

Abstract
Bacterial enoyl-acyl carrier protein reductase (ENR) catalyzes an essential step in fatty acid biosynthesis. ENR is an attractive target for narrow-spectrum antibacterial drug discovery because of its essential role in metabolism and its sequence conservation across many bacterial species. In addition, the bacterial ENR sequence and structural organization are distinctly different from those of mammalian fatty acid biosynthesis enzymes. High-throughput screening to identify inhibitors of Escherichia coli ENR yielded four structurally distinct classes of hits. Several members of one of these, the 2-(alkylthio)-4,6-diphenylpyridine-3-carbonitriles (“thiopyridines”), inhibited both purified ENR (50% inhibitory concentration [IC50] = 3 to 25 μM) and the growth of Staphylococcus aureus and Bacillus subtilis (MIC = 1 to 64 μg/ml). The effect on cell growth is due in part to inhibition of fatty acid biosynthesis as judged by inhibition of incorporation of [14C]acetate into fatty acids and by the increased sensitivity of cells that underexpress an ENR-encoding gene (four- to eightfold MIC shift). Synthesis of a variety of compounds in this chemical series revealed a correlation between IC50 and MIC, and the results provided initial structure-activity relationships. Preliminary structure-activity relationships, potency on purified ENR, and activity on bacterial cells indicate that members of the thiopyridine chemical series are effective fatty acid biosynthesis inhibitors suitable for further antibacterial development.
The synthesis of fatty acids is accomplished through one of the cell's macromolecular biosynthetic pathways, but unlike comparable pathways for DNA, RNA, cell wall, and protein biosynthesis, fatty acid biosynthesis has not been utilized extensively as a target for antibiotic therapy. Significant differences among the structures and organizations of the enzymes catalyzing this pathway in mammals and bacteria suggest that it is feasible to develop highly selective bacterial fatty acid synthesis inhibitors with little likelihood of inhibiting the mammalian counterparts (9, 10). Genes encoding the steps of the pathway have been shown to be essential in several bacterial species (reviewed in reference 9). The efficacy and specificity of triclosan (18) and isoniazid (22) against the enoyl-acyl carrier protein (ACP) reductases (ENRs) in Staphylococcus aureus and Mycobacterium tuberculosis, respectively, confirm the utility of focusing on that particular fatty acid synthesis step as a site of metabolic intervention. The ENR activity in gram-positive species (such as S. aureus) and gram-negative species (including Escherichia coli and Haemophilus influenzae) is encoded entirely by essential, homologous fabI genes. The presence of a structurally unrelated gene, fabK, encoding a functional ENR in pathogenic species such as Streptococcus pneumoniae, Enterococcus faecalis, and Pseudomonas aeruginosa limits the potential spectrum of compounds which inhibit the fabI gene product (7). Nevertheless, new antibiotics with activity against methicillin-resistant S. aureus, vancomycin-resistant S. aureus, and many members of the gram-negative Enterobacteriaceae which carry only the fabI-encoded ENR would be clinically useful.
While the antibacterial activities of triclosan and isoniazid establish the suitability of the target for drug intervention, the compounds themselves are limited in their utility. Isoniazid has been a frontline drug for the treatment of tuberculosis since 1950 but is ineffective against most other species. Triclosan has found wide use in consumer products but has never been used for systemic therapeutic purposes due to toxicity. Consequently, broader-acting, more selective inhibitors are needed. Recently, one research group described two new series of ENR inhibitors (11, 19, 21, 23). Here, we report a new chemically distinct series, and we show that these 2-(alkylthio)-4,6-diphenylpyridine-3-carbonitriles (“thiopyridines”) inhibit bacterial growth through a mechanism that includes ENR inhibition.
MATERIALS AND METHODS
Bacterial strains.
The bacterial strains used in antibacterial activity assays were obtained from the American Type Culture Collection, the E. coli Genetic Stock Center (EGSC; Yale University), the Bacillus Genetic Stock Center (Ohio State University), or the Genome Therapeutics (GENE) collection, as noted: E. coli ATCC 35218, E. coli WO159 (AB1157 [EGSC]; recJ asmB1 ΔrfaC::Kanr), E. coli WO153 (AB1157 [EGSC]; recJ asmB1 ΔtolC::Kanr), B. subtilis NO8 (BD170 [Bacillus Genetic Stock Center]; bmr::CAT), vancomycin-resistant Enterococcus faecium ATCC 700221, methicillin-resistant S. aureus ATCC 700699, and methicillin-sensitive S. aureus CYL316 (17).
Purification of E. coli ENR.
The E. coli fabI gene (AAC74370) was cloned into the pET30a expression vector (Novagen, Inc., Madison, Wis.) and expressed in E. coli BL21(DE3) cells. The purification procedure utilized chromatography with Q-Sepharose, blue resin, and hemagglutinin resin as follows. Cell pellets were suspended in lysis buffer (50 mM KH2PO4 [pH 8.0], 100 mM NaCl, 2 mM EGTA, and 10% glycerol), and cells were broken by passage through a Microfluidics cell disrupter. Lysates were centrifuged, and the supernatant was applied to a Q-Sepharose column preequilibrated in buffer (10 mM Tris-HCl [pH 8.0], 0.1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 100 mM NaCl, 10% glycerol, 0.1% β-mercaptoethanol, and 0.02% Brij 35). ENR was eluted with a NaCl gradient (0.1 to 1 M) in the equilibration buffer. The major peak fractions were pooled and concentrated, dialyzed (10 mM Tris-HCl [pH 7.5], 0.1 mM EGTA, 0.1 mM PMSF, 10% glycerol, 0.1% β-mercaptoethanol, and 0.02% Brij 35), and centrifuged. The supernatant was loaded on a preequilibrated blue resin column (10 mM Tris-HCl [pH 7.5], 0.1 mM EGTA, 1 mM PMSF, 50 mM NaCl, 10% glycerol, 0.1% β-mercaptoethanol, and 0.02% Brij 35). ENR was eluted with the equilibration buffer containing NaCl (gradient of 50 to 1,000 mM), dialyzed in equilibration buffer, and further purified on a hydroxyapatite column (20 mM KH2PO4 [pH 8.0], 0.1 mM EGTA, 0.1 mM PMSF, 10% glycerol, 0.1% β-mercaptoethanol, and 0.02% Brij 35). ENR was eluted with a gradient of KH2PO4 up to 500 mM. The peak fractions were pooled and dialyzed in storage buffer (10 mM morpholinepropanesulfonic acid [MOPS; pH 7.0], 150 mM NaCl, 0.1 mM EGTA, 50% glycerol, 0.02% Brij 35) and then stored at −20°C.
Endpoint assay of ENR and high-throughput screen.
An ENR endpoint assay was engineered utilizing crotonoyl coenzyme A (CoA) as a substrate and measuring the overall decrease in NADH by fluorescence according to the following reaction: crotonoyl-CoA + NADH → butyryl-CoA + NAD+. By comparing sample readings to those of negative (absence of compound) and positive (absence of enzyme) controls, the percent inhibition of enzymatic activity by each compound was determined. The assay was performed in 96-well half-area black plates (Corning). The final concentration of each component in the 50-μl reaction mixture was as follows: sodium phosphate, 100 mM (pH 7.5); NADH, 100 μM; dithiothreitol, 1 mM; FabI, 1.67 μg/ml; crotonoyl-CoA, 0.4 mM (Sigma; C-4146). The assay was initialized by adding all components except substrate to plates containing compounds. After 20 min, the crotonoyl-CoA substrate was added and plates were incubated for 35 min and then read on a Victor2V plate reader (PE Lifescience) with the fluorometry option set at the following parameters: umbelliferone (0.1 s); CW-lamp filter, F355; emission filter, F460. The signal/background ratio of the resulting screening assay was 3 to 4, with a Z′ factor of 0.69 (26). Throughput of the screen with a screening platform constructed in-house was 80 plates/day. Hits were confirmed by using a kinetic assay.
Kinetic assay for ENR and IC50 measurements.
A kinetic assay in microtiter wells was used to confirm the activity of compounds identified by the endpoint assay screen and to measure 50% inhibitory concentrations (IC50s). The final concentrations of each component in each 50-μl reaction mixture are as follows: sodium phosphate, pH 7.5, 100 mM; NADH, 200 μM; dithiothreitol, 1 mM; ENR, 3 μg/ml; crotonoyl-CoA, 0.8 mM. For IC50 measurements, compounds were tested in serial dilution. The reaction was initiated by addition of the substrate, crotonoyl-CoA, and monitored continuously for 5.5 min on a spectrophotometer by measuring absorption at 340 nm. The rate of decrease in the amount of NADH in each reaction well was converted to percentage of inhibition by using SOFTmax PRO software (Molecular Devices, Sunnyvale, Calif.) and the following formula: % of inhibition = 100 × [(rate in the presence of compound − rate of negative control)/(rate of positive control − rate of negative control)]. Reaction in the presence of 2% dimethyl sulfoxide but without inhibitory compounds served as the positive control, and reaction in the presence of all components except ENR served as the negative control.
Synthesis of ENR inhibitors.
Analogs were prepared, from readily synthesized chalcones, in two additional steps. Treatment of the chalcones with 2-cyanoethanethioamide in methanolic sodium methoxide provided the intermediate 1,2-dihydro-4,6-diaryl-2-thioxopyridine-3-carbonitriles (15). Alkylation on sulfur with cesium carbonate in dimethyl formamide and an organic bromide gave the desired 2-(alkylthio)-4,6-diphenylpyridine-3-carbonitriles.
Antimicrobial activity assay.
Assays to determine the MICs of compounds were performed according to NCCLS recommendations (20) in a 100-μl volume in 96-well format, with serial twofold dilutions of the drug concentration over a twofold dilution range of 0.125 to 64 μg/ml unless noted otherwise.
Mode of action studies. (i) Measurement of inhibition of macromolecular biosynthesis.
Macromolecular biosynthesis in S. aureus CYL316 was measured essentially as described previously (12) except that radioactive precursors (acetate, N-acetylglucosamine, uridine, thymidine, or a mixture of amino acids) were 14C labeled and obtained from ICN Biochemicals Inc. Total counts incorporated at 45 min of incubation without inhibitors ranged from 3,000 for acetate, thymidine, and amino acids to >8,000 for N-acetylglucosamine and >11,000 for uridine. In these experiments, the GTC-004061 MIC for S. aureus CYL316 was 8 μg/ml and the triclosan MIC was 0.08 μg/ml.
(ii) Staphylococcus fabI underexpression hypersensitivity assay.
The endogenous S. aureus fabI gene was replaced with a drug resistance marker (Ermr) in a two-step allele-replacement procedure (4). About 1 kb of DNA sequence flanking each side of the S. aureus fabI gene (16) was PCR amplified from genomic DNA, ligated to an erythromycin resistance cassette by crossover PCR (16), and cloned into an allele-replacement suicide vector carrying a kanamycin resistance marker (Kanr) and the sacB gene encoding levansucrase (2). This allele-replacement plasmid was introduced by electroporation into S. aureus host strain CYL316 containing the multicopy plasmid pCL112Δ19 with an int gene which constitutively expresses integrase (17). Selection for Kanr yielded cells containing the entire plasmid by means of a single crossover recombination. Next, a Staphylococcus fabI gene (AAO04309) was cloned by PCR and placed under control of the tetracycline-regulated promoter (xyl/tet) (6) on an integration vector carrying a chloramphenicol resistance marker (Cmr) and an attP site. This plasmid was introduced into the S. aureus host strain CYL316 containing the single crossover allele-replacement construct. Selection for chloramphenicol-resistant, lipase-negative clones resulted in the isolation of strains in which the plasmid had integrated at the attB/L54a site within the lipase gene geh. Insertion at this site inactivated the lipase gene. Propagation in the absence of selection resulted in the loss of the multicopy plasmid pCL112Δ19. Finally, growth in sucrose and in the presence of anhydrotetracycline, the tet promoter inducer, selected for cells that had completed a second crossover event to eliminate the sacB gene and yield a total replacement of the endogenous fabI gene with the Ermr cassette.
This strain with a single copy of the Staphylococcus fabI gene under the regulation of the xyl/tet promoter at the geh locus grew poorly in the absence of the inducer, anhydrotetracycline, indicating that cells are dependent on fabI expression from the xyl/tet promoter for robust growth and that the tet promoter retains some activity even in the absence of inducer. For underexpression studies, cells were streaked and propagated overnight on Mueller-Hinton broth supplemented with 50 μg of anhydrotetracycline/ml. Several similar-sized colonies were resuspended in Trypticase soy broth and added to the MIC microtiter dish wells at a final cell concentration producing an optical density at 600 nm (OD600) of 0.005. Cell growth was measured as OD600 after an overnight incubation (about 20 h).
(iii) Staphylococcus fabI overexpression rescue assay.
The lac repressor regulated by the penicillinase promoter (PPCN) and T7 RNA polymerase regulated by the spac-I promoter (25) were cloned onto an integration plasmid containing the attP site and introduced into the geh locus of S. aureus CYL316 as described above. The Staphylococcus fabI gene (AAO04309) was cloned as a PCR product and placed under the control of the T7 promoter on a replicating shuttle vector, which also contains PPCN driving lacI to ensure adequate repression of T7 RNA polymerase in the absence of isopropyl-β-d-thiogalactopyranoside (IPTG). In the presence of inducer IPTG, excess ENR is produced. For overexpression studies, the cells were grown overnight in the absence of inducer. Several similar-sized colonies were resuspended in Trypticase soy broth and inoculated into the MIC plates at a final cell concentration equivalent to an OD600 of 0.001, with and without 1 mM IPTG. Cell growth was measured as OD600 after an overnight incubation (about 20 h).
RESULTS
Identification of inhibitors of E. coli ENR.
High-throughput screening of a chemical library with an endpoint assay resulted in the identification of four different chemical classes of inhibitors of E. coli ENR (FabI). In particular, GTC-004061 (Fig. 1), a member of one class, inhibited FabI by 98% at 10 μM but failed to inhibit 10 additional bacterial enzymes including kinases, transferases, synthetases, and reductases significantly (i.e., <13% inhibition at 10 μM). Therefore, this compound appears to be a specific inhibitor of ENR. In order to verify the inhibitory activity of this class and explore the structural dependencies of the inhibition, we synthesized several additional analogs of the 2-(alkylthio)-4,6-diphenylpyridine-3-carbonitrile GTC-004061 and determined their IC50s in a kinetic assay of E. coli ENR. The results from a subset of these analogs confirm that many members of this thiopyridine class potently inhibit ENR (Table 1). In addition, the set of compounds provides preliminary information on the relationship between the structure and the enzyme-inhibitory activity. For example, the results in Table 1 illustrate the importance of the carboxylic acid group on R3 for both ENR-inhibitory activity and cell penetration. GTC-268733, an amide analog of GTC-343129, is significantly less active, suggesting that the acid function is an integral part of the minimum pharmacophore for binding to ENR. While the relative position of the acid on the phenyl ring (meta or para) has little effect on activity, a slight extension of the acid group by one carbon (GTC-330346 versus GTC-004061) causes a decrease in activity, possibly due to a steric limitation within the enzyme.
FIG. 1.
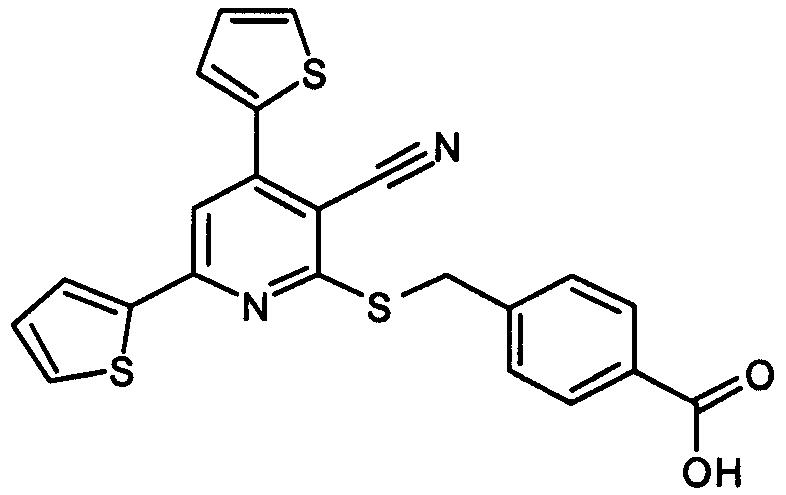
Chemical structure of the thiopyridine compound 2-(alkylthio)-4,6-diphenylpyridine-3-carbonitrile (GTC-004061).
TABLE 1.
Antibacterial and enzyme-inhibitory activities of ENR inhibitors
| GTC no. | R1 R2 R3 | IC50 (μM)a | MIC for S. aureus ATCC 700699 (μg/ml)a |
|---|---|---|---|
| 343129 | 3 | 0.75 | |
| 343131 | 3 | 0.75 | |
| 343130 | 6 | 1.5 | |
| 004061 | 4 | 2 | |
| 330346 | 25 | 4 | |
| 341772 | 25 | 8 | |
| 268963 | 12 | 32 | |
| 268847 | 9 | >64 | |
| 268776 | 12 | >64 | |
| 096296 | 17 | >64 | |
| 268726 | 18 | >64 | |
| 268724 | 33 | >64 | |
| 268925 | 46 | >64 | |
| 268733 | 70 | >64 |
Compounds are ranked by MIC and then by IC50.
Evaluation of antibacterial activity.
Several thiopyridine series analogs were tested for antibacterial activity against four species of bacteria, E. coli, E. faecium, S. aureus, and B. subtilis, by determining the MIC over a twofold dilution compound concentration range of 0.125 to 64 μg/ml (Table 1). Several compounds were active against S. aureus, and the MICs exhibited significant correlation with the IC50s (Table 1). Since some classes of ENR inhibitors are known to be substrates for efflux pumps in gram-negative species (21), we determined the MICs for both wild-type E. coli ATCC 35218 and E. coli WO153 carrying an lpxC mutation enhancing permeability (13) and a deletion of the tolC gene, which is required for most drug efflux from E. coli (14). However, no activity was observed against either E. coli strain at a compound concentration of up to 128 μg/ml (data not shown), suggesting either that the thiopyridine compounds cannot enter the cell or that they are pumped out by a means that does not require tolC.
Three compounds, GTC-004061, GTC-330346, and GTC-341772, inhibited B. subtilis strain NO8 with MICs of 64, 8, and 16 μg/ml, respectively. While B. subtilis contains both a FabI and a FabL ENR, the two exhibit sufficient similarity to suggest that a compound could inhibit both (9). None of the compounds inhibited E. faecium. Compound GTC-004061 was tested against E. faecalis, but no inhibition of growth was observed in studies up to a concentration of 64 μg/ml. This is consistent with the known presence of both a FabI and an unrelated FabK ENR in E. faecalis (8).
Cells that underexpress fabI are hypersensitive to compounds in the thiopyridine series.
Since the correlation between the MICs and the IC50s of this set of thiopyridine analogs indicates that these compounds are inhibiting cell growth by means of their activity on the ENR target, we investigated the cellular mode of action (MOA) of these compounds further. Previous studies have established that bacterial strains underproducing and overproducing ENR are more and less susceptible, respectively, to the ENR inhibitor triclosan than is the parental wild-type strain (3, 21). Accordingly, strains of S. aureus that underexpress fabI under the control of a tet-regulated promoter (1) and that overexpress fabI under the control of a T7 promoter were constructed and tested for sensitivity to thiopyridine compounds as well as to positive- and negative-control compounds.
The growth of cells underexpressing fabI is about fourfold more sensitive to the thiopyridine compound GTC-004061 than is the growth of the overexpressing strain or the wild-type parent (Fig. 2A). The growth of the same fabI-underexpressing strain is also about fourfold more sensitive to the known ENR inhibitor triclosan, while the fabI-overexpressing strain is about threefold less sensitive to triclosan, in comparison to growth of the parental strain in the presence of triclosan (Fig. 2B). These results are consistent with previous reports regarding changes in the triclosan susceptibilities of cells under- and overexpressing the fabI gene (3, 21) and indicate that the engineered constructs are functioning as expected. The growth of fabI-overexpressing and -underexpressing cells in the presence of novobiocin, which acts on a different target, was identical in all three strains (Fig. 2C), indicating that altered expression of fabI does not change the sensitivity of cells to antibiotics in general. The growth of fabI-underexpressing cells in the presence of a second thiopyridine analog, GTC-330346, was also reduced compared to growth of the wild-type strain in the presence of the compound; however, fabI-overexpressing cells did not exhibit reduced sensitivity to GTC-330346 (data not shown). These results indicate that compounds in the thiopyridine series inhibit growth of the underexpressing strain through inhibition of the fabI-encoded ENR.
FIG. 2.

Specific hypersensitivity of fabI-underexpressing cells to thiopyridine series compound GTC-004061. The effects on cell growth of thiopyridine series compound GTC-004061 (A), as well as a positive control, triclosan (B), and a negative control, novobiocin (C), were determined using S. aureus cells underexpressing fabI from a tet-regulated promoter (squares) or overexpressing fabI from a T7 promoter (triangles) and compared to the parental wild-type CYL316 cells (diamonds).
Compounds in the thiopyridine series inhibit fatty acid biosynthesis in bacterial cells.
To further characterize the cellular MOA of the thiopyridine series of compounds, we measured the effect of one of these compounds directly on fatty acid biosynthesis in cells. As shown in Fig. 3, GTC-004061 reduced the incorporation of radiolabeled acetate into fatty acids by 45% at 0.5× MIC in 30 min of incubation compared to about 70% reduction in incorporation by triclosan under the same conditions. Inhibition of the incorporation of radiolabeled amino acids into protein and radiolabeled N-acetylglucosamine into peptidoglycan by both GTC-004061 and triclosan was also detectable, but the degree of inhibition was comparable to, or less than, that observed for fatty acid synthesis. While these results are consistent with inhibition of fatty acid biosynthesis as the MOA of GTC-004061 on these cells, the compound also appears to potently inhibit the incorporation of radiolabeled thymidine and uridine into DNA and RNA, respectively. These data suggest that the antibacterial activity of this compound on wild-type S. aureus cells may be due to multiple MOAs, but comparison with the results from a known membrane potential disrupter (SA-411) (L. L. Ling, X. Liang, X. Puyang, A. C. Arvanites, T. Opperman, and D. T. Moir, Abstr. 43rd Intersci. Conf. Antimicrob. Agents Chemother., abstr. F-1469, 2003) indicates that GTC-004061 is not acting through general nonspecific membrane effects on cells. Unlike GTC-004061, the salicylamide SA-411 inhibits all five macromolecular biosynthetic pathways potently even at a concentration of 0.5× MIC (Fig. 3). This potent inhibition of all pathways has been observed previously for a related class of membrane-perturbing compounds, the salicylanilides (12).
FIG. 3.

Effects of GTC-004061 and controls on macromolecular biosynthesis pathways in S. aureus. The percent inhibition of the biosynthesis of fatty acids, protein, peptidoglycan, DNA, and RNA at 30 min by compounds added at concentrations of 0.5× MIC compared to a control with no drug is shown. Values are the averages of two independent measurements. Triclosan, chloramphenicol, phosphomycin, novobiocin, and rifampin are positive controls for inhibition of the synthesis of fatty acids, protein, peptidoglycan, DNA, and RNA, respectively. The salicylamide SA-411 is a membrane-perturbing control compound.
DISCUSSION
Compounds in the thiopyridine chemical series described here exhibit several features which make them attractive for further development as antibacterials: (i) they are potent inhibitors of E. coli ENR, displaying IC50s in the low-micromolar range, (ii) preliminary structure-activity relationship analysis suggests feasible directions for improving potency, (iii) several compounds in this series exhibit MICs for S. aureus, and (iv) the cellular mechanism of action is due in part to inhibition of the fabI gene product. These features, together with the validation of ENR as an essential enzyme and the target of clinically useful antibiotics such as isoniazid, argue for development of this chemical series into antibacterials.
Spectrum.
While these thiopyridine compounds were initially identified in a high-throughput screen for E. coli ENR inhibitors and they do exhibit potent IC50s for the E. coli enzyme, they do not show activity against E. coli cells. Several of the compounds arrest the growth of S. aureus strains and Bacillus subtilis, but they have no detectable effect on the growth of either wild-type E. coli cells or a strain carrying a deletion of the tolC gene along with the asmB1 allele of lpxC, which enhances the permeability of E. coli to some compounds (13). We examined the susceptibility of the tolC deletion strain because TolC is the outer membrane factor for the acrAB efflux pump (5), and tolC mutants show increased sensitivity to a large number of antibacterial compounds (5, 24). However, mutational analysis has revealed that genes encoding two additional putative outer membrane factors, yjcP and yohG, play roles in drug resistance in E. coli (24). Therefore, the thiopyridine compounds described here may not enter the cell, or they may be pumped out by a tolC-independent mechanism. The activity of these compounds on gram-positive species lacking a fabK gene, such as S. aureus and B. subtilis, is consistent with better accessibility in these cells, which lack an outer membrane. While B. subtilis carries both a fabI gene and a fabL gene, the two gene products share considerable sequence identity, and triclosan is known to inhibit both, although with different potencies (8). It is possible that compounds in the thiopyridine series can be engineered to gain activity on E. coli cells by focusing on structural changes that could contribute to outer membrane permeability.
Target specificity.
Inhibition of cell growth by compounds in the thiopyridine series appears to involve inhibition of ENR as well as an additional target(s). In wild-type cells, direct measurement of the incorporation of radiolabeled precursors into macromolecules indicates that GTC-004061 inhibits fatty acid biosynthesis more potently than it inhibits protein or peptidoglycan synthesis. However, the action of GTC-004061 is not specific for fatty acid biosynthesis in wild-type cells, since it also inhibits DNA and RNA synthesis even more effectively. By contrast, in cells underexpressing the fabI gene, the two thiopyridines, GTC-004061 and GTC-330346, as well as triclosan all exhibit enhanced potency, consistent with specific hypersensitivity of these cells to ENR inhibitors such as the thiopyridines and triclosan but not to novobiocin. Based on these data, we propose that the thiopyridine compounds inhibit both ENR and one or more secondary targets in S. aureus and that reduction in the amount of ENR in fabI-underexpressing cells causes ENR inhibition to become the dominant mechanism of inhibition. This is consistent not only with the macromolecular biosynthesis results but also with the fact that cells overexpressing the fabI gene do not exhibit reduced sensitivity to GTC-004061 and GTC-330346. According to this hypothesis, underexpression of a target is a more sensitive method for evaluating target specificity than is overexpression because it permits detection of target-specific activity even in the presence of some degree of secondary target activity. This capability of the underexpression method to decouple dual target activities, together with the fact that it permits the use of less compound and the use of compounds with moderate MICs, makes it a valuable resource for compound evaluation.
While the thiopyridines appear to inhibit more than one target in S. aureus, considerable evidence indicates that the compounds in this series are not acting as membrane-perturbing agents or general biocides. First, we have tested GTC-004061 in a membrane permeability assay using the dye DiSC35 and including a known membrane-perturbing salicylamide compound as a positive control (Ling et al., 43rd ICAAC). While the salicylamide caused significant leakage of the dye from the cells, GTC-004061 did not (data not shown). Second, the same salicylamide compound inhibited all five macromolecular biosynthetic pathways potently, in contrast to the more selective pattern of inhibition exhibited by GTC-004061 (Fig. 3). Third, GTC-004061 does not significantly inhibit any of 10 other enzymes, including other reductases. Fourth, three compounds in the thiopyridine series are active against both S. aureus and B. subtilis, but they are inactive against E. coli and E. faecium. Such selective growth inhibition properties are not typical of general biocides.
Two classes of ENR inhibitors with antibacterial activity have been described recently by other groups. Heerding et al. (11) reported on 1,4-disubstituted imidazoles with IC50s for E. coli ENR of about 4 μM and MICs for S. aureus of 8 μg/ml. The thiopyridine series described here exhibits very similar potency. Seefeld et al. (23) and Payne et al. (21) reported another chemical series of compounds which exhibit IC50s for E. coli ENR of about 4 μM and MICs for S. aureus of 0.5 μg/ml. These compounds are similar in potency to the thiopyridines against the E. coli enzyme but display slightly higher MICs for S. aureus. In total, this group of inhibitors from three quite different chemotypes, two from previous efforts and the new chemotype reported here, suggests that ENR is a feasible target and that antibacterials directed against this target can be developed.
Acknowledgments
We thank Xiaoling Puyang and Shubha Narayan for excellent technical support. We acknowledge Julia Pinto for helpful discussions and assistance with thiopyridine chemistry. We thank Bruce Rogers for interest in the project and a critical reading of the manuscript.
This work was supported by Genome Therapeutics Corp.
REFERENCES
- 1.Bateman, B. T., N. P. Donegan, T. M. Jarry, M. Palma, and A. L. Cheung. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect. Immun. 69:7851-7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bramucci, M. G., and V. Nagarajan. 1996. Direct selection of cloned DNA in Bacillus subtilis based on sucrose-induced lethality. Appl. Environ. Microbiol. 62:3948-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeVito, J. A., J. A. Mills, V. G. Liu, A. Agarwal, C. F. Sizemore, Z. Yao, D. M. Stoughton, M. G. Cappiello, M. D. Barbosa, L. A. Foster, and D. L. Pompliano. 2002. An array of target-specific screening strains for antibacterial discovery. Nat. Biotechnol. 20:478-483. [DOI] [PubMed] [Google Scholar]
- 4.Fan, F., R. D. Lunsford, D. Sylvester, J. Fan, H. Celesnik, S. Iordanescu, M. Rosenberg, and D. McDevitt. 2001. Regulated ectopic expression and allelic-replacement mutagenesis as a method for gene essentiality testing in Staphylococcus aureus. Plasmid 46:71-75. [DOI] [PubMed] [Google Scholar]
- 5.Fralick, J. A. 1996. Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. J. Bacteriol. 178:5803-5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geissendorfer, M., and W. Hillen. 1990. Regulated expression of heterologous genes in Bacillus subtilis using the Tn10-encoded tet regulatory elements. Appl. Microbiol. Biotechnol. 33:657-663. [DOI] [PubMed] [Google Scholar]
- 7.Heath, R. J., and C. O. Rock. 2000. A triclosan-resistant bacterial enzyme. Nature 406:145-146. [DOI] [PubMed] [Google Scholar]
- 8.Heath, R. J., N. Su, C. K. Murphy, and C. O. Rock. 2000. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 275:40128-40133. [DOI] [PubMed] [Google Scholar]
- 9.Heath, R. J., S. W. White, and C. O. Rock. 2002. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 58:695-703. [DOI] [PubMed] [Google Scholar]
- 10.Heath, R. J., S. W. White, and C. O. Rock. 2001. Lipid biosynthesis as a target for antibacterial agents. Prog. Lipid Res. 40:467-497. [DOI] [PubMed] [Google Scholar]
- 11.Heerding, D. A., G. Chan, W. E. DeWolf, A. P. Fosberry, C. A. Janson, D. D. Jaworski, E. McManus, W. H. Miller, T. D. Moore, D. J. Payne, X. Qiu, S. F. Rittenhouse, C. Slater-Radosti, W. Smith, D. T. Takata, K. S. Vaidya, C. C. Yuan, and W. F. Huffman. 2001. 1,4-disubstituted imidazoles are potential antibacterial agents functioning as inhibitors of enoyl acyl carrier protein reductase (FabI). Bioorg. Med. Chem. Lett. 11:2061-2065. [DOI] [PubMed] [Google Scholar]
- 12.Hilliard, J. J., R. M. Goldschmidt, L. Licata, E. Z. Baum, and K. Bush. 1999. Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob. Agents Chemother. 43:1693-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kloser, A., M. Laird, M. Deng, and R. Misra. 1998. Modulations in lipid A and phospholipid biosynthesis pathways influence outer membrane protein assembly in Escherichia coli K-12. Mol. Microbiol. 27:1003-1008. [DOI] [PubMed] [Google Scholar]
- 14.Koronakis, V., A. Sharff, E. Koronakis, B. Luisi, and C. Hughes. 2000. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405:914-919. [DOI] [PubMed] [Google Scholar]
- 15.Krauze, A., Z. Bomika, A. M. Shestopalov, L. A. Rodinovskaya, J. Pelcers, Y. A. Sharanin, and V. K. Promonenkov. 1981. Synthesis and some reactions of 3-cyanopyridine-2-thiones. Khimiya Geterotsiklicheskikh Soedinenii 3:377-382. [Google Scholar]
- 16.Kuroda, M., T. Ohta, I. Uchiyama, T. Baba, H. Yuzawa, I. Kobayashi, L. Cui, A. Oguchi, K. Aoki, Y. Nagai, J. Lian, T. Ito, M. Kanamori, H. Matsumaru, A. Maruyama, H. Murakami, A. Hosoyama, Y. Mizutani-Ui, N. K. Takahashi, T. Sawano, R. Inoue, C. Kaito, K. Sekimizu, H. Hirakawa, S. Kuhara, S. Goto, J. Yabuzaki, M. Kanehisa, A. Yamashita, K. Oshima, K. Furuya, C. Yoshino, T. Shiba, M. Hattori, N. Ogasawara, H. Hayashi, and K. Hiramatsu. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225-1240. [DOI] [PubMed] [Google Scholar]
- 17.Lee, C. Y., S. L. Buranen, and Z. H. Ye. 1991. Construction of single-copy integration vectors for Staphylococcus aureus. Gene 103:101-105. [DOI] [PubMed] [Google Scholar]
- 18.Levy, C. W., A. Roujeinikova, S. Sedelnikova, P. J. Baker, A. R. Stuitje, A. R. Slabas, D. W. Rice, and J. B. Rafferty. 1999. Molecular basis of triclosan activity. Nature 398:383-384. [DOI] [PubMed] [Google Scholar]
- 19.Miller, W. H., M. A. Seefeld, K. A. Newlander, I. N. Uzinskas, W. J. Burgess, D. A. Heerding, C. C. Yuan, M. S. Head, D. J. Payne, S. F. Rittenhouse, T. D. Moore, S. C. Pearson, V. Berry, W. E. DeWolf, Jr., P. M. Keller, B. J. Polizzi, X. Qiu, C. A. Janson, and W. F. Huffman. 2002. Discovery of aminopyridine-based inhibitors of bacterial enoyl-ACP reductase (FabI). J. Med. Chem. 45:3246-3256. [DOI] [PubMed] [Google Scholar]
- 20.NCCLS. 1997. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 4th ed. Approved standard M7-A4. NCCLS, Wayne, Pa.
- 21.Payne, D. J., W. H. Miller, V. Berry, J. Brosky, W. J. Burgess, E. Chen, W. E. DeWolf, Jr., A. P. Fosberry, R. Greenwood, M. S. Head, D. A. Heerding, C. A. Janson, D. D. Jaworski, P. M. Keller, P. J. Manley, T. D. Moore, K. A. Newlander, S. Pearson, B. J. Polizzi, X. Qiu, S. F. Rittenhouse, C. Slater-Radosti, K. L. Salyers, M. A. Seefeld, M. G. Smyth, D. T. Takata, I. N. Uzinskas, K. Vaidya, N. G. Wallis, S. B. Winram, C. C. Yuan, and W. F. Huffman. 2002. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob. Agents Chemother. 46:3118-3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quemard, A., J. C. Sacchettini, A. Dessen, C. Vilcheze, R. Bittman, W. R. Jacobs, Jr., and J. S. Blanchard. 1995. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 34:8235-8241. [DOI] [PubMed] [Google Scholar]
- 23.Seefeld, M. A., W. H. Miller, K. A. Newlander, W. J. Burgess, D. J. Payne, S. F. Rittenhouse, T. D. Moore, W. E. DeWolf, Jr., P. M. Keller, X. Qiu, C. A. Janson, K. Vaidya, A. P. Fosberry, M. G. Smyth, D. D. Jaworski, C. Slater-Radosti, and W. F. Huffman. 2001. Inhibitors of bacterial enoyl acyl carrier protein reductase (FabI): 2,9-disubstituted 1,2,3,4-tetrahydropyrido[3,4-β]indoles as potential antibacterial agents. Bioorg. Med. Chem. Lett. 11:2241-2244. [DOI] [PubMed] [Google Scholar]
- 24.Sulavik, M. C., C. Houseweart, C. Cramer, N. Jiwani, N. Murgolo, J. Greene, B. DiDomenico, K. J. Shaw, G. H. Miller, R. Hare, and G. Shimer. 2001. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob. Agents Chemother. 45:1126-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yansura, D. G., and D. J. Henner. 1984. Use of the Escherichia coli lac repressor and operator to control gene expression in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 81:439-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang, J. H., T. D. Chung, and K. R. Oldenburg. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67-73. [DOI] [PubMed] [Google Scholar]