

Abstract
Innate immune sensors of foreign nucleic acids are essential for antiviral immunity, but these same sensors can cause autoimmune disease through inappropriate detection of self nucleic acids. The sources of the endogenous RNA and DNA that trigger autoreactive responses include chromatin and ribonucleoproteins that are the targets of autoantibodies in numerous autoimmune diseases, including systemic lupus erythematosus (SLE). In this review, I discuss recent data implicating endogenous retroelements – viruses that make up a substantial fraction of our genomes – as an important source of endogenous nucleic acids that can cause autoimmune disease. Understanding this potentially pathologic role for retroelements and the precise mechanisms by which their genomes are sensed and metabolized has important implications for the diagnosis and treatment of numerous autoimmune disorders.
INTRODUCTION
After over a decade of remarkable progress in our understanding of innate immunity, it is now clear that the primary mechanism for detection of viral infection is the sensing of nucleic acids by a variety of innate immune receptors [1]. Viral genomes are composed of either RNA or DNA, and the numerous sensors that detect each of these nucleic acids can be broadly categorized into two categories. First, several Toll-like receptors are expressed largely by hematopoietic cells and couple nucleic acid detection to a number of essential immune functions: inducible cytokine responses, antigen presentation to lymphocytes, and enhancement of virus-specific antibody responses. Second, intracellular receptors for RNA and DNA are broadly expressed by all nucleated cells and detect the presence of foreign nucleic acids within the infected cell itself. These cell-intrinsic sensors activate a type I interferon (IFN) response, which alerts neighboring cells to the presence of infection and drives the induction of hundreds of antiviral genes that serve to prevent viral replication and spread. The two classes of nucleic acid receptors communicate with each other to coordinate the protective antiviral response, although the nature of this crosstalk is only beginning to be understood. In the case of viral infection of non-hematopoietic cells, the cell-intrinsic sensors are the first receptors to be triggered. These activate the IFN response, as well as other signals that recruit professional antigen presenting like dendritic cells and monocytes to the site of infection. The recruited cells phagocytose local dead cells and debris, and use TLRs to sample this cargo for foreign nucleic acids. The activated APCs then migrate to the draining lymphoid organs, where they present antigens from infected cells to T cells and B cells. Finally, B cells use nucleic acid-sensing TLRs to synergize with their antigen receptors to drive robust antibody responses to particles containing both foreign antigen and nucleic acids – the hallmarks of viral particles [2]. In parallel, the IFN response in the context of foreign antigens augments the differentiation of effector and memory T cells [3].
The events described above illustrate the stepwise progression of productive antiviral responses, with each step under the control of innate immune nucleic acid receptors. This allows for the continued “quality control” of the adaptive immune response such that the most robust and long-lived antiviral responses require both foreign antigens and foreign nucleic acids. However, it is now clear that endogenous (non-infectious) nucleic acids can feed into this coordinated response. Chronic detection of these nucleic acids can overcome lymphocyte tolerance and drive T and B cell responses to abundant self antigens, particularly those like chromatin and ribonucleoproteins that contain T cell epitopes, B cell epitopes, and TLR ligands. Indeed, autoantibody responses to these nucleic acid-protein complexes are diagnostic for and underlie the pathology of a number of IFN-associated autoimmune diseases, including SLE and Sjogrens syndrome.
The contribution of TLRs to SLE and related autoimmune disorders was first recognized a decade ago [4], and this realization has led to the development of new classes of therapeutics that aim to eliminate the early events of disease through inhibition of TLRs or type I IFNs. However, if we draw parallels to the coordinated model summarized above, we can envision a scenario in which accumulation of endogenous nucleic acids within cells could trigger an inappropriate cell-intrinsic antiviral response that would lead to autoimmunity by precisely the same stepwise program that protects us from infection. Indeed, the recent confluence of numerous independent lines of inquiry - the genetics of rare human diseases, basic innate immunity research, and study of the interaction between host cells and human immunodeficiency virus (HIV) - has revealed a remarkable example of cell-intrinsic initiation of autoimmunity. In this case, the nucleic acids are of viral origin, but they are derived from the endogenous retroelements that comprise over a third of the nucleotide content of the human genome.
RETROELEMENTS AND AUTOIMMUNITY
Endogenous retroelements are so-called because they replicate through a “copy and paste” mechanism that involves reverse-transcription of a viral RNA transcript into a double-stranded DNA molecule that can insert into a unique genomic site. Retroelements come in three flavors, two of which are autonomous in that an individual, functional element encodes all of the activities required for its own retrotransposition. First, the long terminal repeat (LTR) retrotransposons are derived from ancient, infectious retroviruses that infected the germline and then lost the ability to spread from cell to cell, although the mechanics of reverse transcription and integration resemble those of extant infectious retroviruses like murine leukemia virus (MLV) and HIV. Reverse transcription of LTR retroviral RNA is primed by the 3′ end of a cellular tRNA that binds to an 18 nucleotide complementary sequence in the viral RNA genome called the primer binding site. Second, the long interspersed nuclear elements (LINEs) are non-LTR viruses ~6 kilobases in length that are as ancient as the oldest multicellular organisms. LINEs encode two proteins that coordinate their retrotransposition through a process called target-primed reverse transcription (TPRT), which utilizes a nick in genomic DNA to synthesize a strand of DNA complementary to a LINE RNA molecule. Thus, unlike the LTR viruses, LINE reverse transcription occurs at the precise genomic site of intended retrotransposition. Third, the non-autonomous retrotransposons, including short interspersed nuclear elements (SINEs), are quintessential “selfish genes” that do not encode any proteins of their own and harness the LINE reverse transcriptase to mediate retrotransposition in trans. An excellent recent review describes in detail the origins and biology of these retroelements, as well as the numerous ways in which they have sculpted our genomes [5]. Retrotransposons have been extremely successful throughout evolution, and the human genome contains millions of these elements. Although most of them are inactive owing to the acquisition of deletions and mutations over evolutionary time, dozens of LTR retroelements and ~150 LINEs are intact and potentially competent for retrotransposition. Consequently, the retroelement RT enzymes are among the most abundant protein coding genes, and their activities can give rise to nucleic acid species that are detected by the same receptors that sense infectious viruses, as discussed below.
Biologists have explored whether and how retroelements contribute to autoimmune disease for over two decades. Much of the historical focus on this connection has revolved around their capability to produce neo-antigens to drive autoreactive lymphocyte responses [6,7]. For example, activation of an endogenous retrovirus would result in the production of new proteins. Because of their abrupt appearance in a tissue, these proteins could bypass or override tolerance mechanisms that delete lymphocytes specific for constitutive self antigens. This antigen-based mechanism has been proposed in the context of multiple sclerosis [8], SLE [9], Sjogrens syndrome [10], and other autoimmune disorders in which specific retroviruses may give rise to the targeted autoantigens. Although the proteins of retroelements could in principle serve as antigens, there is no clear evidence for an autoimmune disease caused by autoreactive lymphocyte responses to endogenous retroviral antigens.
More recently, an alternative model has emerged that reveals the reverse-transcription products of retroelements – not the proteins encoded by them – as potentially important triggers of certain autoimmune diseases. This model has its roots in the 2006 discovery of a cell-intrinsic antiviral response triggered by detection of intracellular DNA [11,12]. This pathway, termed the interferon stimulatory DNA (ISD) pathway, is analogous to that activated by the RNA sensors RIG-I and MDA5, and even shares some common signaling components with the RNA-activated antiviral response, including tank-binding kinase 1 (TBK1) and the interferon regulatory factor 3 (IRF3) transcription factor. The sensor(s) of the ISD pathway remain enigmatic, but the recent identification of an ISD-specific signaling molecule called STING (stimulator of IFN genes; [13,14]) has allowed a genetic dissection of the role of the ISD pathway in host defense and autoimmunity. Importantly, the existence of the ISD pathway considerably broadens the types of pathogens that could be detected within infected cells, including DNA viruses [15] and intracellular bacteria [11,16]. In the context of retroviruses and retroelements, this is particularly relevant since their capped, polyadenylated genomic RNAs are essentially indistinguishable from cellular transcripts and are not thought to trigger the RIG-I or MDA5 pathways that detect 5′ triphosphate RNAs or double-stranded RNA structures, respectively. However, the production of a RNA-DNA hybrid and ultimately a double-stranded DNA genome through reverse transcription offers an opportunity for the host cell to distinguish retroviral replication intermediates from its own nucleic acids.
We developed a biochemical screen to identify key components of the ISD pathway, and we found an enzyme called 3′ repair exonuclease 1 (Trex1; [17]), which was first described over 40 years ago as the most abundant 3′ –> 5′ DNA exonuclease in cells [18]. Microarray data of the ISD pathway revealed that Trex1 is an IFN-inducible gene, which immediately suggested an important role in antiviral defense. Perhaps most importantly, Yanick Crow and his colleagues identified loss of function TREX1 mutations in a rare, type I IFN-associated human autoimmune disease called Aicardi-Goutieres Syndrome (AGS; [19]). Interestingly, Trex1 mutations also cause familial chilblain lupus and are strongly associated with some SLE cases [20–22]. Using Trex1-deficient mice as a model of AGS and related diseases, we found that Trex1 is an essential negative regulator of the STING-dependent ISD pathway [17,23]. Thus, Trex1 serves to metabolize the DNA ligands that trigger the ISD pathway, and in the absence of functional Trex1 these ligands accumulate, triggering cell-intrinsic initiation of IFN-dependent autoimmunity. We developed a method to extract and identify this accumulated DNA, and we found that DNA derived from endogenous retroelements was much more abundant in Trex1-deficient cells compared to control cells [17]. Moreover, Trex1 potently blocked retrotransposition of model retroelements by metabolizing their reverse-transcribed DNA [17]. Based on the these findings and the emerging evidence linking Trex1 mutations to specific human diseases, we predicted that the other genes mutated in AGS would be anti-retroviral enzymes, that the STING-dependent ISD pathway would play a role in anti-retroviral defense, and that retroelements would be the underlying cause of certain autoimmune diseases [17]. As discussed below, independent evidence is accumulating to support this retroelement hypothesis.
AGS GENES ARE ANTI-RETROVIRAL ENZYMES
In line with the anti-retroviral role of Trex1 described in the context of retroelements [17], Lieberman and colleagues found that Trex1 also metabolizes the reverse-transcribed cDNA of HIV [24]. In the absence of Trex1, HIV cDNA accumulates within infected cells, and this triggers a STING-dependent innate immune response [24]. Thus, Trex1 sets a threshold for the innate immune response to retroviral infection. Interestingly, HIV encodes its own antagonists of antiviral immunity that target IRF3 for degradation [25,26]. Together, the evasion of host defense by HIV and the metabolism of its reverse transcription intermediates by Trex1 may explain why the cell-intrinsic innate immune response to HIV infection was only recently characterized [27–30].
In addition to Trex1, there are several other human genes that are mutated in AGS patients. Crow and colleagues identified loss of function alleles in an IFN-inducible gene called SAMHD1 as the cause of ~15% of AGS cases [31]. At the time, very little was known about this gene, but shortly after the identification of the AGS mutations, two groups reported that SAMHD1 is an essential HIV restriction factor in human myeloid cells [32,33]. Interestingly, the lentiviral accessory factor Vpx, which is encoded by some primate lentiviruses, targets SAMHD1 for proteasomal degradation, thus relieving this restriction. Similarly, knockdown of SAMHD1 in human myeloid cells renders them highly susceptible to HIV infection. SAMHD1 is a deoxnucleoside triphosphohydrolase that removes the phosphates from dNTPs, thereby depleting the pool of available nucleotides that can be used for reverse transcription [34,35]. SAMHD1 therefore prevents the formation of the immunostimulatory nucleic acid species that are likely degraded by Trex1 (Figure 1). As dNTPs are also essential for genomic DNA replication, it will be interesting to determine how SAMHD1 is specifically targeted to sites of reverse transcription as opposed to sites of scheduled genomic DNA replication.
Figure 1.
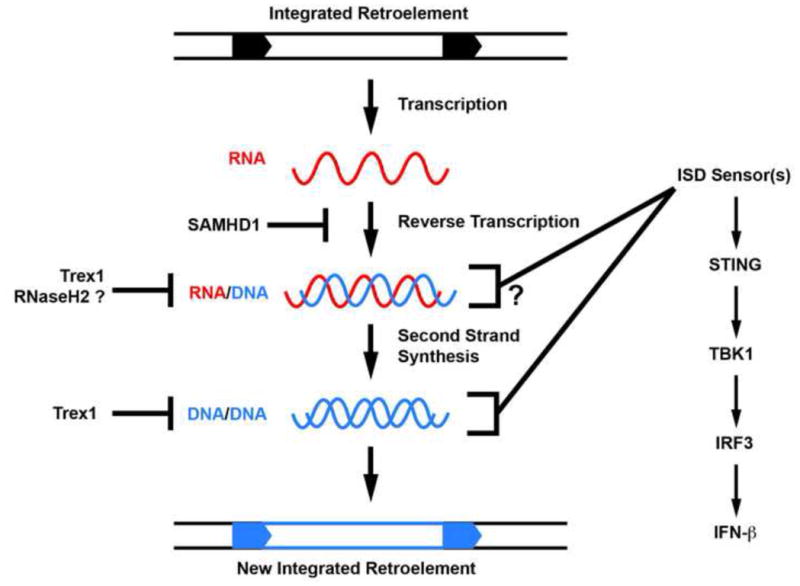
AGS genes cooperate to metabolize reverse transcription products of endogenous retroelements. A schematic of the retroviral reverse transcription process is shown. SAMHD1, an AGS gene and HIV-1 restriction factor, prevents the initiation of reverse transcription by controlling dNTP availability to RT enzymes. A speculative role for RNaseH2 in degrading the RNA strand of RNA/DNA hybrids is depicted. Trex1 can block retrotransposition by metabolizing the reverse-transcribed DNA of retroelements. Failure to metabolize these viral nucleic acids may trigger the ISD pathway through accumulation of RNA/DNA hybrids or reverse-transcribed DNA.
At the same time that the human TREX1 mutations were first reported, Crow, Jackson, and colleagues described AGS-causing mutations in each of the three subunits of the RNaseH2 enzyme [36]. RNaseH2 is an endonuclease that cleaves the phosphodiester backbone of RNA in the context of RNA-DNA hybrids, thus leading to the proposition that RNaseH2 may contribute to metabolizing the obligate intermediates of reverse transcription (Figure 1). However, a recent characterization of RNAseH2-deficient mice revealed an unexpected and essential role for RNAseH2 in the removal of misincorporated ribonucleotides during genomic DNA replication [37,38]. These single ribonucleotides are introduced into elongating DNA by replicative DNA polymerases at a rate of around one ribonucleotide per 7,000 bases. Because the phosphodiester bonds of RNA are ~100,000-fold more sensitive to spontaneous hydrolysis than those of DNA, failure to remove these misincorporated ribonucleotides leads to extensive DNA damage, resulting in early embryonic lethality in the knockout mice. Interestingly, this early lethality can be significantly delayed by crossing RNaseH2B-deficient mice to p53 knockout mice, and RNaseH2B/p53 double knockout fibroblasts grow in culture, unlike plain RNaseH2B-null fibroblasts [37]. This important study reveals a key role for RNaseH2 in maintaining genome integrity, and suggests that misincorporated ribonucleotides are the most common genomic lesion in mammalian cells.
It is interesting to compare the clear phenotype of RNaseH2 deficiency in mice to that of AGS in humans. AGS patients suffer from autoimmune disease, and they have no evidence for the cancer susceptibility and “accelerated aging” that characterize other diseases caused by excessive DNA damage or inadequate DNA repair [39,40]. Importantly, and unlike the AGS alleles of Trex1 and SAMHD1 [19,34,35,41], the AGS-causing mutations in RNaseH2 do not grossly impair enzymatic activity [42]. Thus, an important question arises: do the AGS mutations cause a more mild insufficiency in RNaseH2 activity for removal of misincorporated ribonucleotides that allows for normal development but causes autoimmunity? Or do the AGS mutations impact a different (perhaps anti-retroviral) function of RNaseH2 that cannot be revealed in knockout mice because of the severity of the null allele?
Together, the identification of several human AGS genes unites these enzymes in a potentially common antiviral pathway (Figure 1; [19,31,36]). The clearly defined anti-retroviral functions of two of these genes [17,24,32,33] lend support to the retroelement hypothesis and suggest interesting new avenues for designing better therapies for AGS, which is currently untreatable and incurable.
IMPLICATIONS FOR TISSUE-SPECIFIC AUTOIMMUNITY
If retroelements are involved in AGS and related diseases, then reverse transcriptase inhibitors may hold promise as a potential therapy to ameliorate disease. Indeed, Wabl and colleagues have found that treatment of Trex1-deficient mice from conception with a cocktail of three FDA-approved anti-retroviral drugs dramatically reduces mortality and ameliorates tissue inflammation [43]. This fascinating study immediately suggests that similar drugs may be of therapeutic value in humans, and it will be important to identify which drugs are mediating the effect and whether there are other existing RT inhibitors that work as well or even better. More broadly, it will be important to understand which RT enzyme(s) are associated with disease, and whether these RT activities are expressed in a tissue-specific manner that coincides with the affected organs in humans and mice [23,39]. There are three reverse transcriptase activities encoded in mammalian genomes: telomerase, the LTR retroviral RT enzymes, and the LINE RT enzymes. Telomerase is unlikely to be involved because of its highly restricted substrate specificity and its activity in all dividing cells. The LTR retroviral RT is a potential candidate, but the requirement for a tRNA primer and its cognate binding site in the retroviral RNA genome also severely limits the scope of RNAs that can be reverse transcribed by this enzyme. The LINE RT makes the most logical sense because it is capable of reverse-transcribing any polyadenylated RNA, including the LINE genomic RNAs, the SINE genomic RNAs, and cellular mRNAs that are destined to become processed pseudogenes [5]. Modern genomics approaches will enable the more definitive exploration of these questions, through identification of intact RT enzymes and retroelements in the genome and then determination of which of these enzymes are expressed in a given tissue. Understanding the expression dynamics of the retroelement sequences in our own genome will provide important insights into basic mechanisms of disease and will refine approaches to block retroelement activity to ameliorate autoimmunity.
CONCLUSIONS
The potential involvement of endogenous retroelements in autoimmune disease has recently gained support from a confluence of independent lines of investigation, suggesting that an approach integrating retroelement biology, innate immunity, and host defense against retroviruses may yield important new insights into the origins and pathological progression of certain autoimmune diseases. In the future, if the relevant RT activities can be unequivocally identified and inhibited in a safe and complete manner, the drugs to treat AGS and related diseases may already exist.
Highlights.
Endogenous retroelements are a source of viral nucleic acids within our own genomes
Aicardi-Goutieres Syndrome genes encode anti-retroviral enzymes
Failure to metabolize retroelement cDNAs can drive IFN-mediated autoimmunity
Acknowledgments
I apologize to colleagues whose work I was unable to cite because of space considerations. I am grateful to all the members of my lab for discussions, and to Hannah Volkman for help with the figure. We are supported by the National Institute of Allergy and Infectious Disease (AI084914, 5U54AI057141-08), the European Union (FP7/2007-2013) grant agreement number 241779 (NIMBL: http://www.NIMBL.eu), the Lupus Research Institute, and the Rita Allen Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 2.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 3.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 5.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 6.Colmegna I, Garry RF. Role of endogenous retroviruses in autoimmune diseases. Infect Dis Clin North Am. 2006;20:913–929. doi: 10.1016/j.idc.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Balada E, Ordi-Ros J, Vilardell-Tarres M. Molecular mechanisms mediated by human endogenous retroviruses (HERVs) in autoimmunity. Rev Med Virol. 2009;19:273–286. doi: 10.1002/rmv.622. [DOI] [PubMed] [Google Scholar]
- 8.Perron H, Germi R, Bernard C, Garcia-Montojo M, Deluen C, Farinelli L, Faucard R, Veas F, Stefas I, Fabriek BO, et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult Scler. 2012 doi: 10.1177/1352458512441381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perl A, Nagy G, Koncz A, Gergely P, Fernandez D, Doherty E, Telarico T, Bonilla E, Phillips PE. Molecular mimicry and immunomodulation by the HRES-1 endogenous retrovirus in SLE. Autoimmunity. 2008;41:287–297. doi: 10.1080/08916930802024764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sander DM, Szabo S, Gallaher WR, Deas JE, Thompson JJ, Cao Y, Luo-Zhang H, Liu LG, Colmegna I, Koehler J, et al. Involvement of human intracisternal A-type retroviral particles in autoimmunity. Microsc Res Tech. 2005;68:222–234. doi: 10.1002/jemt.20234. [DOI] [PubMed] [Google Scholar]
- 11.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- *13.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. See annotation to ref. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *14.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. Refs 13 and 14 identified STING/MITA, which is the first essential and unique signaling component of the ISD pathway. [DOI] [PubMed] [Google Scholar]
- 15.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe. 2012;11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *17.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. This study established the first links between Trex1, retroelement cDNA metabolism, the ISD pathway, and AGS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindahl T, Gally JA, Edelman GM. Properties of deoxyribonuclease 3 from mammalian tissues. J Biol Chem. 1969;244:5014–5019. [PubMed] [Google Scholar]
- *19.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. See annotation for Ref 36. [DOI] [PubMed] [Google Scholar]
- 20.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 21.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcon-Riquelme ME, Gallant CJ, Boackle SA, Criswell LA, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12:270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23.Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, Stetson DB. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. This study demonstrated an essential role for the STING-dependent ISD pathway in a mouse model of AGS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *24.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. This study found that Trex1 metabolizes HIV-1 cDNA and sets a threshold for activation of STING-dependent antiviral responses to HIV infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. 2008;373:85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doehle BP, Chang K, Rustagi A, McNevin J, McElrath MJ, Gale M., Jr Vpu Mediates Depletion of Interferon Regulatory Factor 3 during HIV Infection by a Lysosome-Dependent Mechanism. J Virol. 2012;86:8367–8374. doi: 10.1128/JVI.00423-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M., Jr Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol. 2009;83:10395–10405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, Greene WC. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. 2010;143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manel N, Littman DR. Hiding in plain sight: how HIV evades innate immune responses. Cell. 2011;147:271–274. doi: 10.1016/j.cell.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–832. doi: 10.1038/ng.373. See annotation to Ref 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **32.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. See annotation to Ref 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **33.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. Refs 32 and 33 demonstrate that SAMHD1, an enzyme that is mutated in AGS, is a key restriction factor for HIV-1 in myeloid cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powell RD, Holland PJ, Hollis T, Perrino FW. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011;286:43596–43600. doi: 10.1074/jbc.C111.317628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- *36.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916. doi: 10.1038/ng1842. Refs 19, 31, and 36 describe five genes that are mutated in AGS. These important studies provide the framework for understanding the role of nucleic acid metabolism in preventing autoimmunity, as well as the basis for exploring the interrelationship of AGS genes. [DOI] [PubMed] [Google Scholar]
- **37.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–1022. doi: 10.1016/j.cell.2012.04.011. This paper, together with Ref. 38*, identifies an essential role for RNaseH2 in removing misincorporated ribonucleotides from genomic DNA, and raises important questions about how this process may relate to AGS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *38.Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med. 2012;209:1419–1426. doi: 10.1084/jem.20120876. See annotation to Ref 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thoms KM, Kuschal C, Emmert S. Lessons learned from DNA repair defective syndromes. Exp Dermatol. 2007;16:532–544. doi: 10.1111/j.1600-0625.2007.00559.x. [DOI] [PubMed] [Google Scholar]
- 41.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J Biol Chem. 2008;283:31649–31656. doi: 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perrino FW, Harvey S, Shaban NM, Hollis T. RNaseH2 mutants that cause Aicardi-Goutieres syndrome are active nucleases. J Mol Med. 2009;87:25–30. doi: 10.1007/s00109-008-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **43.Beck-Engeser GB, Eilat D, Wabl M. An autoimmune disease prevented by anti-retroviral drugs. Retrovirology. 2011;8:91. doi: 10.1186/1742-4690-8-91. In this study, a cocktail of anti-retroviral drugs used to treat HIV-1 infection dramatically rescued Trex1-deficient mice from mortality and substantially ameliorated autoimmune pathology, providing the first demonstration of a potential treatment for AGS and related diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]