

Summary
Twist is a key transcription activator of epithelial-mesenchymal transition (EMT). It remains unclear how Twist induces gene expression. Here we reported a mechanism by which Twist recruits BRD4 to direct WNT5A expression in basal-like breast cancer (BLBC). Twist contains a “histone H4 mimic” GK-X-GK motif that is di-acetylated by Tip60. The di-acetylated Twist binds the second bromodomain of BRD4, whose first bromodomain interacts with acetylated H4, thereby constructs an activated Twist/BRD4/P-TEFb/RNA-PolII complex at the WNT5A promoter and enhancer. Pharmacologic inhibition of the Twist-BRD4 association reduced WNT5A expression and suppressed invasion, cancer stem cell (CSC)-like properties, and tumorigenicity of BLBC cells. Our study indicates that the interaction with BRD4 is critical for the oncogenic function of Twist in BLBC.
Introduction
Recruitment and activation of RNA-PolII at gene promoters are two key steps required for a productive transcription (Zhou et al., 2012). Following RNA-PolII recruitment to a gene promoter, TFIIH phosphorylates the serine 5 of the heptapeptide repeats in the C-terminal domain (CTD) of RNA-PolII, resulting in initial synthesis of short RNA species. However, RNA-PolII pauses in the proximal-promoter and requires a second phosphorylation event on serine 2 of the CTD that is carried out by the pause release factor P-TEFb, a complex composed of CDK9 and cyclin T1/2. Importantly, the recruitment of P-TEFb to RNA-PolII is mediated, in part, by BRD4 (Jang et al., 2005).
BRD4 is a member of the BET (bromodomain and extra terminal domain) family proteins that are characteristic of two tandem bromodomains (BDs) located in the N-terminus. The BDs of BET proteins recognize acetylated-lysine residues in nucleosomal histones (Filippakopoulos et al., 2012), facilitating the recruitment of transcriptional proteins to chromatin. Recent studies have shown that pharmacologic inhibition of BRD4 with BET-specific BD inhibitors effectively blocks MYC expression in multiple myeloma (Delmore et al., 2011), Burkitt's lymphoma and acute myeloid leukemia (Dawson et al., 2011; Zuber et al., 2011). However, many mechanistic questions about BRD4 functions as a chromatin regulator in gene transcription are still unanswered. For example, how does BRD4 interact and work with transcription factors at the target gene promoter and enhancer sites? Whether and how do the two BDs in BRD4 function differently in gene transcription?
Breast cancer is a heterogeneous disease that can be divided into four major subtypes based on gene expression profiling: luminal A, luminal B, ErbB2, and basal-like. Basal-like breast cancer (BLBC) is characterized by the lack of expression of estrogen receptor (ER), progesterone receptor (PR) and epidermal growth factor receptor 2 (HER2) and positive expression of basal markers [Cytokeratin (CK) 5/6 and CK14] (Rakha et al., 2008). The absence of effective targeted therapies and poor response to standard chemotherapy often results in a rapidly fatal clinical outcome for this disease. Notably, BLBC has activated the epithelial-mesenchymal transition (EMT) program, which provides cells with increased plasticity and stem cell-like properties required during embryonic development, tissue remodeling, wound healing and metastasis (Thiery et al., 2009).
Twist and Snail are two key members of EMT-activating transcriptional factors. During mesoderm development in Drosophila, Snail functions as a transcriptional repressor to prevent expression of genes that belong to ectoderm; whereas Twist serves as a transcriptional activator to induce mesodermal gene expression (Leptin, 1991). A complete loss of all mesodermal characteristics occurs only when both Snail and Twist are absent. These results suggest that Snail and Twist work synergistically, controlling distinct sets of genes, to coordinate EMT induction and mesoderm formation (Zeitlinger et al., 2007). We previously showed that Snail interacts with several transcriptional repressive complexes to suppress gene expression. However, the mechanism underlying gene transcriptional activation by Twist has remained elusive. In this study, we sought to identify Twist-interacting proteins and determine the mechanism by which Twist controls gene transcriptional activation in EMT and BLBC.
Results
BRD4-BD2 interacts with lysine-acetylated Twist
We sought to identify Twist-interacting proteins from a stable HeLa S3 cell line expressing Flag-Twist. Affinity protein purification, followed by SDS-PAGE and analysis by mass spectrometry, revealed the presence of BRD4 and TRRAP/EP400 (data not shown). To validate the interaction between Twist and BRD4, we co-expressed HA-Twist and Flag-BRD4 in HEK293 cells in the presence or absence of the histone deacetylase inhibitor Trichostatin (TSA). After immunoprecipitating Twist, we detected the associated BRD4, and vice versa (Figure 1A and Figure S1A). Although similar amounts of Twist were immunoprecipitated from cells with and without TSA treatment, Twist was more acetylated and interacted with more BRD4 in cells treated with TSA. In addition, immunoprecipitation with a pan-acetylated-lysine (pan-AcK) antibody pulled down Twist and BRD4 in cells treated with TSA. Similar observations were made in Twist-expressing HeLa S3 cells (Figure 1B and Figure S1B). We further confirmed the interaction between the endogenous Twist and BRD4 and acetylation of the endogenous Twist in four BLBC cell lines, both of which were substantially enhanced with TSA treatment (Figure 1C and Figure S1C). The Twist-BRD4 interaction is specific because Twist did not associate with other BET members (BRD2, BRD3 and BRDT) or lysine specific demethylase 1 (LSD1), and BRD4 did not associate with TCF4 (Figure S1D). The increased Twist-BRD4 interaction by TSA could not be due to an altered sub-cellular localization of these two proteins as TSA did not affect their localization (Figure S1E).
Figure 1. BD2 of BRD4 is required for its interaction with acetylated Twist.

(A) HA-Twist and Flag-BRD4 were co-expressed in HEK293 cells. After treating cells with TSA (2 μM) for 12 hr, Twist, BRD4 and acetylated Twist were immunoprecipitated with HA, Flag and pan-acetylated-lysine (pan-AcK) antibodies, respectively, and analyzed by Western blotting.
(B) HeLa cells stably expressing Flag-Twist were treated with TSA as in (A), Flag-Twist, endogenous BRD4 and acetylated Twist were immunoprecipitated and examined by Western blotting.
(C) Cells were treated as described in (A), endogenous Twist, BRD4 and acetylated Twist were immunoprecipitated and examined by Western blotting.
(D) Schematic depiction of the functional domains of BRD4 and deletion constructs used (top panel). ET stands for extra-terminal domain. Flag-tagged wild-type (WT) or deletion mutants of BRD4 were co-expressed with HA-Twist in HEK293 cells. After immunoprecipitated with HA or Flag antibody, the bound BRD4 or Twist was examined by Western blotting.
(E) Schematic diagram showing the double bromodomain (BD1+BD2) of BRD4 and individual BD constructs used (top panel). Flag-BD1WT, BD1YN, BD2WT and BD2YN were co-expressed with HA-Twist in HEK293 cells treated with TSA as in (A). Twist and BDs were immunoprecipitated with HA and Flag antibodies, respectively, and analyzed by Western blotting.
(F) HA-Twist and Flag-BRD4 were co-expressed in HEK293 cells treated with TSA as in (A). Twist and BRD4 were immunoprecipitated with HA and Flag antibodies, respectively, in the presence or absence of JQ1 (1 μM) and analyzed by Western blotting.
(G) Cells were treated with TSA as in (A), endogenous Twist and BRD4 were immunoprecipitated with Twist and BRD4 antibodies, respectively, in the presence or absence of JQ1 (1 μM) and examined by Western blotting.
See also Figure S1
We next generated BRD4 deletion constructs and co-expressed them with Twist in HEK293 cells. We found that only N-terminal fragments containing both BDs, but not other regions of BRD4, retained the ability to interact with Twist (Figure 1D). When BD1 or BD2 was co-expressed with Twist in HEK293 cells, only BD2WT, but not BD1WT, bound to Twist (Figure 1E and Figure S1F). Mutation of the conserved tyrosine and asparagine residues in the acetyl-lysine binding pocket of BD2 to alanine (BD2YN) reduced its binding to Twist. The Twist-BRD4 interaction was readily disrupted when JQ1, a BET-specific BD inhibitor, was added to the immunoprecipitation reaction (Figure 1F-1G and Figure S1G-S1H). Similarly, MS417, a BET-specific BD inhibitor with approximately 10-fold higher binding affinity than JQ1 (Zhang et al., 2012), effectively blocked the Twist-BRD4 interaction in four BLBC cell lines (Figure S1I-S1J). These results indicate that the Twist-BRD4 interaction is mediated by the BD2 of BRD4 binding to lysine-acetylated Twist.
Twist di-acetylation at K73 and K76 by Tip60 is required for Twist-BRD4 interaction
The N-terminal half of Twist contains an acidic segment and two lysine/arginine-rich basic motifs that share high sequence similarity to histones H2B and H4, respectively (Figure S2A). We generated Twist deletion constructs DL1 (residues 15-202), DL2 (residues 31-202), and DL3 (residues 47-202) and co-expressed them individually with Flag-BRD4 in HEK293 cells. DL1 retained whereas DL2 and DL3 lost interaction with BRD4 (Figure 2A and Figure S2B). Surprisingly, in contrast to DL1, DL2 and DL3 also completely lost acetylation (Figure 2B and Figure S2C). Because the first 30 N-terminal residues in Twist do not contain lysine, the loss of acetylation in DL2 and DL3 suggests that the N-terminal region is critical for Twist acetylation. In our mass spectrometry analysis, the NuA4 histone acetyltransferase complex proteins, including TRRAP and EP400 (Doyon and Cote, 2004), were identified as Twist association partners. We postulated that Tip60, the acetyltransferase of NuA4 complex, was responsible for Twist acetylation. Indeed, when TwistWT, DL1, DL2 and DL3 were co-expressed with Tip60 in HEK293 cells, we found that TwistWT and DL1, but not DL2 or DL3, interacted with Tip60 (Figure 2C and Figure S2D). Endogenous Twist-Tip60 interaction was confirmed in three BLBC cell lines, which was markedly enhanced by TSA treatment (Figure S2E). We further observed that ectopic expression of Tip60 in BT549 and SUM1315 cells resulted in enhanced acetylation of Twist and the association of Twist with BRD4 even in the absence of TSA, whereas knockdown of Tip60 yielded opposite effects even in the presence of TSA (Figure 2D and Figure S2F).
Figure 2. Twist di-acetylation at K73/K76 by Tip60 is required for interaction with BRD4.

(A) Schematic diagram showing the domain organization of Twist, with deletion and mutation constructs used (top panel). HA-tagged WT or deletion mutants of Twist were co-expressed with Flag-BRD4 in HEK293 cells treated with or without TSA. Twist and BRD4 were immunoprecipitated with HA and Flag antibodies, respectively, and analyzed by Western blotting.
(B) HA-tagged WT or deletion mutants of Twist were expressed in HEK293 cells treated with TSA. Twist and acetylated Twist were immunoprecipitated with HA and pan-AcK antibodies, respectively, and analyzed by Western blotting.
(C) Flag-tagged WT or deletion mutants of Twist were co-expressed with HA-Tip60 in HEK293 cells treated with TSA. Twist and Tip60 were immunoprecipitated with Flag and HA antibodies, respectively, and analyzed by Western blotting.
(D) Ectopic expression of Tip60 or knockdown of endogenous Tip60 was performed in BT549 and SUM1315 cells. After endogenous Twist was immunoprecipitated, acetylation of Twist and the bound BRD4 were examined by Western blotting.
(E) HA-tagged WT and mutant Twist were expressed in HEK293 cells, acetylated Twist was immunoprecipitated with HA and pan-AcK antibodies, respectively, and examined by Western blotting.
(F) HA-tagged WT or mutant Twist was co-expressed with Flag-tagged BD1 and BD2 in HEK293 cells treated with TSA. Twist and BDs were immunoprecipitated with HA and Flag antibodies, respectively, and examined by Western blotting.
(G) HA-tagged WT or mutant Twist was co-expressed with Flag-tagged BD2 in HEK293 cells treated with TSA. Twist, BRD4, and acetylated Twist were immunoprecipitated with HA, Flag, and pan-AcK antibodies, respectively, and analyzed by Western blotting.
(H) Determination of Tip60 catalyzed acetylation sites in Twist by mass spectrometry. Peptides contain K73 and K76 acetylations are shown in the top and bottom panels, respectively.
See also Figure S2
Twist contains five lysine residues (K33, K38, K73, K76 and K77) in its N-terminal region, which are highly conserved among different species (Figure S2A). Point mutation of K33R, K73R and K76R showed a reduced level of acetylation compared to that of TwistWT (Figure 2E and Figure S2G). TwistWT, K33R, K38R and K77R showed similar interaction with BD2 whereas K73R and K76R exhibited clearly weaker binding to BD2 (Figure 2F and Figure S2H). The K73R/K76R double mutant showed an almost complete loss of acetylation and interaction with BD2 (Figure 2G and Figure S2I). We further confirmed the Tip60-mediated Twist acetylation on K73/K76 by mass spectrometry analysis (Figure 2H and Figure S2J). There are 12 nuclear acetyltransferases (HAT) divided into three major groups: (1) the GNAT family (e.g., PCAF); (2) the MYST family (e.g., Tip60); and (3) the p300/CBP family (e.g., p300 and CBP) (Rekowski and Giannis, 2010). To examine whether other HATs can also acetylate Twist, we knocked down the expression of p300, CBP, PCAF or Tip60 individually in BT549 and SUM1315 cells (Figure S2K). We found that knockdown of Tip60, but not p300, CBP or PCAF suppressed Twist acetylation at K73/K76. Taken together, these data support our contention that Tip60 is the major HAT responsible for the K73/K76 acetylation on Twist and that di-acetylation of Twist is required for its association with BRD4-BD2.
Histone H4 mimicry in Twist is responsible for its interaction with BRD4
Di-acetylations of the N-terminal tail of H4 at K5 and K8 are often required for the interaction of H4 with BDs of BET family proteins (Filippakopoulos et al., 2012; Moriniere et al., 2009). The Twist sequence at K73 and K76 shares high similarity to the N-terminal tail of H4 at K5 and K8 (Figure 3A). To investigate this “histone mimicry” in the interaction between Twist and BRD4, we performed a pulldown study using biotinylated H4 and Twist peptides and lysate of HEK293 cells expressing BRD4-BD2. We observed that biotinylated H4-K5ac/K8ac peptide (residues 1-21) was bound to BD2 and that this interaction was disrupted by non-biotinylated H4-K5ac/K8ac peptide (lane 2 vs lane 1, Figure 3B). This interaction was also markedly reduced by a Twist-K73ac/K76ac peptide (residues 61-80) but not by the un-acetylated corresponding peptide (lanes 3 and 4 vs lane 1, Figure 3B). Similarly, the interaction of a biotinylated Twist-K73ac/K76ac peptide with BD2 was disrupted by a Twist-K73ac/K76ac, but not un-acetylated, peptide (lanes 7 and 8 vs lane 5, Figure 3B). Notably, acetylated H4 peptide also disrupted the acetylated Twist and BD2 association (lane 6 vs lane 5, Figure 3B), indicating that di-acetylated K5/K8 in H4 and di-acetylated K73/K76 in Twist function similarly as a recognition motif for BRD4-BD2.
Figure 3. K73ac/K76ac Twist and BRD4 interaction is critical for the function of Twist.
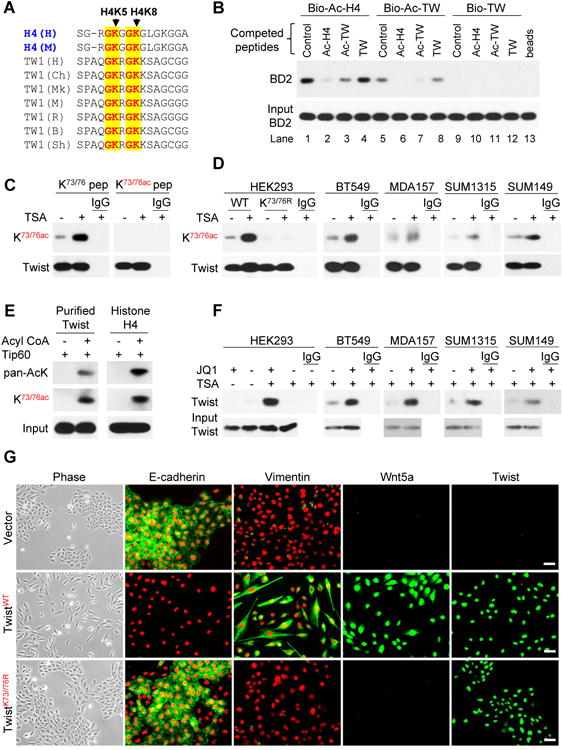
(A) Sequence alignment between K5/8 of histone H4 and K73/76 of Twist. H, M, Ch, Mk, R, B, and Sh stand for human, mouse, chimpanzee, monkey, rat, bovine, and sheep, respectively.
(B) The indicated biotinylated peptides were mixed with lysates from HEK293 cells expressing BD2 without or with indicated non-biotinylated competing peptides, the bound BD2 was analyzed by Western blotting after pull-down of the biotinylated peptides.
(C) HA-Twist was expressed in HEK293 cells treated with or without TSA. The immunoprecipitated Twist was analyzed on Western blots using indicated antibody in the presence of Twist-K73/K76 or Twist-K73ac/K76ac peptides.
(D) HA-tagged or endogenous Twist was immunoprecipitated from cells treated with or without TSA using HA and Twist antibodies, respectively, and analyzed by K73ac/K76ac antibody.
(E) Purified human Twist or histone H4 was incubated with purified Tip60 in the absence or presence of acetyl-CoA. The acetylation of Twist and histone H4 was examined by pan-AcK and anti-K73ac/K76ac antibodies.
(F) K73ac/K76ac Twist was immunoprecipitated with K73ac/K76ac antibody in the presence or absence of JQ1. The bound Twist was examined by Western blotting.
(G) HMLE cells expressing the vector or the WT or K73/76R Twist were examined for morphological changes indicative of EMT by phase-microscopy and the expression of E-cadherin, vimentin, Wnt5a, and Twist (green) by immunofluorescent staining. Nuclei stained with DAPI (red). Scale bar, 50 μM.
See also Figure S3
We then developed a specific antibody against Twist-K73ac/K76ac. Twist recognition by this antibody was disrupted by a Twist-K73ac/K76ac peptide, but not the corresponding non-acetylated peptide (Figure 3C and Figure S3A). This antibody recognized immunoprecipitated TwistWT, but not TwistK73R/K76R that harbored mutated K73 and K76 (Figure 3D and Figure S3B). In line with our contention that Tip60 acetylates Twist at K73/K76, both Twist and H4 acetylated by purified Tip60 in vitro were recognized by this Twist-K73ac/K76ac antibody and a pan-acetylated antibody (Figure 3E). Furthermore, this antibody readily detected endogenous Twist-K73ac/K76ac that was immunoprecipitated from four BLBC cell lines (Figure 3D and Figure S3B). Although immunoprecipitation of the endogenous Twist by this antibody was weak, it was robustly increased by the addition of JQ1 to the binding buffer, indicating that the K73ac/K76ac site is masked by binding to BRD4 in cells (Figure 3F).
We further characterized the functional importance of this di-acetylation-dependent Twist-BRD4 interaction in HMLE cells. Ectopic expression of TwistWT resulted in an induction of EMT as indicated by the downregulation of E-cadherin and upregulation of vimentin (Figure 3G and Figure S3C). While localized in the nucleus (Figure S3D), TwistK73R/K76R expression failed to induce EMT, indicating that the interaction with BRD4 is critical for the function of Twist.
Molecular basis of BRD4 binding to lysine-acetylated Twist
To determine the molecular basis of di-acetylation-dependent Twist-BRD4 association, we characterized binding of the two BDs of BRD4 to a series of Twist peptides (residues 68-79) bearing no, single or double acetylated lysine at K73 and K76 by NMR titration. As shown in 2D 1H-15N HSQC spectra (Figure S4A), BRD4-BD2 exhibited substantially more extended chemical shift perturbations upon binding to the single-acetylated, and even more to the di-acetylated Twist peptides, than those produced by the BRD4-BD1, confirming that the BRD4-BD2 is largely responsible for BRD4 association with the di-acetylated Twist. The preferred recognition of Twist-K73ac/K76ac by BRD4-BD2 was supported in a fluorescence anisotropy competition binding study using a fluorescein-labeled H4K5ac/K8ac peptide as an assay probe, yielding Ki of 800 μM and >3,000 μM for BRD4-BD2 and BRD4-BD1, respectively (Figure S4B). Furthermore, BRD4-BD1 and other BDs, including those from CBP and PCAF, showed almost no interaction with the single- or di-acetylated Twist peptides (data not shown).
We next solved the three-dimensional structure of BRD4-BD2 bound to Twist-K73ac/K76ac peptide using NMR spectroscopy to determine the molecular basis of this selective interaction (Figure 4A-4B, Figure S4C and Table S1). As revealed in the 3D structure, the Twist-K73ac/K76ac peptide is bound in the protein across an elongated cavity formed between the ZA and BC loops of this left-handed four-helical bundle structure. Specifically, acetylated-K73 is bound in the canonical acetyl-lysine binding site, forming a hydrogen bond between its carbonyl oxygen and the side-chain nitrogen of the conserved Asn433. Acetylated-K76 is recognized, next to K73ac, by the BD2 in a small hydrophobic cavity that is lined with Trp374, Val380, Leu385, and Val439. While the overall recognition of the di-acetylated-K73/K76 in Twist by the BD2 is similar to that of the di-acetylated K5/K8 in H4 by the BD1 of BRD4, several additional interactions observed in the former complex explain its selectivity. For instance, the imidazole nitrogen atom of His437 of BD2 is within hydrogen bond distance to the backbone carbonyl oxygen of the K73ac (Figure 4A-4B). Notably, within this highly conserved acetyl-lysine binding pocket, His437 in BRD4-BD2, which corresponds to Asp144 in BRD4-BD1, is unique. Asp144 was not engaged in any interaction with the H4 peptide as shown in the crystal structure of the BD1/H4-K5ac/K8ac peptide complex (Filippakopoulos et al., 2012), explaining the failed binding of BRD4-BD1 to Twist-K73ac/K76ac. To further examine the role of His437 in BRD4/Twist association, we engineered two point mutants by switching His437 and Asp144 in the two BDs, generating BD2-H437D and BD1-D144H mutants. Remarkably, we found that BD2-H437D almost completely lost its ability to bind to the di-acetylated Twist, whereas BD1-D144H gained binding ability for the acetylated Twist (Figure 4C), confirming the important function of His437 in the Twist-K73ac/K76ac recognition.
Figure 4. The structural and molecular basis of Twist-K73ac/K76ac recognition by BRD4.

(A) Stereo ribbon diagram of the 3D solution structure of the BRD4-BD2 bound to a di-acetylated K73ac/K76ac Twist peptide (yellow). Side chains of key residues engaged at the protein/peptide interactions are depicted and color-coded by atom type.
(B) Surface electrostatic potential (left) or Space-filled (right) representation of the BRD4-BD2/Twsit-K73ac/K76ac complex structure highlights His437 (red) at the acetyl-lysine binding site that is responsible for the BRD4-BD2' specificity of this molecular recognition.
(C) HA-tagged Twist was co-expressed with Flag-tagged WT or mutant BD1 and BD2 in HEK293 cells. Twist and BDs were immunoprecipitated with HA and Flag antibodies, respectively, and the bound BDs and Twist were analyzed by Western blotting.
(D) HA-tagged Twist was co-expressed with Flag-tagged BD1, BD2, BD1+BD2 in HEK293 cells. After immunoprecipitated Twist, BDs and H4, the association and acetylation of these molecules were examined by Western blotting.
See also Figure S4 and Table S1
We observed additional intermolecular interactions in the complex structure that contribute to the selectivity of BRD4-BD2/Twist recognition. For instance, the methyl group of Ala70 of Twist interacts with the aromatic side-chain of the conserved Tyr432 in BRD4-BD2, whereas side-chains of Ser78 of Twist and Glu438 of the BD2 form electrostatic interactions. Importantly, both Ala70 and Ser78 in Twist are located outside the di-acetylation GK-X-GK motif and are not conserved in H4. Ala70 has no corresponding residue in H4 whereas the corresponding residue for Ser78 in H4 is Leu10, which could not form a favorable interaction with Glu438 in the BD2, or even Asp145 in the BD1. Collectively, our structural insights provide a detailed understanding of the molecular basis for the selective recognition of Twist-K73ac/K76ac by the BRD4-BD2.
Histone H4 and Twist synergistically interact with BRD4
Because single BD2 of BRD4 can interact with H4 or Twist, we examined their interactions in a cellular context by expressing Twist or TwistK73R/K76R with single or double BDs of BRD4 in HEK293 cells. After immunoprecipitation of Twist, BDs, or H4 individually, the association and acetylation of the other two molecules were analyzed by Western blotting (Figure 4D and Figure S4D). First, we immunoprecipitated Twist and examined the presence of other two molecules (left panel, Figure 4D). We found that TwistK73R/K76R did not associate with any of the BDs (left panel, lanes 7-9, Figure 4D) whereas Twist associated with BD2 and BD1+BD2 but not BD1. Notably, the associated BD1+BD2 also contained H4 (left panel, lanes 4-6, Figure 4D). Second, we immunoprecipitated BD1, BD2, or BD1+BD2 and examined the presence and acetylation of Twist and H4 (middle panel, Figure 4D). BD1 associated with H4 but not Twist, whereas BD2 interacted with both Twist and H4, indicating that Twist and H4 can compete for interaction with BD2 (middle panel, lanes 4-6, Figure 4D). BD1+BD2 also associated with Twist and H4. Intriguingly, the amount of H4 associated with single BD (BD1 or BD2) is similar to that with double BDs in BD1+BD2 (middle panel, lanes 2 and 3 vs lane 1, Figure 4D), suggesting that only one BD in BD1+BD2 binds to H4. In the presence of Twist, the binding of BD1+BD2 to H4 did not alter (middle panel, lane 4 vs lane 1, Figure 4D). Because Twist interacts with BD2 but not BD1, H4 likely only interacts with BD1 when BD1+BD2, Twist and H4 are all present. Consistent with this contention, the amount of Twist associated with BD1+BD2 was more than that with BD2 (middle panel, lane 4 vs lane 5, Figure 4D), where Twist and H4 competed for the binding to the BD2. Lastly, we immunoprecipitated H4 and examined the association of Twist and BDs (right panel, Figure 4D). We found that levels of BD1, BD2 and BD1+BD2 appeared to be equivalent, suggesting that H4 interacted equally with BD1, BD2 or BD1+BD2 and only one BD interacted with H4 in BD1+BD2. In the presence of Twist, the immunoprecipitated BD2 was reduced (right panel, lane 5 vs lane 2, Figure 4D), suggesting that Twist and H4 compete for the interaction with single BD2. However, when the double BDs (BD1+BD2) were present, only one BD was engaged in interaction with H4, since the intensity of immunoprecipitated BD1+BD2 was about equal to that of Twist (right panel, lane 1 vs lane 4, Figure 4D). Consistent with the results from immunoprecipitated Twist, the immunoprecipitated BD1+BD2 by H4 contained Twist, re-affirming the association of three protein molecules in cells. Taken together, these results indicate that Twist and H4 can simultaneously interact with the double BDs of BRD4, in which BD1 binds to H4 whereas BD2 associates with Twist. This distinct binding selectivity of the two BDs of BRD4 is supported by our structural analysis.
Twist-BRD4 interaction is required for WNT5A expression
To identify the transcriptional target of the Twist-BRD4 complex, we performed cDNA microarray analysis of HMLE and luminal T47D cells that have undergone Twist-mediated EMT (Figure 3G and Figure S5A). We reasoned that genes that are transcriptionally active in Twist/HMLE and Twist/T47D cells but are down-regulated by JQ1 in these cells but not in vector control cells are likely targets of the Twist-BRD4 complex (Figure 5A). Among the 29 overlapping genes, WNT5A is noted to encode a critical ligand of both canonical (controlling pluripotency) and non-canonical (regulating motility and planar cell polarity) Wnt pathways. Upregulation of Wnt5a is correlated with an aggressive phenotype in melanoma as well as breast, lung and prostate tumors (Witze et al., 2008). We thus selected WNT5A as an example to characterize the transcriptional mechanism of Twist. We noticed that TwistWT but not TwistK73R/76R induced Wnt5a expression (Figure 3G and Figure S3C). Similarly, TwistWT induced EMT and Wnt5a expression in T47D cells (Figure S5A). In addition, TWIST expression positively correlates with WNT5A expression in eight microarray datasets from human breast cancer (Figure S5B). Using Twist and Wnt5a antibodies that detect Twist and Wnt5a, respectively, in xenograft tumors derived from SUM1315 cells, which express high levels of Twist and Wnt5a, but not MCF7 cells, which express low levels of Twist and Wnt5a (Figure S5C), we found that Twist is also positively correlated with Wnt5a expression in breast cancer specimens, with both increased expression found predominantly in ER-breast cancer (Figure S5D). Further, in fourteen breast cell lines (Figure 5B), both the mRNA and protein levels of Twist and Wnt5a were found to be largely correlated, with elevated expressions found in BLBC cell lines. BRD4 expression is relatively constant among normal breast, luminal and BLBC cell lines (Figure 5B). Consistently, no significant difference in BRD4 mRNA was found between ER+ and ER- breast cancers from a 477 sample microarray dataset (Figure S5E).
Figure 5. Twist positively correlates with Wnt5a expression in breast cancer.

(A) Gene expression profiling analysis (left panel) was used to identify potential Twist target genes. Common Twist target genes between HMLE and T47D cells were shown in the heat-map (right panel).
(B) The mRNA and protein levels of Twist, Wnt5a and BRD4 were analyzed by RT-PCR and Western blotting.
(C) The Effects of stable shRNA knockdown of endogenous Twist in SUM1315 cells that were transiently transfected with WT or mutant (KR) Twist on the expression of Wnt5a and various molecules was evaluated by Western blotting.
(D) Wnt5a expression in five BLBC cell lines with knockdown of Twist and/or BRD4 was analyzed by Western blotting. NTC stands for non-target control siRNA.
See also Figure S5
We generated a clone of SUM1315 cells with stable knockdown of Twist. Twist-knockdown reduced the mesenchymal phenotype as these cells were clustered together; cells also gained expression of epithelial markers and reduced the expression of mesenchymal markers (Figure 5C and Figure S5F). Ectopic expression of TwistWT, but not TwistK73R/K76R, restored the mesenchymal phenotype in these cells. Twist-knockdown also resulted in suppression of Wnt5a expression. Ectopic expression of TwistWT, but not TwistK73R/K76R, recovered Wnt5a expression (Figure 5C) and restored the invasiveness and mammosphere formation in these cells (Figure S5G). These results are in line with observations in HMLE cells (Figure 3G) and indicate that the Twist-BRD4 interaction is critical in maintaining mesenchymal phenotype/characteristics in BLBC cells. Consistently, JQ1 suppressed Wnt5a expression in both Twist/HMLE and Twist/T47D EMT cells (Figure S5H). In addition, knockdown of Twist or BRD4 in five BLBC cell lines resulted in reduced Wnt5a expression; double knockdown of these two molecules almost completely abolished Wnt5a expression (Figure 5D). The downregulation of Wnt5a by BRD4-knockdown is specific, because knockdown of other BET members did not alter Wnt5a expression (Figure S5I). In addition, knockdown of BRD4 did not change the expression of either epithelial or mesenchymal markers (Figure S5J). The downregulation of Wnt5a correlated with inhibition of invasiveness in BLBC cells; addition of recombinant Wnt5a partially restored invasiveness (Figure S5K). Collectively, these results indicate that the Twist-BRD4 interaction is likely conserved in HMLE and BLBC cells for EMT and that this interaction is required for the expression of Wnt5a, which may represent as a bona fide target of Twist for promoting tumorigenicity in BLBC.
Twist-BRD4 interaction is required for the recruitment of BRD4 at the WNT5A super-enhancer
To delineate how the Twist-BRD4 complex activates WNT5A expression, we constructed a Twist-Gal4 fusion protein by fusing Twist N-terminal residues 1-100 to Gal4 DNA-binding domain (DBD). We also generated several N-terminal deletion mutants of Twist fused with Gal4-DBD, including DL1-Gal4, DL2-Gal4, DL3-Gal4 and KR-Gal4 (Figure S6A). When these Twist-Gal4 constructs were co-expressed with the Gal4-luciferase reporter, Gal4-luciferase activity was moderately increased by about 2-fold compared to the control; co-expression of BRD4 with the Twist-Gal4 fusion constructs that contain the N-terminal region required for Tip60-mediated acetylation (i.e. TW and DL1) greatly enhanced luciferase activity to approximately 8-fold, suggesting that the N-terminal half of Twist contains transactivation activity and its interaction with BRD4 boosts this activity.
The WNT5A promoter contains two Twist-responsive E-boxes, conserved in human and mouse, and located at -160 bp and -67 bp from transcriptional starting site (TSS) (Figure 6A). We cloned the human WNT5A promoter (-2000 bp upstream of translation starting site) and generated several deletion and E-box mutants of the promoter-luciferase constructs, including Luc1 (-2000 bp), Luc2 (-760) and LucEM (-760, two E-box mutations). As expected, Twist alone induced Luc1 and Luc2 promoter luciferase activity; co-expression of BRD4 further enhanced the Twist-induced Luc1 and Luc2 promoter luciferase activities (left panel, Figure 6A). In addition, mutation of each E-box (E1M & E2M) in this region reduced whereas mutation of both E-boxes (EM) completely abolished Twist-BRD4-mediated activation of the WNT5A promoter luciferase activity, suggesting that both E-boxes are required for Twist-BRD4 induced transcriptional activation. TwistK73R/K76R mutant partially decreased WNT5A promoter luciferase activity and was completely insensitive to BRD4-mediated transcriptional activation (right panel, Figure 6A). BRD4-mediated enhancement of Twist transcriptional activity is specific because other BET members did not possess this capability and treatment with JQ1 or MS417 disrupted this BRD4-mediated enhancement (Figure S6B).
Figure 6. The Twist-BRD4 complex directly activates WNT5A transcription.

(A) Schematic depiction of the WNT5A promoter and WNT5A reporter luciferase constructs used (top panel). Enhancement of Wnt5a luciferase activity by co-expression of Twist and BRD4 in HEK293 cells.
(B) Twist, BRD4, H4ac4, RNA-PolII and P-TEFb (CDK9) association at the WNT5A promoter as assessed by ChIP. SP-specific primer; CP-control primer (5 kb downstream of 3′-UTR).
(C) Twist, BRD4, H4ac and H3K27ac association at the WNT5A enhancer as assessed by ChIP SP-specific primer (22 kb upstream of TSS); CP-control primer (5 kb downstream of 3′-UTR).
(D) Effects of Twist-knockdown or JQ1 treatment on the association of Twist, BRD4, RNA-PolII and P-TEFb (CDK9) and H4ac4 at the WNT5A promoter as assessed by ChIP in SUM1315 cells. SP and CP primers used are same as in (B).
(E) (Left panel) Assessing effects of transient expression of HA-Twist and/or Flag-BRD4 on their association with RNA-PolII and P-TEFb (CDK9) in HEK293 cells. (Right panel) Assessment of endogenous BRD4 knockdown effect on the association of Twist, BRD4, RNA-PolII and P-TEFb (CDK9) in SUM1315 cells.
(F) SUM1315 cells were serum starved for overnight followed by stimulation with FGF, TNFα, or EGF plus insulin for 3 hr in the absence or presence of JQ1 (right panel). Twist was immunoprecipitated and K73ac/K76ac of Twist and the association of Tip60 and BRD4 were analyzed. Expression of Wnt5a, Twist, BRD4 and Tip60 were also examined by Western blotting.
(G) BLBC cells were treated with JQ1, expression of c-Myc and Wnt5a was analyzed by Western blotting.
(H) Invasion (left) and tumorsphere-formation assays (right) of cells treated as in (G) were examined in the absence or present of recombinant Wnt5a (100 ng/ml). Statistical analysis (mean ± SD) from three independent experiments with duplicates was shown.
For A, B, C, and D, statistical analysis (mean ± SD) from three separate experiments in triplicates was shown.
See also Figure S6
Chromatin immunoprecipitation (ChIP) analysis revealed that Twist, BRD4 and acetylated H4 associated at the WNT5A promoter in BT549 and SUM1315 cells, together with P-TEFb and RNA-PolII (Figure 6B). A recent study indicated that BRD4 preferentially occupied a small subset of super-enhancers in transcriptional active key oncogenes that are critical for proliferation and survival of tumor cells (Loven et al., 2013). Intriguingly, WNT5A is one of these key oncogenes that contain BRD4-associated super-enhancer, which covers exon1, promoter and a region up to 30 kb upstream of TSS in WNT5A genomic sequence in Chromosome 3. To examine whether Twist and BRD4 also bind the WNT5A super-enhancer in BLBC, we designed two sets of ChIP primers that are 22 and 28 kb upstream of TSS. ChIP experiments indicated that Twist and BRD4 indeed occupied the WNT5A enhancer together with H3K27ac, a mark of active enhancer (Figure 6C). Knockdown of Twist or JQ1 treatment inhibited the association of BRD4 at the WNT5A enhancer (data not shown). Knockdown of Twist or JQ1 treatment also reduced the presence of BRD4, P-TEFb and RNA-PolII at the WNT5A promoter (Figure 6D). However, JQ1 treatment did not affect the association of Twist at the WNT5A promoter, suggesting that Twist is required for the recruitment of the BRD4/P-TEFb/RNA-PolII complex to the WNT5A promoter. Consistent with these observations, TwistKR, which could not interact with BRD4 and failed to rescue Wnt5a expression (Figure 5C), was unable to recruit the BRD4/P-TEFb/RNA-PolII complex to the WNT5A promoter (data not shown). In addition, ectopic expression of BRD4 increased Twist interaction with P-TEFb and RNA-PolII whereas knockdown of BRD4 reduced the association of Twist with P-TEFb and RNA-PolII (Figure 6E). Our results indicate that Twist recruits BRD4, and together with P-TEFb and RNA-PolII at the WNT5A promoter/enhancer to activate transcription.
The direct transcriptional activation of WNT5A by the Twist-BRD4 complex prompted us to investigate the stimuli responsible for Twist acetylation and WNT5A expression. We found that several stimuli, including TNFα and EGF plus insulin, could induce Twist acetylation at K73/K76 (Figure S6C). TNFα and EGF/insulin treatments greatly enhanced the interaction of Twist with BRD4 and with Tip60, increased K73ac/K76ac of Twist, and promoted Wnt5a expression (left panel, Figure 6F); JQ1 blocked the interaction of Twist with BRD4 and thus suppressed Wnt5a expression (right panel, Figure 6F). Consistent with these findings, TNFα or EGF/insulin treatments greatly enhanced the association of Twist, BRD4, P-TEFb and RNA-PolII at the WNT5A promoter (Figure S6D). Knockdown of Twist suppressed the association of BRD4 and Twist at the WNT5A promoter, however, JQ1 treatment did not inhibit the binding of Twist at the WNT5A promoter (Figure S6E). These data suggest that the association of BRD4 at the WNT5A promoter is mediated by Twist.
Although JQ1 was reported to reduce c-Myc expression, we noticed that JQ1 (1 μM) caused a decrease of c-Myc expression only in one of five examined BLBC cell lines (Figure 6G). However, JQ1 reduced Wnt5a expression in all cell lines. The low sensitivity to JQ1 in Hs578T cells is likely due to the remarkably high expression levels of Twist and Wnt5a in this particular cell line (Figure 5B). Increased JQ1 concentration resulted in Wnt5a downregulation in a dose-dependent manner in this cell line (Figure S6F). The downregulation of Wnt5a by JQ1 correlated with its inhibition of invasion and tumorsphere-formation of these cells; addition of recombinant Wnt5a could partially restore this inhibitory effect (Figure 6H and Figure S6F-S6G). Together, these data indicate that the Twist-BRD4 interaction, enhanced by extracellular signals, is required for the recruitment of P-TEFb/RNA-PolII complex to the WNT5A super-enhancer for transcription of WNT5A, which executes, at least in part, the oncogenic function of Twist. JQ1 disrupts this interaction and thereby suppresses WNT5A expression in BLBC.
The Twist-BRD4-Wnt5a axis is critical for tumorigenicity in breast cancer
To further examine the oncogenic role of the Twist-BRD4-Wnt5a axis and explore the therapeutic potential of BET-specific inhibitors for targeting this axis in BLBC in vivo, we established two Wnt5a-knockdown clones in SUM1315 cells. Knockdown of Wnt5a inhibited the non-canonical Wnt pathway, exemplified by the downregulation of JNK phosphorylation (Figure 7A). Wnt5a-knockdown also suppressed the canonical Wnt/β-catenin pathway, indicated by the downregulation of β-catenin and the suppression of Akt/GSK-3β phosphorylation. These effects were further confirmed by β-catenin reporter assay (Figure S7A). Although Wnt5a-knockdown did not alter the expression of epithelial or mensenchymal markers, it reduced the expression of several pluripotent molecules (CD44, Sox2 and Oct4). Consistently, Wnt5a-knockdown suppressed invasion and tumorsphere-formation in these cells (Figure 7B).
Figure 7. The Twist-BRD4-Wnt5a axis is critical for tumorigenicity in vitro and in vivo.
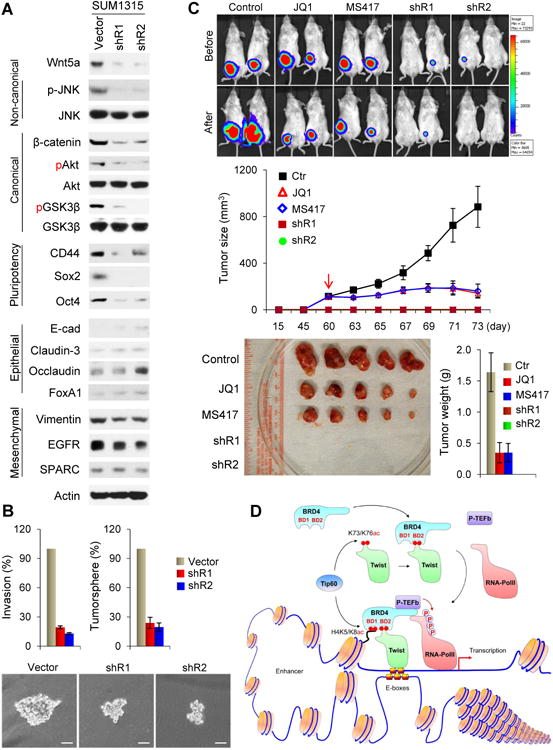
(A) Expression of various molecules in SUM1315 cells with Wnt5a-knockdown.
(B) Invasion and tumorsphere-formation in SUM1315 cells with Wnt5a-knockdown. Data are presented as a percentage of vector control values (mean ± SD in three separate experiments in duplicates). Representative pictures of tumorsphere are shown in the bottom panel, Scale bar, 100 μM.
(C) Vector control and Wnt5a-knockdown SUM1315 cells were injected into the mammary fat pad of NOD-SCID mice. When tumors from mice injected with control cells reached 100 mm3, mice were divided into three groups and treated with JQ1 (50 mg/Kg), MS417 (20 mg/Kg), or solvent control, respectively. The size of tumor was recorded by bioluminescence imaging before or after 2-wk treatment. Tumor weight was also measured. Data are represented as a mean ± SEM from 5 mice.
(D) A proposed model illustrating the interaction of Twist and BRD4 at the enhancer/promoter of WNT5A, which leads to the transcriptional activation of WNT5A expression in EMT and BLBC.
See also Figure S7
In vivo studies were performed by injecting SUM1315 vector control cells or Wnt5a-knockdown clones into the mammary fat pads of NOD-SCID mice. When control tumors were approximately 100 mm3, mice were divided into three groups to receive daily treatments of JQ1 (50 mg/Kg), MS417 (20 mg/Kg), or solvent control for two weeks. We found that knockdown of Wnt5a completely inhibited tumor growth in SUM1315 cells, and that both JQ1 and MS417 treatments significantly inhibited tumor growth (Figure 7C). The growth inhibitory effect of JQ1 and MS417 correlated with suppression of Wnt5a expression and downregulation of proliferative marker Ki67 in these tumors (Figure S7B). These results suggest that Wnt5a is critical for the tumorigenicity of BLBC. BET-specific inhibitors suppress the tumorigenicity of BLBC by inhibiting the Twist-BRD4 interaction and WNT5A expression.
Discussion
Our study provides several mechanistic insights into how Twist and BRD4 function cooperatively to activate gene transcription in EMT and BLBC. First, we show that Twist uses a unique mechanism for recruiting BRD4 in gene transcription (Figure 7D). Although BRD4 is the key transcriptional regulator, it lacks specific DNA binding motif. How BRD4 and its associated transcriptional complex are recruited to gene-specific promoters/enhancers remains elusive. We found that Twist contains an “H4-mimic” GK-X-GK motif and becomes di-acetylated by Tip60, which also acetylates multiple lysine residues in histone H4 including K5 and K8. By binding to BRD4-BD2 via the K73ac/K76ac motif, Twist recruits BRD4 to target gene promoters/enhancers through the recognition of and interaction with E-boxes by its bHLH domain. Once localized in the chromatin, BRD4-BD1 binds with acetylated H4-K5ac/K8ac to facilitate the docking of the BRD4 complex on promoters/enhancers and thereby activates pause release factor P-TEFb to phosphorylate and release RNA-PolII for WNT5A transcription.
Our study demonstrates that the two BDs of BRD4 have distinct functions and binding specificities for acetylated proteins in transcription. Although single BD1 or BD2 of BRD4 is individually capable of interacting with acetylated H4 in vitro, only BD1 is engaged in the binding with acetylated H4 in the tandem BD1+BD2. This is consistent with the observation that single BD1 of Brdt binds to acetylated histone H4 nearly as well as Brdt (full-length; contains BD1+BD2) (Moriniere et al., 2009), and that BRD4-BD1 specifically recognizes acetylation marks on H4, whereas BRD4-BD2 has broad binding specificity for di-acetylated substrates (Filippakopoulos et al., 2012). In line with this contention, only BRD4-BD2 interacts with Twist-K73ac/K76ac. We found that charged amino acid residues (D144 in BD1, H437 in BD2) surrounding the acetyl-lysine-binding pocket of BDs contributed to the binding specificity of BD1 and BD2. Additional residues beyond the di-acetylation motif further contribute to Twist's association with BRD4-BD2. Notably, it has recently been reported that BRD4 is phosphorylated by CK2 on several Ser residues in the C-terminal region of BD2, and that these phosphorylations were suggested to affect the interaction of BRD4 with acetylated histones and transcriptional cofactors (Wu et al., 2013). Although single BD2 of BRD4 can interact with either acetylated H4 or Twist-K73ac/K76ac individually, the tandem BD1+BD2 of BRD4 apparently form a ternary complex with two acetylated proteins in that di-acetylated H4 is bound to BD1 and di-acetylated Twist bound to BD2. These results suggest that BRD4 utilizes its tandem BDs as an integration platform to cooperatively interact with H4 and Twist in assembling the integrated transcriptional complex containing P-TEFb and RNA-PolII at target gene promoters/enhancers. Notably, several transcription-associated proteins that contain tandem binding modules have been shown to engage in combinatorial recognition of different post-translational modifications (PTMs) in histones for the assembly of transcriptional complexes (Zeng et al., 2010). For example, the tandem PHD-BD module in BPTF specifically recognizes a combination of H3K4me3 and H4K16ac in gene activation (Ruthenburg et al., 2011). Our results not only support this notion but also extend the functionality of these tandem binding modules in directing gene transcriptional activation as exemplified by the tandem BDs of BRD4 in bridging histone and non-histone transcription factor.
Notably, these histone-mimic sequences contain lysine/arginine-rich resides, which are often viewed conventionally as a nuclear-localization signal (NLS), as in the case of Twist. However, TwistKR mutant, which cannot be acetylated but still resides in the nucleus, fails to interact with BRD4 and is unable to induce EMT and WNT5A expression. Our results suggest that theses lysine/arginine-rich “potential” NLS motifs in transcription factors may have previously un-recognized histone-mimic functions. Consistent with our findings, histone-mimic sequences are deployed by influenza non-structural protein 1 (NS1) in inhibiting human transcription elongation complex in the anti-virus response and by HP1 in forming HP1-chromatin complex (Canzio et al., 2013; Marazzi et al., 2012). We believe that PTMs on histone mimic sequences present in non-histone proteins likely play an important role, via conserved molecular mechanisms as seen with those PTMs in histone, in governing the assembly and function of transcriptional complexes in chromatin.
Second, our study demonstrates that Twist is a transcriptional activator responsible for WNT5A expression in BLBC. Twist has been shown to bind to the E-cadherin promoter to repress transcription in a way similar to that of Snail. However, this contradicts the role of Twist in development, where it acts as a transcriptional activator to upregulate mesoderm-specific genes in Drosophila. When the bHLH domain of Twist was replaced with Gal4-DBD, we found that the Twist-Gal4-DBD fusion was sufficient to activate gene expression, indicating that Twist functions as a transcriptional activator. We further show that Twist recruits BRD4 and the associated P-TEFb and RNA-PolII to the WNT5A promoter/enhancer to directly activate WNT5A expression, which is required for invasion and the maintenance of CSC-like properties of BLBC. Notably, Wnt5a is induced in epithelial cells during EMT and required for maintenance of CSC-like properties in the resulting mesenchymal cells (Scheel et al., 2011). In addition, Wnt5a expression is required for the loss of cell-cell contacts allowing cells to migrate to the edge of wounds, and is also necessary for intestinal epithelial stem cells to regenerate damage tissues during wound healing and tissue repair (Miyoshi et al., 2012). The correlated expression of Twist and Wnt5a in BLBC supports our contention that the Twist-BRD4-Wnt5a signaling axis plays a critical role in the development and progression of BLBC.
Third, our study indicates that the Twist-BRD4 interaction represents a druggable target for treating BLBC. Although Twist is highly expressed in BLBC, the absence of a clear ligand-binding domain in Twist creates a formidable obstacle toward developing small molecules that inhibit its activity as a transcription factor. We found that BET-specific BD inhibitors disrupted the Twist-BRD4 interaction and resulted in significant Wnt5a reduction, leading to inhibition of invasion and tumorigenicity of BLBC cells in vitro and in vivo. Based on our mechanistic understanding of Twist-BRD4 interaction in gene transcription, we predict that selective chemical inhibition of BRD4/H4 interaction would result in a broad inhibition of BRD4 functions as chromatin regulator in gene transcription, whereas selective inhibition of the BRD4/transcription factor association might affect specific transcription factor's ability in their target gene activation. BD inhibitors selectively target BD2 over BD1 of BRD4 are needed to address these questions; they will also further functionally validate the effectiveness and therapeutic benefits of targeting BRD4 for treating BLBC.
Experimental Procedures
Protein Purification and Mass Spectrometry Analysis
We generated a clone of HeLa S3 cells with stable expression of Flag-Twist (Li and Zhou, 2011). After enriching the nuclear extracts from 40 liters of suspension culture, we carried out affinity protein purification with Flag affinity columns. The final eluted immunocomplexes were separated on SDS-PAGE and the bound proteins were excised from the gel and subjected to Nano-LC-MS/MS mass spectrometry analysis (Applied Biomics; Hayward, CA). To identify the acetylated lysine residues on Twist, the acetylated Twist was digested with trypsin and the tryptic peptides were analyzed by LC-MS/MS using an LTQ Velos Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled with a Nano-LC Ultra/cHiPLC-nanoflex HPLC system (Eksigent, Dublin, CA) through a nano-electrospray ionization source (Li et al., 2013). Tandem MS/MS data were acquired using CID fragmentation of selected peptides during the information-dependent acquisition. The LC-MS/MS results were subjected to protein identification and acetylation sites determination using ProteomeDiscoverer 1.3 software (Thermo Fisher Scientific, Waltham, MA) and MASCOT server.
Protein Structure Analysis by NMR
The NMR spectral collection, analysis, and structure determination of the BRD4-BD2 with Twist-K73ac/K76ac were performed as previously reported (Zhang et al., 2012). Briefly, NMR samples contained a protein/peptide complex of 0.5 mM in a 100 mM sodium phosphate buffer, pH 6.5 that contains 5 mM perdeuterated DTT and 0.5 mM EDTA in H2O/2H2O (9/1) or 2H2O. All NMR spectra were collected at 30°C on NMR spectrometers of 800, 600 or 500 MHz. The 1H, 13C and 15N resonances of the protein in the complex were assigned by triple-resonance NMR spectra collected with a 13C/15N-labeled and 75% deuterated BRD4-BD2 bound to an unlabeled Twist peptide (Clore and Gronenborn, 1994). The distance restraints were obtained from 3D 13C- or 15N-NOESY spectra. Protein structures were calculated with a distance geometry-simulated annealing protocol using X-PLOR (Brunger, 1993) that was aided with iterative automated NOE assignment by ARIA for refinement (Nilges and O'Donoghue, 1998). Structure quality was assessed by Procheck-NMR (Laskowski et al., 1996). The structure of the protein/ligand complex was determined using intermolecular NOE-derived distance restraints that were obtained from 13C-edited (F1), 13C/15N-filtered (F3) 3D NOESY spectra.
Immunoprecipitation, Immunoblotting, Immunofluorescence, Immunohistochemical Staining, RT-PCR, and Chromatin Immunoprecipitation (ChIP)
Detail methods provided in the Supplemental Experimental Procedures.
Tumorigenesis Assay
All procedures were approved by the Institutional Animal Care and Use Committee at the University of Kentucky College of Medicine and conform to the legal mandates and federal guidelines for the care and maintenance of laboratory animals. Animals were maintained and treated under pathogen-free conditions. Female NOD-SCID mice (6–8 weeks old; Taconic, Germantown, NY) were injected with breast cancer SUM1315 (2×106 cells/mouse) cells via mammary fat pad, and mice had three groups: vector control and Wnt5a-knockdown stable clones. Tumor growth was monitored with caliper measurements. When tumors were approximately 1.0 cm in size, mice were euthanized and tumors excised. Data were analyzed by Student's t test; p < 0.05 was considered significant.
Statistical Analysis
Data are presented as mean ± SD. A Student's t test (two-tailed) was used to compare two groups (p < 0.05 was considered significant) unless otherwise indicated.
Supplementary Material
Highlights.
Twist contains a “histone H4 mimic” GK-X-GK motif that is di-acetylated by Tip60
BRD4 forms a bivalent link with acetylated H4 at BD1 and di-acetylated Twist at BD2
The Twist-BRD4 complex controls WNT5A expression in EMT and basal-like breast cancer
BET inhibitors suppress tumorigenicity by disrupting the Twist-BRD4 interaction
Significance.
BLBC is associated with an aggressive clinical history, development of recurrence, distant metastasis and shorter patient survival. BLBC contains abundant EMT transcription factor Twist and possesses many CSC-like characteristics, suggesting that Twist-activated EMT program confers growth advantages to BLBC. However, the absence of a clear ligand-binding domain in Twist creates a formidable hurdle toward developing inhibitors that can suppress its function. We found that Twist interacts with and recruits the BRD4/P-TEFb/RNA-PolII transcription complex to WNT5A super-enhancer for gene activation. BET-specific inhibitors disrupted the Twist-BRD4 interaction and resulted in significant Wnt5a reduction, leading to inhibition of invasion and tumorigenicity of BLBC in vitro and in vivo. Our study indicates that targeting the Twist-BRD4 interaction provides an effective approach for treating BLBC.
Acknowledgments
We thank Cathy Anthony for critical reading and editing of this manuscript. We also thank Dr. Jing Chen for technical assistance on mass spectrometry analysis. We are grateful to Dr. James E. Bradner for providing JQ1, Dr. Robert A. Weinberg for HMLE cells, Dr. Michael Rosenblatt for SUM1315 cells, and Dr. Bruno Amati for Tip60 antibodies as valuable reagents for this study. This work was supported in part by grants from the National Institute of Health (CA125454 to B.P. Zhou, P20CA1530343 to B.M. Evers, and HG004508 and CA87658 to M.-M. Zhou), American Cancer Society (RSG13187 to YW), and Nature Science Foundation of China (91129303 to JD). We acknowledge the University of Kentucky Proteomics Core that is partially supported by grants from the National Cancer Institute (P30CA177558) and the National Institute of General Medical Sciences (P20GM103486).
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists
Accession Numbers: Microarray data of Twist expression in HMLE and T47D cells with or without JQ1 treatment was deposited at the Gene Expression Omnibus database with the accession number GSE53222. Structure factors and coordinates for the second bromodomain of BRD4 in complex with K73ac/K76ac-di-acetylated Twist peptide are deposited at the Protein Data Bank under PDB ID code 2MJV, and the NMR spectral data are deposited at the BioMagResBank (BMRB) under BMRB accession number 19738.
Supplementarl Information: Supplemental information includes seven figures and Supplementary Experimental Procedures and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brunger AT. X-PLOR version 3.1: A system for X-ray Crystallography and NMR (version 3.1 edit) Yale University Press; New Haven, CT: 1993. [Google Scholar]
- Canzio D, Liao M, Naber N, Pate E, Larson A, Wu S, Marina DB, Garcia JF, Madhani HD, Cooke R, et al. A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature. 2013;496:377–381. doi: 10.1038/nature12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM. Multidimensional heteronuclear nuclear magnetic resonance of proteins. Methods in enzymology. 1994;239:349–363. doi: 10.1016/s0076-6879(94)39013-4. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Cote J. The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev. 2004;14:147–154. doi: 10.1016/j.gde.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Leptin M. twist and snail as positive and negative regulators during Drosophila mesoderm development. Genes Dev. 1991;5:1568–1576. doi: 10.1101/gad.5.9.1568. [DOI] [PubMed] [Google Scholar]
- Li J, Zhou BP. Activation of beta-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer. 2011;11:49. doi: 10.1186/1471-2407-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhou Q, Sunkara M, Kutys ML, Wu Z, Rychahou P, Morris AJ, Zhu H, Evers BM, Huang C. Ubiquitylation of phosphatidylinositol 4-phosphate 5-kinase type I gamma by HECTD1 regulates focal adhesion dynamics and cell migration. J Cell Sci. 2013;126:2617–2628. doi: 10.1242/jcs.117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazzi I, Ho JS, Kim J, Manicassamy B, Dewell S, Albrecht RA, Seibert CW, Schaefer U, Jeffrey KL, Prinjha RK, et al. Suppression of the antiviral response by an influenza histone mimic. Nature. 2012;483:428–433. doi: 10.1038/nature10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriniere J, Rousseaux S, Steuerwald U, Soler-Lopez M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461:664–668. doi: 10.1038/nature08397. [DOI] [PubMed] [Google Scholar]
- Nilges M, O'Donoghue S. Ambiguous NOEs and automated NOE assignment. Prog NMR Spectroscopy. 1998;32:107–139. [Google Scholar]
- Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568–2581. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- Rekowski M, Giannis A. Histone acetylation modulation by small molecules: a chemical approach. Biochim Biophys Acta. 2010;1799:760–767. doi: 10.1016/j.bbagrm.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ, Allis CD. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–940. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Witze ES, Litman ES, Argast GM, Moon RT, Ahn NG. Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science. 2008;320:365–369. doi: 10.1126/science.1151250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Zinzen RP, Stark A, Kellis M, Zhang H, Young RA, Levine M. Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev. 2007;21:385–390. doi: 10.1101/gad.1509607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–262. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, Rusinova E, Gerona-Nevarro G, Moshkina N, Joshua J, et al. Down-regulation of NF-kappaB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem. 2012;287:28840–28851. doi: 10.1074/jbc.M112.359505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu Rev Biochem. 2012;81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.