

Abstract
Three-dimensional (3D) matrices have significant advantages compared to conventional two-dimensional (2D) matrices for studying cell adhesion, migration, and tissue organization. Cellular behavior is dependent on the surrounding matrix environment for signaling and induction of biological responses (Carletti, et al., 2011; Pampaloni, et al., 2007; Vlodavsky, 1999). 2D cultures induce an artificial polarity in cultured cells between upper and lower surfaces not present normally in the in vivo environment. No longer nonpolar, many aspects of cellular behavior are altered (Beacham, et al., 2007; Grinnell and Petroll, 2010; Yamada and Cukierman, 2007). In addition, 2D models lack the physical properties of 3D matrix, such as topography, stiffness, and dimensionality. To begin to mimic the 3D environment of in vivo connective tissue extracellular matrix (ECM), collagen gels have been used widely (see Unit 10.3). Culture of cells in collagen gels results in a bipolar fibroblast morphology that resembles the in vivo phenotype (Friedl and Brocker, 2000; Even-Ram and Yamada, 2005; Grinnell and Petroll, 2010). Although more physiological, 3D collagen gels lack the complex biochemical and physical microenvironment present in an in vivo ECM that regulates cellular physiological properties (Beacham, et al., 2007).
A variety of methods to create a more in vivo-like ECM have been published (Yamada and Cukierman, 2007). Adding critical ECM components to 3D collagen matrices, including fibronectin, hyaluronan, link protein and glycosaminoglycans, can more accurately mimic the structural microenvironment of the native ECM (Friedl and Brocker, 2000). Other ECM models use cultured cell lines, such as fibroblasts, to derive an ECM lattice through secretion of an organized ECM (Beacham, et al., 2007). Different cell lines have been chosen to generate a specific microenvironment for study of particularly types of cellular behavior (Kutys and Yamada, 2013). For example, cultured bovine corneal endothelial cell lines produce an ECM mimicking an in vivo subendothelium, and the EHS tumor cell line produces a matrix that can be extracted to produce Matrigel, which simulates basement membrane molecular complexity including laminin, collagen IV and nidogen (Beacham, et al., 2007; Friedl and Brocker, 2000). To simulate a physiological environment even more closely, 3D matrices derived from mouse tissue slices from which cells were extracted have reportedly provided successful ECM replicas for studying in vivo cellular behavior (Cukierman, et al., 2001).
Because of the important roles of the extracellular microenvironment on normal and tumor cells, we have developed protocols to produce cell-free (decellularized) 3D matrices from cryostat sections of normal and tumor human tissues. These extracted matrices can be used as a 3D tissue culture environment to analyze effects of various 3D matrices on normal and tumor cell responses and behavior. Using human pancreas and breast tissue samples, we have successfully prepared cell-free 3D ECM models, used them as cell culture substrates for a human breast cancer cell line, MDA-MB-231, and then performed immunofluorescence staining to characterize intracellular structures. A frequently observed difference between normal and tumor tissue-derived ECM environments involves the amount of deposited fibrillar collagen (Provenzano, 2008). Tumor tissues from both breast and pancreas often contain substantially more collagen than normal adjacent tissue, and this protocol preserves this difference in cell-free 3D matrices from these tissues (Vidi, et al., 2013). This 3D culture system we describe using cell-free 3D matrix provides an approach to studying cellular behavior and migratory mechanisms associated with cancer.
The basic protocol describes methods for successfully extracting cells and cellular debris from human tissue cryostat sections to obtain a clean, cell-free 3D ECM for plating cell lines (Figure 1). Cellular structures can be analyzed by chemical fixation, permeabilizing the cells to permit antibody penetration into the cytoplasm, and immunofluorescence staining protocols, as described in the following support protocol. We provide additional support protocols for embedding human tissues, sectioning human tissue, hematoxylin and eosin (H&E) staining, and trichrome staining techniques. H&E as well as Trichrome staining are used as quality control measures.
NOTE: Use sterile and aseptic techniques when handling live cell lines.
NOTE: Observe universal standard precautions when handling human tissue. Wear personal protective equipment (gloves, laboratory coat, eye protection, etc.).
Keywords: three-dimensional (3D) extracellular matrix, cell extraction, human tissue, acellular matrix, H&E staining, trichrome staining, immunostaining, collagen
Basic Protocol: EXTRACTION OF HUMAN TISSUE SECTIONS TO GENERATE A CELL-FREE 3D MATRIX
Prior to extraction, human tissues must first be embedded in OCT, followed by cryostat sectioning. Embedding of snap-frozen human tissue fragments, or excised blocks of tissue, is described in Support Protocol 2. The embedded tissues are then sectioned at a thickness of 12 μm using a cryostat adjusted to optimal cutting temperature (see Support Protocol 3). The cryostat sections are captured on positively charged microslides and stored at −80° until extraction. Use of human tissues from living patients requires formal informed consent, e.g., by the NIH or an institutional review board. Handling of human tissues requires strict observance of standard universal precautions throughout entire protocol. The laboratory where human specimens are used requires Biosafety Level 2 certification.
This protocol provides guidelines for complete extraction of cells from human tissue cryostat sections to yield a cell-free 3D matrix. In this protocol, both human pancreas and mouse tissues are used. Other normal and tumor human tissues can presumably be extracted successfully using the same, or a slightly modified, protocol. Mouse non-tumor organ tissues (breast, pancreas, and kidney) have also been successfully extracted by us using a slightly modified protocol as indicated in notes for the basic protocol.
MATERIALS
Extraction buffer (see recipe)
Phosphate-buffered saline (PBS; APPENDIX 2A)
10 U/ml DNase I recombinant, RNase free (Roche) in PBS +/+ (see recipe)
Plastic slide box (holds 100 slides), e.g., from Ted Pella
12 μm thick cryostat sections of human tissue samples on positively charged slides (see Support Protocol 2)
100 × 15 mm plastic petri dish (BD Falcon)
1000 μl pipette tips (without filters)
Cell Extraction
Prepare cell extraction buffer (see recipe) fresh on day of extraction. Warm on a water bath to 37° for 20–30 min.
-
Remove slide box containing cryostat sections of human tissue samples from a −80° freezer.
Human tissue samples had first been embedded in OCT upon arrival and stored at −80° (see Support Protocol 2). Tissue blocks in OCT were then sliced at 12 μm thickness using a cryostat and captured on positively charged microslides (see Support Protocol 3). Place each slide horizontally into a separate 100 × 15 mm plastic petri dish with tissue slices facing up. Place the dishes on the platform of an orbital rocker (e.g., a VWR DW-150 Waver) set to the lowest setting (~50 cycles/min, ~15° tilt angle).
-
Extract matrices two times for 15 min each with 10 ml of cell extraction buffer while rocking at room temperature.
When rocking slides on the rocker, make sure the slides are completely submerged in extraction buffer and not floating on top of the buffer. For aspiration of the extraction buffer between extractions, we recommend attaching a short glass Pasteur pipette (e.g., 6-inch) to the hose of a vacuum aspiration system and then fit the glass pipette tip with a 1000 μl plastic filter-less disposable pipette tip. Adjust vacuum to a low setting to provide gentle aspiration to avoid damaging or detaching the tissue section from the slide. If extracting mouse tissue, only one cell extraction buffer wash of 15 min is needed. Total extraction time for mouse tissue is 60 min compared with 75 min for human tissue. Continue to extract human matrices nine times for 5 min each with 10 ml aliquots of cell extraction buffer while continuing to rock at room temperature.
Aspirate remaining cell extraction buffer and wash matrices three times for 10 min each with 15 ml aliquots of PBS while continuing to rock at room temperature.
-
Aspirate all liquid from the plastic petri dish. A clear layer of gel-like DNA will cover the matrices on the slide. Carefully aspirate off this clear layer without contacting or disrupting the human tissue sample.
This step requires slow, deliberate motions and attention to detail to avoid detaching the tissue sections. Samples from different types of tissues may vary in their propensity to detach from the slide. Reduce this problem by keeping the pipette tip further away from the surface of the tissue sample to remove the DNA gel without disrupting the tissue. This DNA gel removal step is critical to permit successful adhesion to the ECM of plated cells in subsequent steps (see Support Protocol 1), so remove as much excess DNA as possible during this step. Following the aspiration of DNA, wash matrices one time for 30 min with 15 ml of PBS while continuing to rock at room temperature.
To remove additional DNA, treat the matrices with 10 U/ml DNase I (see recipe). Add 10 ml of the DNase I solution to each plastic petri dish and rock at room temperature for 30 min.
After DNase I treatment, aspirate remaining liquid. As described above, very carefully aspirate off any remaining pieces of the residual clear DNA gel without dislodging the tissue sample.
-
Add 15 ml of sterile PBS to each plastic petri dish and tightly seal each dish using Parafilm. Allow the matrices to wash overnight while rocking at room temperature.
This step is necessary to wash away residual Triton X-100 that may remain in the extracted tissue matrix. Residual Triton X-100 may interfere with immunofluorescence staining. An inverted phase-contrast microscope can be used to examine the 3D matrix in order to determine how much of the matrix still remains on the slide.
-
Following overnight washing, matrices are ready for cell plating (see Support Protocol 1).
If cell plating is not done immediately following cell extraction, store the matrices as follows. In a cell culture hood, transfer each slide containing cell-extracted matrix to a new sterile 100 × 15 mm plastic petri dish. Add 15 ml of sterile PBS +/+ and seal each dish with Parafilm. Dishes may be stored at 4° for up to a week. Longer storage may be possible and will require the addition of antibiotics.
Support Protocol 1: PLATING OF CELLS, FIXATION, PERMEABILIZATION, AND IMMUNOFLUORESCENCE STAINING
This support protocol describes use of the human breast cancer cell line, MDA-MB-231, for immunofluorescence analysis after cell culture on extracted 3D human tissue matrix. The MDA-MB-231 line is widely used in cancer research, and it displays high invasiveness and ability to metastasize. In addition, these cells are suitable for transfection assays[1]. Other normal or malignant cell lines can be substituted.
The MDA-MB-231 cell line is routinely cultured in Dulbecco’s modified Eagle medium (DMEM) with high glucose and glutamine (HyClone) supplemented with 10% fetal bovine serum. Cultured MDA-MB-231 cells should never be allowed to reach complete confluence while maintaining stock cultures. When cells reach 70–80% confluence, they are routinely subcultured at a 1:5 dilution. The following support protocol describes plating, fixation with paraformaldehyde, permeabilization, and immunofluorescence staining. For the examples shown we used rhodamine phalloidin and anti-collagen antibody, but other antibodies can be substituted depending on subject of interest. Examples of such staining are shown in Figure 2.
Figure 2.

Immunofluorescence staining of MDA-MB-231 cells plated on cell extracted human pancreas tumor tissue. Cells were labeled for actin with Rhodamine Phalloidin (Invitrogen) (A, D) and extracted tissue collagen was immunolabeled with primary anti-collagen type I mouse antibody and secondary anti-mouse antibody conjugated to AlexaFluor 647 (B, E). Maximum intensity projections of z-stacks collected are shown; thickness of matrices can be easily quantified from z-stack depths. Collagen I images were processed with low pass filter using MetaMorph software. C and F represent overlay of actin and collagen images. Scale bar equals 10 μm.
MATERIALS
Cell culture of MDA-MB-231 cells cultured in 75 cm2 cell culture flask
Cell culture medium containing fetal bovine serum (FBS; see recipe)
DMEM with glutamine and high glucose (HyClone)
0.05% trypsin-EDTA (1x) (Gibco)
Penicillin/streptomycin (10,000 Units/ml penicillin and 10,000 μg/ml streptomycin, Gibco)
Fixation solution containing 4% paraformaldehyde (see recipe)
Phosphate-buffered saline (PBS; APPENDIX 2A)
1X phosphate buffered saline (+calcium/+magnesium) (HyClone)
Permeabilizing solution (see recipe)
Blocking solution (see recipe)
0.05% Tween 20 in PBS (see recipe)
Fluoro-gel with anti-fading agent (Electron Microscopy Sciences 17983-20)
Primary antibody solution (see recipe)
Secondary antibody solution (see recipe)
Extracted human tissue matrices on positively charged slides (see Basic Protocol)
100 × 15 mm style plastic petri dish (BD Falcon)
37°C, 10% CO2 humidified incubator
50 ml plastic conical centrifuge tube
20 μl pipetter
1000 μl pipetter
Immunostain Moisture Chamber (Ted Pella #21049)
24 × 60 no. 1 premium cover glass slips (Fisher Scientific 12-548-5P)
Clear nail polish
Plating MDA-MB-231 Cell Line
Pre-warm the DMEM/10% FBS, DMEM (serum-free), and trypsin at 37° and defrost the aliquot of penicillin/streptomycin (penstrep).
Place the extracted human tissue matrices in a tissue culture hood along with media and pipettes.
In the culture hood, place each slide with extracted matrix into a new 100 × 15 mm plastic petri dish. Add 15 ml of pre-warmed DMEM (serum-free) to each dish. Incubate the dishes in a 37°C, 10% CO2 humidified incubator, where they will remain until the cells are ready for plating.
Place a flask of MDA-MB-231 at 80% confluence in the hood. Detach the cells by aspirating media and adding 12 ml of pre-warmed 0.05% trypsin-EDTA (1x). Monitor cell detachment using an inverted phase contrast microscope.
When the cells become rounded (after 1–2 min), aspirate the trypsin from the flask and wash the flask with 10 ml DMEM/10% FBS. Using the 10 ml DMEM/10% FBS just added to the flask, carefully detach the cells by flushing the surface of the flask multiple times by pipetting up and down. Collect the entire cell suspension and transfer to a 50 ml conical centrifuge tube. Add 10 ml of DMEM/10% FBS for a total volume of 20 ml. Using a clinical tabletop centrifuge, centrifuge the cell suspension at ~400 x g at room temperature for 5 min.
Following centrifugation, aspirate the liquid, leaving the cell pellet. Add 7 ml of DMEM (serum-free) to the tube and gently pipet the cell suspension up and down until the cells are re-suspended without clumps.
Using a sterile pipette tip, transfer an aliquot to a cell counter.
Count the cells and calculate the amount of cells required to plate 13 ml of cell suspension in medium with cell concentration of 3.3 × 104 cells/ml for each plastic petri dish.
Re-centrifuge the cells as in step 5.
Aspirate the remaining liquid and re-suspend the cell pellet in the calculated volume of DMEM (serum-free) containing penstrep diluted 100-fold (e.g., two plastic petri dishes would use 26 ml of DMEM (serum-free) and 260 μl of penstrep).
Remove the matrices from the incubator. Aspirate the DMEM (serum-free) and transfer the slides to new sterile 100 × 15 mm style plastic petri dishes.
Add 13 ml of re-suspended cells in DMEM (serum-free) containing penicillin/streptomycin to each plastic petri dish; the slide with extracted matrices should be completely submerged. Cover and place the dish into an incubator.
Allow the cells to attach to the matrices for 4 hours in a 37°C, 10% CO2 humidified incubator. After the 4-hour incubation period, view the slides under an inverted phase contrast microscope to confirm cell attachment and spreading.
Fixation and Permeabilization of Cells Plated on Human 3D Matrices
After the cell incubation gently aspirate cell culture medium.
-
To each slide in their individual plastic petri dishes, add 10 ml of warmed fixation solution (37°C).
Complete this fixation step rapidly after removing from the incubator to prevent morphological changes. We have found it most efficient to pre-aliquot new plastic petri dishes with warmed fixation solution followed by submersion of each slide handled with forceps. Fix slides for 30 min at room temperature.
Wash slides with three changes of PBS for 10 min each at room temperature.
Permeabilize cell membranes with 10 ml of the permeabilization solution for 10 min at room temperature.
Wash once for 5 min with PBS.
Wash overnight with sterile PBS at room temperature in petri dishes sealed with Parafilm.
Immunofluorescence Staining
Fill enough humidification chamber wells with water, one for each slide stained.
Transfer the slides from the plastic petri dishes to the humidification chamber. Aspirate excess liquid from slide.
-
Block non-specific protein-binding sites on the slides using 500 μl of blocking solution per slide. Place Parafilm on top of the slide and allow to block for 30 min at room temperature.
For most efficient staining, Parafilm pieces should be cut to approximately the same size as the slide. Using forceps, very carefully lay flattened Parafilm piece on top of solution. Do not press down, just lay the Parafilm on top. This will prevent dripping off the edge of the slide. Aspirate. Wash once with 500 μl per slide of 0.05% Tween 20 solution for five min.
Apply 500 μl primary antibody to each slide and cover with Parafilm. Allow to stain for 1 hour at room temperature.
Aspirate. Wash three times with 500 μl per slide of 0.05% Tween 20 solution for ten minutes per wash. Use Parafilm for each wash and place humidification chamber with slides on circular rocker (VWR DW-150 Waver) set to lowest speed (~50 cycles/min, ~15° tilt angle).
Apply 500 μl secondary antibody to each slide and cover with Parafilm. Allow to stain for 45 min at room temperature.
Aspirate. Remove each slide from the humidification chamber and place into a 100 × 15 mm plastic petri dish. Wash three times for 10 minutes with 15 ml of 0.05% Tween 20 solution.
Wash twice for ten minutes with 15 ml of PBS.
Aspirate liquid from the slide and mount slides using Fluoro-gel and glass cover slips avoiding the formation of air bubbles.
Allow mounting medium to solidify for one hour at room temperature.
Seal the slide edges with clear nail polish to prevent drying and store slides at 4°C.
Support Protocol 2: EMBEDDING TISSUE IN O.C.T
Upon arrival from a source (e.g., National Disease Research Interchange, NDRI; Cooperative Human Tissue Network, CHTN or company Asterand), human tissue biospecimen samples must be handled using standard universal precautions. Frozen human tissue samples arrive in a snap-frozen state and are immediately placed at −80° for storage purposes. O.C.T. compound (optimal cutting temperature) medium is the chosen method for embedding the tissues. Compared with formalin-fixed, paraffin embedding methods, OCT prevents cross-linking of the tissue sample, undesirable for plating and examination of cell behavior. Induced cross-links change the morphology of the ECM, interfering with behavior and migratory mechanisms of re-plated cells on the acellularized tissue ECM (Scicchitano, et al., 2006). The following procedure can be used or modified for other human tissue types, and we have also used this procedure for mouse tissue.
MATERIALS
2 inch-diameter, 4 inch long solid copper cylinder to promote rapid freezing
Human tissue samples, such as pancreas or breast (e.g., from National Disease Research Interchange, NDRI; Cooperative Human Tissue Network, CHTN; Asterand)
22 × 22 mm Peel-A-Way disposable plastic tissue embedding molds (Electron Microscopy Sciences, Cat. #70182)
O.C.T. compound medium (Tissue-Tek)
Positively charged slides (Daigger Superfrost Plus/Colorfrost Plus microslides)
Embedding Tissues in O.C.T
Place small pellets or powdered dry ice in two containers. In one container, surround the copper cylinder with dry ice, while keeping it upright with the top surface level exposed. In the second container, level the dry ice and cover with a piece of aluminum foil. Allow the copper cylinder and aluminum foil to cool. Spraying the surface of the dry ice with 70% ethanol may expedite the cooling process.
Remove the human tissue sample from the −80°C freezer and place on dry ice.
-
Arrange Peel-A-Way disposable plastic tissue embedding molds for easy access. Apply a layer of O.C.T. to each of the molds sufficient to cover the bottom.
If there are any bubbles in the layer of O.C.T., use the 1000 μl pipette for removal by gently aspirating a tiny volume to pick up each bubble. It is critical to prepare molds without, or with minimal bubbles. Bubbles interfere with and create challenges during cryo-sectioning (see support Protocol 3) of the tissue samples. Fill an extra disposable plastic tissue embedding mold with excess O.C.T. at room temperature until it is almost full.
Take one mold with a thin O.C.T. layer and position it on top of the copper cylinder. Allow the O.C.T. to freeze completely.
Remove the first human tissue sample from its packaging. Make sure to handle the tissue using standard universal precautions.
Using tweezers to grip the tissue, dip the tissue into the excess O.C.T. at room temperature until it is completely submerged.
Remove the mold with the thin frozen O.C.T. layer from the copper cylinder and position the tissue sample in the center of it.
Quickly fill this tissue sample mold with O.C.T. medium until the tissue sample is completely covered.
-
Using the 1000 μl pipette, carefully remove all of the bubbles from the mold by gently aspirating a small volume to pick up each bubble. This step is extremely important for proper cryo-sectioning (see Support Protocol 3).
Some samples create more, smaller bubbles than others for reasons that were not clear. Try to remove as many or all of the bubbles if you can. Once all of the bubbles are removed to the best of your ability, place the mold on the aluminum foil-covered dry ice container. Allow it to completely freeze and solidify.
-
Repeat steps 5 through 11 for all of the tissue samples. Be careful to label each sample properly and keep a record of all samples.
It is recommended to label the sides of the plastic mold containing sample prior to freezing. Use a permanent, water resistant marker. Once all of the samples are completely frozen, wrap them individually (or in pairs, e.g., tumor + normal) and store them at −80°C.
Support Protocol 3: CRYOSTAT TISSUE SECTIONING
The Basic Protocol requires prior sectioning of human tissue samples using a research-quality cryostat, such as the Leica CM3050 S Cryostat. A tissue thickness of 12 μm was chosen for practicality in sectioning various tissues while providing optimal tissue thickness for observing cells plated in the final extracted 3D extracellular matrix environment. Familiarity with the cryostat instruction manual is imperative for troubleshooting purposes. Tissue samples with high fat content, such as breast tissue, will require maximum, or near maximum, temperature settings on the cryostat machine (e.g., −50°C). Tissue samples containing less fat, such as some breast tumor or pancreas samples, will have a higher optimal cutting temperature. Cryo-sectioning is a technique requiring patience, delicate movements, and attention to detail. Prior practice is encouraged to become familiar with the machine.
MATERIALS
Cryostat (Leica CM3050 S)
Specimen disc, 30 mm
Low-profile disposable blades, 80 mm × 8 mm × 0.25 mm
O.C.T. embedded human tissue sample molds (see Support Protocol 2)
Daigger Superfrost Plus/Colorfrost Plus 75 × 25 mm pre-cleaned microslides (positively charged)
Plastic slide box (holds 100 slides), e.g., from Ted Pella
O.C.T compound medium (Tissue-Tek)
Paint brush, soft round tip, no. 4 width
Tweezers (e.g., Dumont no. 5)
Tissue Cryo-sectioning
-
The day before planned slicing, pre-set temperature settings on the cryostat based on the tissue sample to be sliced. Place specimen disc, paint brush and tweezers inside the machine chamber so they equilibrate with the chamber temperature. Also, insert a new blade into the machine.
Consult the cryostat instruction manual for suggestions on tissue-specific temperature settings. The day before you intend to do sectioning, set both the chamber temperature (CT) and object temperature (OT) a bit lower (colder) than what you believe to be the optimal slicing temperature. It is easier for the machine to increase (warmer) temperature quickly rather than drop (colder) temperature. Always make sure to use a new blade for optimal sectioning. The following morning, pre-chill slide box on dry ice.
Remove O.C.T. embedded human tissue sample from −80°C freezer.
Place the tissue block with mold inside the cryostat chamber and allow its temperature to equilibrate with the chamber temperature (e.g., 20 min).
Label positively charged slides with appropriate information for cataloging tissue slices using a sharpened pencil. Arrange the slides on top of the cryostat so they easily accessible.
Peel away disposable mold from the tissue block. Retain the original plastic mold for tissue identification purposes.
Pour a small amount of O.C.T. compound on the specimen disc (e.g., 3 drops). Immediately place the tissue block on top of the platform as the O.C.T compound begins to solidify due to the cold chamber temperature. Arrange the tissue block in the orientation you expect to slice.
Allow the O.C.T. compound on the tissue block to solidify completely and secure the disc and tissue block on the slicing arm.
Slice away excess O.C.T. compound to expose the tissue sample.
-
Set the machine to 12 μm thickness. Arrange the anti-curl shield properly. Begin slicing.
Practice and patience will be needed to perfect sectioning technique. Adjusting the temperature controls and repositioning the mold platform as well as anti-curl shield are all pertinent to optimal cutting. In addition, adjusting automatic or manual slicing speed is critical for obtaining desirable slices. Referencing the machine instruction manual is helpful in preventing common technique issues such as splintered sections, curled, wrinkled, or torn sections. Once an adequate section is obtained, pick up a positively charged slide, invert it, and with a quick and fluid motion capture the section on the slide by direct physical contact with the tissue section.
Allow the slide to sit at room temperature for 2–3 minutes to ensure adhesion to the slide.
Place the slide in the cooled slide box.
Repeat steps 11–14 for additional sections of the same tissue block, or steps 4–14 for new tissue blocks.
Store all tissue slides in the slide box at −80°C until needed for subsequent use.
Support Protocol 4: HEMATOXYLIN & EOSIN STAIN
H&E staining is a common histology technique used for identifying various components of a tissue sample. The two dyes utilized are hematoxylin, a basic dye, and eosin, an acidic dye. Hematoxylin dyes nuclei present in a tissue sample dark blue, while eosin dyes components of the tissue such as the cytoplasm and collagen shades of red, orange and bright pink. We experimented with different ranges of exposure time to the various dyes. Human breast and pancreas tissue were stained successfully with the following protocol modified from procedures of the Rosen Lab, Baylor College of Medicine[2]. All steps are completed at room temperature. Examples are shown in Figure 3.
Figure 3.

H&E stained 12 μm sections of intact human pancreas. (A and B) Normal pancreas; (C and D) Pancreatic adenocarcinoma tumor; (A and C) scale bar in C equals 3 mm and corresponds to A as well; (B and D) 10x magnification, scale bar in D equals 300 μm and corresponds to B as well.
MATERIALS
Phosphate-buffered saline (PBS; APPENDIX 2A)
Hematoxylin solution, Gill no. 3 (Sigma)
Acid ethanol (see recipe)
Acidified eosin Y (see recipe)
90% ethanol
100% ethanol
Xylene (previously used solution is acceptable)
Glass staining dish with cover (Wheaton Science Products)
Glass staining rack, holds 10 slides (Wheaton Science Products)
Permount histological mounting medium (Fisher Scientific)
24 × 60 no. 1 premium cover glass slips (Fisher Scientific 12-548-5P)
H&E Staining of Human Tissue Slices for Nuclei, Cytoplasm, and Collagen
Arrange slides to be stained in the staining rack.
-
Submerge slide rack completely into staining dish containing PBS and wash two times at room temperature for 3 min each.
Volume of liquid in glass staining slide dish will vary depending on specific glass dish used. We used approximately 150 ml of liquid. It is critical that the entire slide be submerged in liquid for this and subsequent steps, so complete submersion should govern the volumes to add to each dish for each step of the protocol. -
Place slides in dish containing hematoxylin solution, Gill no. 3 for 2 min.
Hematoxylin may be re-used. Store it in a dark container or cover any clear container with aluminum foil because the stain is light sensitive. If needed, filter the stain to remove particles before use. Dip slides once in container of distilled water (dH2O).
Place slides in a container of regular tap water for 5 min.
Dip slides twelve times rapidly into a container of acid ethanol.
Place slides in tap water two times for 1 min each.
Place slides in dH2O for 2 min.
-
Place slides in acidified eosin Y solution for 2 min.
Acidified eosin Y may be re-used. If needed, filter the stain before use. Place slides into 90% ethanol solution for 5 min.
Place slides in 100% ethanol for 5 min.
-
Place slides in xylene for 10 min.
Always use the chemical hood for handling xylene. -
Mount slides using Permount histological mounting medium and glass cover slips.
Try to eliminate as many bubbles as possible that may accumulate under the slide using a pipette. However, be careful not to press down on the cover slip too hard in order to avoid distorting the sample. Allow the mounting medium to solidify for approximately one hour.
Store slides at 4°C.
Support Protocol 5: MASSON’S TRICHROME STAIN
Trichrome staining is a common histological procedure utilizing three main colors. It is the most commonly used stain to identify fibrous collagen. Nuclei are stained black, cytoplasm and muscle fibers are stained red, and collagen is stained blue. We use this protocol for staining of collagen in human tissue samples, comparing normal and tumor tissues. The following protocol is the ‘standard staining protocol’ provided with Masson’s Trichrome Stain Kit (Richard-Allan Scientific). All steps are completed at room temperature unless otherwise noted to ensure more intense staining. Examples of Trichrome staining are shown in Figure 4.
Figure 4.

Trichrome-stained 12 μm sections of intact human pancreas. (A and B) Normal pancreas; (C and D) Pancreatic adenocarcinoma tumor; (A and C) scale bar in C equals 3 mm and corresponds to A as well; (B and D) 10x magnification, scale bar in D equals 300 μm and corresponds to B as well.
MATERIALS
-
Masson’s Trichrome Stain Kit (purchased from Thermo Scientific/Richard-Allan Scientific Product #010, Catalog #87019)
Weigert’s iron hematoxylin, part A
Weigert’s iron hematoxylin, part B
Biebrich scarlet-acid fuchsin solution
Aniline blue stain solution
Phosphotungstic-phosphomolybdic acid solution
Bouin’s solution
1% acetic acid solution
Working Weigert’s iron solution (see recipe)
100% ethanol
Xylene (previously used is fine)
Permount histological mounting medium (Fisher Scientific)
Glass staining dish with cover (Wheaton Science Products)
Glass staining rack, holds 10 slides (Wheaton Science Products)
24 × 60 no. 1 premium cover glass slips (Fisher Scientific 12-548-5P)
Trichrome Staining of Cryostat Human Tissue Sections for Collagen
-
Pre-heat water bath to 56°C and pre-warm Bouin’s solution.
A glass staining dish along with a glass staining rack should be used throughout this procedure. We used 150 ml of each solution in glass dishes, but the volume will vary depending on the dish size. It is critical that the slides be completely submerged. Bouin’s solution contains formaldehyde fixative, so no initial fixation procedure is necessary prior to this step. Avoid heating the water bath above 56°C because excessive heating may cause detachment of tissue sections from the slide. Always handle Bouin’s solution in a chemical fume hood because heating increases the release of hazardous fumes. Bouin’s solution should be discarded after each use following chemical waste disposal procedures. Place slides in a glass staining rack into dH2O for 10 min.
Transfer slides to Bouin’s solution for 1 hour in a chemical fume hood.
Rinse the slides in tap water for 5 min, removing the yellow color.
-
Stain slides in working Weigert’s iron solution for 10 min.
Working Weigert’s iron solution can be re-used for up to 10 days after the solution is made. Transfer slides to tap water for 10 min.
-
Stain slides in Biebrich scarlet-acid fuchsin solution for 10 min.
Biebrich scarlet-acid fuchsin solution can be saved and re-used. Filter as necessary. Rinse slides in dH2O for 30 sec.
-
Transfer slides to phosphotungstic-phosphomolybdic acid solution for 5 min.
The phosphotungstic-phosphomolybdic acid solution should be discarded after each use. -
Stain slides in aniline blue stain solution for 10 min.
Aniline blue stain solution can be re-used and filtered as necessary. Nevertheless, it should be replaced if the stain becomes diluted following repeated use. Transfer slides to 1% acetic acid solution for 1 min.
Rinse slides in dH2O for 30 sec.
Dehydrate slides in two changes of 100% ethanol for 1 min each.
-
Place slides in xylene for 10 min.
Always handle xylene in a chemical fume hood. -
Mount slides using Permount histological mounting medium and glass cover slips.
Try to eliminate as many bubbles as possible that may accumulate under the slide using a pipette. However, be careful not to press down on the cover slip too hard in order to avoid distorting the sample. Allow the mounting medium to solidify for one hour.
Store slides at 4°.
REAGENTS AND SOLUTIONS
Use deionized or distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX
Acid ethanol
70% Ethanol containing:
0.25% (v/v) HCl (36.5–38.0% from Sigma H1758-500ML).
Store 6 to 12 months at room temperature
Acidified eosin Y
Eosin Y solution, aqueous (Sigma) containing:
0.5% (v/v) glacial acetic acid
Store 6 to 12 months at room temperature; filter if needed before use
Blocking solution
1.25 ml phosphate-buffered saline (PBS; APPENDIX 2A) containing:
1 drop M.O.M. mouse IgG blocking reagent (Vector Laboratories M.O.M. Kit)
2% normal donkey serum (Jackson ImmunoResearch)
Make fresh at room temperature and do not store
Culture medium with fetal bovine serum
500 ml DMEM with glutamine, high glucose, and phenol red (HyClone)
50 ml fetal bovine serum
Store up to 2 months at 4°
DNase I recombinant 10 U/ml, RNase free (Roche) in PBS +/+
PBS +/+ containing:
0.1% DNase I recombinant 10 U/ml, RNase free (Roche)
Make fresh at room temperature and do not store
Extraction Buffer
Phosphate-buffered saline (PBS; APPENDIX 2A) containing:
0.5% (v/v) Triton X-100 (Sigma T-9284)
20 mM NH4OH
Make fresh at room temperature and do not store
Fixation Solution
Phosphate-buffered saline (PBS; APPENDIX 2A) containing:
4% (v/v) paraformaldehyde (16% diluted stock solution, EM grade, e.g., Electron Microscopy Sciences)
5% (w/v) sucrose
Filter-sterilize
Store up to two months at room temperature.
PBS +/+
Phosphate-buffered saline (PBS; APPENDIX 2A) containing:
1 mM CaCl2
1 mM MgSO4
Store 6 to 12 months at room temperature
PBS supplemented with antibiotics
Lonza 1X Phosphate-buffered saline stock solution, 500 ml (PBS; APPENDIX 2A) containing:
5 ml concentrated stock solution of penicillin (10,000 Units/ml) and streptomycin (10,000 μg/ml) (Gibco)
Store 1 month at room temperature
Permeabilizing Solution
Phosphate-buffered saline (PBS; APPENDIX 2A) containing:
0.5% (v/v) Triton X-100
Primary antibody solution
Phosphate-buffered saline (PBS; APPENDIX 2A) containing:
1:12.5 M.O.M. protein concentrate (Vector Laboratories M.O.M. Kit)
1:40 rhodamine phalloidin (Invitrogen)
1:200 monoclonal anti-collagen type I antibody produced in mouse (Sigma Cat. #C2456)
Make fresh at room temperature and do not store
Secondary antibody solution
Tween 20 in PBS containing:
1:20 normal donkey serum (Jackson ImmunoResearch)
1:400 anti-mouse AlexaFluor 647 (Vector Laboratories)
Make fresh at room temperature and do not store
Tween 20 in PBS
Phosphate-buffered saline (PBS; APPENDIX 2A) containing:
0.05% (v/v) Tween 20 detergent (Sigma)
Store up to 3 months at room temperature
COMMENTARY
Background Information
Most of our knowledge about the mechanisms and regulation of cell behavior and migration is based on studies performed using two-dimensional (2D) petri surfaces and 2D extracellular matrices. Recent approaches using various types of three-dimensional (3D) matrix have started to provide knowledge based on more-physiological 3D physical and biochemical environments (Cukierman, et al., 2001; Yamada and Cukierman, 2007; Artym and Matsumoto, 2010; Friedl and Wolf, 2010; Tibbitt and Anseth, 2012; Doyle et al., 2013). 3D culture systems are providing new insights into in vivo-like cell interactions with the extracellular matrix (ECM). Striking phenotypic differences have been identified between cells in 2D and 3D environments in terms of cell morphology, adhesion, migration, and signaling. For example, cell attachment of human fibroblasts can be enhanced by a factor of five, and their cell migration rate is stimulated by a factor of 1.5 in 3D versus 2D environments (Cukierman et al., 2001). Recent studies reveal that cell behavior can be modulated by multiple signals from the 3D ECM microenvironment as cells sense differences in matrix composition, dimensionality, stiffness, fibrillar architecture, and porosity (Doyle, et al., 2013; Friedl and Wolf, 2010; Haycock, 2011).
Various 3D ECM research models have been generated using matrix produced by cultured cell lines (fibroblast, epithelial) from which cells are extracted using detergent, or by using purified or synthetic polymeric materials including Matrigel and collagen gels (e.g., see Units 10.3, 10.4, 10.9, 10.16, 19.17, 10.18, and 25.2). These 3D model systems reconstruct defined 3D environments to be able to analyze cell behavior in a more physiological setting than 2D cell culture. In order to understand cellular mechanisms related to cancer and metastasis, another approach would be to study cell interactions with microenvironments derived from normal versus tumor human tissue samples. That is, thin tissue sections placed on glass slides will retain biochemical constituents, including signaling molecules and complex in vivo ECM composition, as well as the organization of ECM found in vivo. Cues from such a 3D environment in culture can influence cell adhesion, growth, migration and invasion, and other aspects of cell behavior (Cukierman, et al., 2001; Takezawa, et al., 2002; Ning, 2012).
Using these types of approaches to compare the regulatory effects on normal and tumor cells of the ECM structure and composition, such as collagen deposition, of normal and tumor ECM microenvironments from human patients may provide insights to the roles of these cellular interactions in tumor progression and cancer biology. This Unit provides detailed methods for using human-tissue derived 3D matrices as a new model system for experimentally characterizing the molecular and physical mechanisms of cellular responses when interacting with 3D ECM ex vivo.
Critical Parameters and Troubleshooting
Human Tissue
Human tissue samples can be obtained from the National Disease Research Interchange (NDRI), as well as from the Cooperative Human Tissue Network (CHTN) or company Asterand. Plan ahead and order tissue samples early, since some types of tissues are in short supply.
O.C.T. Embedding
O.C.T. embedding and cryostat sectioning are necessary compared to other methods such as paraffin embedding. This method avoids crosslinking of the tissue and preserves the original tissue morphology and molecular composition. During embedding, all bubbles should be removed from the O.C.T. mold prior to freezing the mold. Bubbles that remain will interfere with successful sectioning. From our experience, we suggest using aspiration with a 1000 μl pipette to remove as many bubbles as possible from the mold.
Cryostat Tissue Sectioning
The technique of sectioning tissue molds with a cryostat requires practice and familiarity with the instrument. Sectioning with the cryostat and capturing slices free of cuts, ripples, and rolling can be very difficult. Familiarity with machine’s instruction manual is highly recommended. For tissues (both human and mouse) that contain a high percentage of fat (e.g., breast), the cryostat should be set to the coldest CT (chamber temperature) and OT (cutting temperature) temperatures the day before slicing. It is much easier to warm up the machine as needed, rather than trying to chill it further. Slices of 12 μm thickness are recommended for the procedures described in this experimental protocol. Other thicknesses of tissue will probably require modifications of the extraction and other protocols. We highly recommend the use of positively charged slides for the capture of human tissue. Other types of slides, e.g., silanyzed slides, do not provide the strong polarity-based attraction of tissue to slide needed to prevent washing of tissue sections from the slide during the multiple extraction and extensive washing steps needed to prepare cell-free 3D matrix.
Cellular extraction
It is critical to prepare the extraction buffer fresh on the day of the extraction to ensure an appropriate concentration of volatile ammonia. Due to the viscous nature of Triton X-100, it is highly suggested to create a 10% stock solution of Triton X-100 to be used in preparing for each extraction experiment. This stock solution can be kept tightly sealed for up to one year at room temperature. During the extraction protocol, it is extremely important to remove completely by aspiration the clear gel-like layer of DNA that appears on top of the tissue. If this DNA remains, it will inhibit cell attachment to the tissue. To carry out successful aspiration of the DNA, we recommend attaching a 1000 μl filter-less pipette tip to a short glass Pasteur pipette, which is then connected to a vacuum aspiration system. Use careful and deliberate motions to remove the DNA without touching the surface of the tissue. Hold the pipette tip close enough to the tissue to capture the DNA but far enough away to avoid dislodging the tissue section from the slide. This step of the procedure is often trying for even the most experienced researcher; the tissue type can often affect how easily tissue section becomes dislodged. For example, sections of human breast tissue are more easily detached from the slide, apparently because their predominant non-polar fatty composition prevents secure adherence to the positively charged slide. Overnight washing at room temperature is necessary to completely remove any residual Triton X-100 that had been present in the extraction buffer because Triton X-100 can interfere with immunofluorescence staining and viability of re-plated cells.
When extracting mouse tissue, we found that the first 15 min cell extraction buffer wash can be eliminated, therefore decreasing the total extraction time to one hour while still providing complete extraction of cellular material from the 3D ECM. Incomplete extraction (see example in Figure 5) will result from failure to follow the described extraction times and appropriate amounts of washes.
Figure 5.
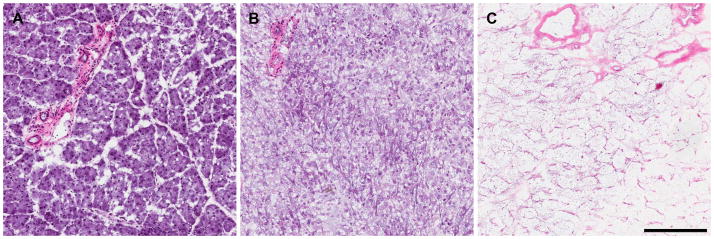
Representative images of H&E stains of partially and fully extracted normal pancreas tissue. (A) Intact mouse pancreas tissue. (B) Incompletely extracted mouse pancreas tissue with residual hematoxylin staining of cell debris. (C) Completely extracted mouse pancreas tissue leaving only 3D ECM. Scale bar equals 200 μm.
Cell Plating
MDA-MB-231 cells are cultured in DMEM with 10% FBS. We suggest sub-culturing this cell type at a 1:5 dilution ratio approximately every three days, allowing to cells to reach only 70–80% confluence. Handle the cells gently, e.g., avoiding overly vigorous pipetting and centrifugation. Cells should be incubated at 37° with 10% CO2 in a well-humidified tissue culture incubator. Cells to be used for plating on extracted human tissue should be cultured to 70–80% confluence. Cells dissociated with trypsin-EDTA are placed into DMEM with 10% FBS to halt protease activity initially, but we perform all remaining steps in serum-free DMEM.
Although some protocols use a hydrophobic histological blocking pen to outline a small area on a slide for plating limited numbers of cells or for immunofluorescence staining in small volumes of solution, we recommend against this approach. The blocking pen contains ingredients that interfere with cell attachment, ability to spread on tissue, and capacity for immunofluorescence staining. We instead suggest using an individual 100 × 15 mm plastic petri dish per slide with the addition of 13 ml of 3.3 × 104 cells/ml in DMEM with penicillin and streptomycin. We find that four hours is needed for successful cell attachment and spreading on our extracted 3D tissue sections.
Fixation/Permeabilizing
To prevent morphological changes in cells that have attached to the extracted tissue sections, take care to add fixation solution immediately to each slide after removal from the tissue culture incubator. The permeabilizing solution contains Triton X-100, so it is critical to perform an overnight wash to remove completely any residual detergent from the extracted matrices. As noted above, any remaining Triton X-100 may prevent successful immunofluorescence staining.
Immunofluorescence Staining
As noted above, to prevent interference with staining do not use a blocking pen during this procedure. We found that placing a piece of Parafilm on top of the liquid on the slide during the blocking, primary and secondary antibody staining, and washing steps helps to keep solutions spread out in a thin layer on the slide while avoiding evaporation. Our support protocol focuses on actin and collagen staining, and staining for other molecules may require modifications in concentrations of primary and secondary antibodies.
H&E and Trichrome Staining
In our experience, we recommend adhering to the time suggestions and other details in these staining protocols for optimal staining of sections. If acid is not added to the eosin, tissue affinity for the stain is greatly decreased and may result in little to no pink stain. In the H&E staining protocol, hematoxylin and eosin may be re-used, storing the hematoxylin in a dark or light-filtered container. Filter stains using filter paper if needed to remove particulates. The acidified ethanol, 90% ethanol and 100% ethanol may be re-used and filtered as necessary. When handling Bouin’s solution, always wear gloves and work under a chemical hood. We recommend discarding the Bouin’s solution after each use. The aniline blue stain solution and Biebrich scarlet-acid fuchsin solution can be filtered and re-used. Working Weigert’s iron hematoxylin solution can be re-used for up to ten days. We followed the standard staining Trichrome protocol provided with the purchased Richard-Allan Scientific Masson’s Trichrome Stain Kit (Catalog number 87019). The alternative microwave staining protocol provided has not been tested by us. For both H&E and Trichrome staining, if small volumes are being used to stain 1 to 4 slides, it is not recommended to re-use any of the solutions. Dilution of the stain is apt to occur, resulting in subsequent variation in stain color.
Anticipated Results
Basic Protocol
Cellular extraction of human or mouse tissue slices will leave behind an ex vivo 3D extracellular matrix that effectively simulates the acellular in vivo environment. The ex vivo ECMs can be used to study cell morphology and signaling, as well as cell behavior, such as proliferation or invasion.
Support Protocol 1
The cells plated on extracted 3D matrix should adhere and spread. Fixation and permeabilizing will prepare the cells for immunofluorescence staining by fixing the morphology and preparing the cells for staining by making the plasma membrane and other membranes permeable to antibodies. Immunofluorescence staining of cells permits confocal microscope analysis of specific cellular proteins.
Support Protocol 2
Embedding tissue in O.C.T. followed by freezing at −80° in a mold will provide a block of tissue in O.C.T. for slicing by a cryostat. Tissue samples remain in their original physiological state (e.g., no crosslinking).
Support Protocol 3
Slicing of tissue molds with a cryostat will produce 12 μm-thick human or mouse tissue sections adherent to positively charged slides ready for storage at −80° until use. The tissue sections should be free of tears, ripples, folds, and other negative features.
Support Protocol 4
H&E staining will result in tissue samples with nuclei stained dark purple, and tissue components including the cytoplasm and collagen will be varying hues of neon pink, orange, and possibly red.
Support Protocol 5
Trichrome staining will show tissue samples with nuclei stained black, cytoplasm and muscle fibers stained red and collagen stained blue.
Time Considerations
Basic Protocol
Cellular extraction requires approximately 4 hours followed by overnight washing. After the overnight wash, extracted tissue slices may be kept in a dish covered and sealed with Parafilm in sterile PBS for up to 3 weeks at 4°C. Longer storage may be possible and will require the addition of antibiotics.
Support Protocol 1
Once at 70–80% confluence, resuspension by trypsinizing, plating, and culturing cells requires approximately 6 hours followed by approximately 1.5 hours for fixation and permeabilizing. After permeabilizing, there is an overnight wash. After the overnight wash, cell plated on extracellular matrices from human or mouse tissue may be stored covered and sealed with Parafilm in sterile PBS with penicillin and streptomycin for up to 2 weeks at 4°C.
Support Protocol 2
The time required for this protocol depends on the number of samples to be embedded. For an experienced researcher, approximately 2 hours will be needed to embed 20–30 samples.
Support Protocol 3
The time required for this protocol depends on the number of tissue sections (slices) and on the number of different sample molds/blocks to be sectioned. For someone experienced with the cryostat, collecting 20 tissue slices each from 6 different molds may take anywhere from 2 to 4 hours.
Support Protocol 4
Total staining in this protocol requires approximately 2 hours from start to finish.
Support Protocol 5
The longest step in this staining protocol is the 1 hour incubation in Bouin’s solution at the beginning of the procedure. The remainder takes approximately 1.5 hours.
Figure 1.

Intact and extracted normal and tumor tissue. Figure comparing intact and extracted, normal and tumor, human pancreas tissue sections, along with normal mouse breast tissue. Left panels: H&E stained images display nuclei and tissue components. Note that nuclei are no longer detected following tissue extraction. Right panels: Trichrome stained images of extracted slices show tumor samples have much more collagen than paired normal samples. 20x magnification and scale bar equals 200 μm.
Acknowledgments
The project described was supported by Pathway to Independence Award K99CA129205 from NCI (V. Artym), NIH/NIDCR Intramural DE 000719 (C. Campbell, V. Artym), and Bucks County Board of Associates plus NCI/NIH grant CA113451 (E. Cukierman).
Footnotes
http://www.atcc.org/products/all/HTB-26.aspx
Website for information on MDA-MB-231 human breast cancer epithelial cell line.
http://www.bcm.edu/mcb/rosenlab/index.cfm?pmid=12983
Website for H&E protocol used as a basis and further modified.
Contribution
Contributed by Catherine B. Campbell and Vira V. Artym, National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD; and Edna Cukierman, Fox Chase Cancer Center, Philadelphia, PA.
Literature Cited
- Artym VV, Matsumoto K. Imaging Cells in Three-Dimensional Collagen Matrix. Current Protocols in Cell Biology. 2010;10:1820. doi: 10.1002/0471143030.cb1018s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beacham DA, Amatangelo MD, Cukierman E. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Current Protocols in Cell Biology. 2007:10.9.1–10.9.21. doi: 10.1002/0471143030.cb1009s33. [DOI] [PubMed] [Google Scholar]
- Carletti E, Motta A, Migliaresi C. Scaffolds for tissue engineering and 3D cell culture. Methods in Molecular Biology. 2011;695:17–39. doi: 10.1007/978-1-60761-984-0_2. [DOI] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Doyle AD, Petrie RJ, Kutys ML, Yamada KM. Dimensions in cell migration. Current Opinion in Cell Biology. 2013 doi: 10.1016/j.ceb.2013.06.004. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even-Ram S, Yamada KM. Cell migration in 3D matrix. Current Opinion in Cell Biology. 2005;17:524–532. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Friedl P, Bröcker EB. The biology of cell locomotion within three-dimensional extracellular matrix. Cellular and Molecular Life Sciences. 2000;57:41–64. doi: 10.1007/s000180050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. Journal of Cell Biology. 2010;188(1):11–9. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell F, Petroll WM. Cell motility and mechanics in three-dimensional collagen matrices. Annual Review of Cell and Developmental Biology. 2010;26:335–61. doi: 10.1146/annurev.cellbio.042308.113318. [DOI] [PubMed] [Google Scholar]
- Haycock JW. 3D cell culture: a review of current approaches and techniques. Methods in Molecular Biology. 2011;695:1–15. doi: 10.1007/978-1-60761-984-0_1. [DOI] [PubMed] [Google Scholar]
- Kutys M, Yamada KM. Regulation of cell adhesion and migration by cell-derived matrices. Experimental Cell Research. 2013 doi: 10.1016/j.yexcr.2013.05.030. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning L, Zhang Y, Chen X, Luo J, Li X, Yang Z, Qin T. Preparation and characterization of decellularized tendon slices for tendon tissue engineering. Journal of Biomedical Materials Research Part A. 2012;100A:1448–1456. doi: 10.1002/jbm.a.34083. [DOI] [PubMed] [Google Scholar]
- Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissues. Nature Reviews Molecular Cell Biology. 2007;8:839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Medicine. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scicchitano MS, Dalmas DA, Bertiaux MA, Anderson SM, Turner LR, Thomas RA, Mirable R, Boyce RW. Preliminary comparison of quantity, quality, and microarray performance of RNA extracted from formalin-fixed, paraffin-embedded, and unfixed frozen tissue samples. Journal of Histochemistry & Cytochemistry. 2006;54(11):1229–1237. doi: 10.1369/jhc.6A6999.2006. [DOI] [PubMed] [Google Scholar]
- Takezawa T, Takenouchi T, Imai K, Takahashi T, Hashizume K. Cell culture on thin tissue sections commonly prepared for histopathology. The FASEB Journal. 2002;16(13):1847–9. doi: 10.1096/fj.02-0405fje. [DOI] [PubMed] [Google Scholar]
- Tibbitt MW, Anseth KS. Dynamic microenvironments: the fourth dimension. Science Translational Medicine. 2012;4(160):160ps24. doi: 10.1126/scitranslmed.3004804. [DOI] [PubMed] [Google Scholar]
- Vidi PA, Bissell MJ, Lelièvre SA. Three-dimensional culture of human breast epithelial cells: the how and the why. Methods in Molecular Biology. 2013;945:193–219. doi: 10.1007/978-1-62703-125-7_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I. Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells. Current Protocols in Cell Biology. 1999:10.4.1–10.4.14. doi: 10.1002/0471143030.cb1004s01. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130(4):601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]