but the largest single commonality is the genetic effect shared between adult ASPD symptoms and retrospective assessments of juvenile CD that contain no genetic contribution from the prospective CD interviews. This finding underscores not only the relative lack of consistency between ratings of the same construct by different raters but also the dangers of reading too much into retrospective reports of child behavior or concurrent behavior assessed by a single informant.
Future directions necessarily include: (1) the need for replication in other population samples; (2) comparable analyses of antisocial behavior in females; (3) specification of the shared environment; and (3) the search for those genes and environments that underlie the most persistent forms of antisociality that have serious adverse consequences for those individuals and for the people they inevitably effect.
Acknowledgments
This work was supported by grants MH-55557, MH-62368 (J.L.S.) and MH-068521 (L.J.E.) from the National Institute of Mental Health, and DA-016977 and DA-018673 (H.H.M.) from the National Institute of Drug Abuse. We thank Dr Michael Neale for his generosity in making the Mx program freely available and Dr Greg Carey for his statistical input on model fitting. We also acknowledge the work of Theresa Martin, Sandra Lee Muzik, Susan Williams and Bonnie Dedeian in the collection of these data, and Jefferson Lyttle for his editorial contributions to this manuscript.
Footnotes
DECLARATION OF INTEREST
None.
References
- Angold A, Prendergast M, Cox A, Harrington RC, Simonoff E, Rutter M. The Child and Adolescent Psychiatric Assessment (CAPA) Psychological Medicine. 1995;25:739–753. doi: 10.1017/s003329170003498x. [DOI] [PubMed] [Google Scholar]
- Barkley RA, Fischer M, Edelbrock C, Smallish L. The adolescent outcome of hyperactive children diagnosed by research criteria: III. Mother–child interactions, family conflicts and maternal psychopathology. Journal of Child Psychology and Psychiatry. 1991;32:233–255. doi: 10.1111/j.1469-7610.1991.tb00304.x. [DOI] [PubMed] [Google Scholar]
- Carey G. Cholesky problems. Behavior Genetics. 2005;35:653–665. doi: 10.1007/s10519-005-5355-9. [DOI] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Cleveland HH, Wiebe RP, Rowe DC. Sources of exposure to smoking and drinking friends among adolescents: a behavioral-genetic evaluation. Journal of Genetic Psychology. 2005;166:153–169. [PubMed] [Google Scholar]
- DiLalla LF, Gottesman II. Heterogeneity of causes for delinquency and criminology: lifespan perspectives. Development and Psychopathology. 1989;1:339–349. [Google Scholar]
- Eaves LJ, Last KA, Martin NG, Jinks JL. A progressive approach to non-additivity and genotype-environmental covariance in the analysis of human differences. British Journal of Mathematical and Statistical Psychology. 1977;30:1–42. [Google Scholar]
- Eaves LJ, Silberg J. Modulation of gene expression by genetic and environmental heterogeneity in the timing of a developmental milestone. Behavior Genetics. 2003;33:1–6. doi: 10.1023/a:1021060430942. [DOI] [PubMed] [Google Scholar]
- Eaves L, Silberg J, Foley D, Bulik C, Maes H, Erkanli A, Angold A, Costello EJ, Worthman C. Genetic and environmental influences on the relative timing of pubertal change. Twin Research and Human Genetics. 2004;7:471–481. doi: 10.1375/1369052042335278. [DOI] [PubMed] [Google Scholar]
- Eaves LJ, Silberg JL, Meyers JM, Maes H, Simonoff E, Neale MC, Pickles A, Reynolds C, Erickson M, Heath AC, Loeber R, Rutter M, Hewitt JK. The main effects of genes and environment on major behavioral problems of adolescence in the Virginia Twin Study of Adolescent Behavioral Development. Journal of Child Psychology and Psychiatry. 1997;38:965–980. doi: 10.1111/j.1469-7610.1997.tb01614.x. [DOI] [PubMed] [Google Scholar]
- Elander J, Rutter M, Simonoff E, Pickles A. Explanations for apparent late onset criminality in a high-risk sample of children followed-up in adult life. British Journal of Criminology. 2000a;40:497–509. [Google Scholar]
- Elander J, Simonoff E, Pickles A, Holmshaw J, Rutter M. Longitudinal study of adolescent and adult conviction rates among children referred to psychiatric services for behavioural or emotional problems. Criminal Behaviour and Mental Health. 2000b;10:40–59. [Google Scholar]
- Faraone SV, Biederman J, Chen WJ, Milberger S, Warburton R, Tsuang MT. Genetic heterogeneity in ADHD: gender, psychiatric comorbidity, and maternal ADHD. Journal of Abnormal Psychology. 1995;104:334–345. doi: 10.1037/0021-843X.104.2.334. [DOI] [PubMed] [Google Scholar]
- Hinshaw SP. Conduct disorder in childhood: conceptualization, diagnosis, comorbidity and risk status for antisocial function in adulthood. Progress in Experimental Personality and Psychopathology Research. 1994;15:3–44. [PubMed] [Google Scholar]
- Jacobson KC, Prescott CA, Kendler KS. Sex differences in the genetic and environmental influences on the development of antisocial behavior. Development and Psychopathology. 2006;14:395–416. doi: 10.1017/s0954579402002110. [DOI] [PubMed] [Google Scholar]
- Jinks JL, Fulker DW. Comparison of the biometrical genetical, MAVA and classical approaches to the analysis of human behavior. Psychological Bulletin. 1970;73:311–349. doi: 10.1037/h0029135. [DOI] [PubMed] [Google Scholar]
- Lahey BB, Piacentini JC, McBurnett K, Stone P, Hartdagen S, Hynd G. Psychopathology in the parents of children with conduct disorder and hyperactivity. Journal of the American Academy of Child and Adolescent Psychiatry. 1988;27:163–170. doi: 10.1097/00004583-198803000-00005. [DOI] [PubMed] [Google Scholar]
- Lange K, Westlake J, Spence MA. Programs for pedigree analysis: III. Variance components by the scoring method. Annals of Human Genetics. 1976;39:485–491. doi: 10.1111/j.1469-1809.1976.tb00156.x. [DOI] [PubMed] [Google Scholar]
- Little RJA, Rubin DB. Statistical Analysis with Missing Data. Wiley; New York: 1987. [Google Scholar]
- Lyons MJ, True WR, Eisen SA, Goldberg J, Meyers JM, Farone SV, Eaves LJ, Tsuang MT. Differential heritability of adult and juvenile antisocial traits. Archives of General Psychiatry. 1995;53:906–915. doi: 10.1001/archpsyc.1995.03950230020005. [DOI] [PubMed] [Google Scholar]
- Maughan B, Rowe R, Messer J, Goodman R, Meltzer H. Conduct disorder and oppositional defiant disorder in a national sample: developmental epidemiology. Journal of Child Psychology and Psychiatry. 2006;45:609–620. doi: 10.1111/j.1469-7610.2004.00250.x. [DOI] [PubMed] [Google Scholar]
- Miles DR, Carey G. The genetic and environmental architecture of human aggression. Journal of Personality and Social Psychology. 1997;72:207–217. doi: 10.1037//0022-3514.72.1.207. [DOI] [PubMed] [Google Scholar]
- Moffitt TE. Juvenile delinquency and attention deficit disorder: boys’ developmental trajectories from age 3 to age 15. Child Development. 1990;61:893–910. doi: 10.1111/j.1467-8624.1990.tb02830.x. [DOI] [PubMed] [Google Scholar]
- Moffitt TE. Adolescent-limited and life-course-persistent antisocial behavior: a developmental taxonomy. Psychological Review. 1993;100:674–701. [PubMed] [Google Scholar]
- Moffitt TE. Life-course-persistent and adolescence-limited antisocial behavior: a 10-year research review and a research agenda. In: Lahey BB, Moffitt TE, Caspi A, editors. Causes of Conduct Disorder and Juvenile Delinquency. Guilford Press; New York: 2003. pp. 49–75. [Google Scholar]
- Moffitt TE, Caspi A. Childhood predictors differentiate life-course-persistent and adolescent-limited antisocial pathways among males and females. Development and Psychopathology. 2001;13:355–375. doi: 10.1017/s0954579401002097. [DOI] [PubMed] [Google Scholar]
- Moffitt TE, Caspi A, Dickson N, Silva PA, Stanton W. Childhood onset versus adolescent onset antisocial conduct in males: natural history from age 3 to 18. Development and Psychopathology. 1996;8:399–424. [Google Scholar]
- Neale MC, Boker SM, Xie G, Maes HH. Mx: Statistical Modeling. 6. Department of Psychiatry, Virginia Commonwealth University; Richmond, VA: 2003. [Google Scholar]
- Neale MC, Cardon LR. Methodology for Genetic Studies of Twins and Families. Kluwer Academic; Dordrecht: 1992. [Google Scholar]
- Odgers CL, Caspi A, Poulton R, Harrington HTM, Broadbent JM, Dickson N, Sears MR, Hancox B, Moffitt TE. Conduct problems subtypes in males predict differential adult health burden. Archives of General Psychiatry. doi: 10.1001/archpsyc.64.4.476. in press. [DOI] [PubMed] [Google Scholar]
- Rutter M, Giller H, Hagell A. Antisocial Behavior by Young People. Cambridge University Press; Cambridge: 1998. [Google Scholar]
- Rutter M, Maughan B, Meyer J, Silberg J, Simonoff E, Taylor E. Heterogeneity of antisocial behavior: causes, continuities, and consequences. In: Dienstbier R, Osgood DW, editors. Nebraska Symposium on Motivation: Motivation and Delinquency. Vol. 44. University of Nebraska Press; Lincoln, NE: 1997. pp. 45–118. [PubMed] [Google Scholar]
- SAS Institute. The SAS Software for Windows for the PC: Version 8. SAS Institute, Inc.; Cary, NC: 2000. [Google Scholar]
- Silberg J, Meyers JM, Pickles A, Simonoff E, Eaves L. Heterogeneity among juvenile antisocial behaviors: findings from the Virginia Twin Study of Adolescent Behavioral Development. In: Bock GR, Goode JA, editors. Genetics of Criminal and Antisocial Behavior (Ciba Foundation Symposium no 194) Wiley; Chichester: 1996. pp. 76–85. [DOI] [PubMed] [Google Scholar]
- Silberg JL, Neale MC, Rutter M, Eaves LJ. Genetic and environmental influences on the temporal association between early anxiety and later depression in girls. Biological Psychiatry. 2001;49:1040–1049. doi: 10.1016/s0006-3223(01)01161-1. [DOI] [PubMed] [Google Scholar]
- Taylor J, Iacono WG, McGue M. Evidence for a genetic etiology for early-onset delinquency. Journal of Abnormal Psychology. 2000;109:634–643. doi: 10.1037//0021-843x.109.4.634. [DOI] [PubMed] [Google Scholar]