

Abstract
The utility of the tethered aminohydroxylation (TA) has been demonstrated by synthesis of the complex β-amino acid residue of microsclerodermins A and B. The TA provided a regio- and stereoselective functionalization of a complex homoallylic alcohol. The route includes late-stage introduction of the aliphatic side chain via a cuprate addition and cross metathesis, a tactic designed to render the synthesis applicable to other microsclerodermins.
The widespread occurrence of vicinal amino alcohols means that efficient and reliable methods for their preparation are important. A powerful approach lies in the direct oxidation of an olefin by introduction of oxygen and nitrogen groups via an aminohydroxylation.1,2 However, one drawback to alkene aminohydroxylation is the unpredictable regiochemical outcome; this can be highly dependent on the nature of the alkene substrate.2 We recognized that tethering the nitrogen source to an allylic alcohol would ensure complete control of regioselectivity in a subsequent osmium catalyzed aminohydroxylation, a tethered aminohydroxylation (TA). Since our early reports,3 we have disclosed significant improvements to the process through discovery of new nitrogen reoxidants for TA, using N-sulfonyloxycarbamates4 and N-aroyloxycarbamates.5,6 The use of such N–O based oxidants avoids the use of t-BuOCl (a common reoxidant in aminohydroxylation) and prevents any unwanted alkene chlorination. Thus, the TA can be used for the regio- and stereoselective aminohydroxylation of substituted alkene substrates in high yields and with low catalyst loadings.
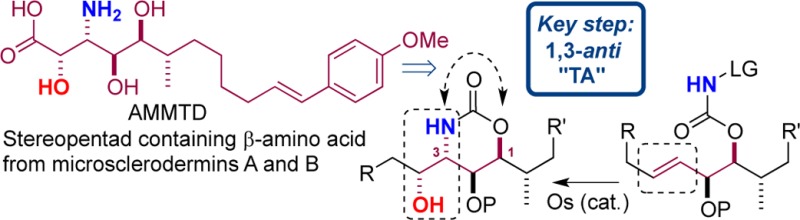
In order to examine the applicability of the TA reaction in the synthesis of complex targets,7 we selected a β-amino acid from the microsclerodermin family, namely a polyhydroxylated amino acid containing five contiguous stereocenters, named AMMTD (Figure 1).
Figure 1.
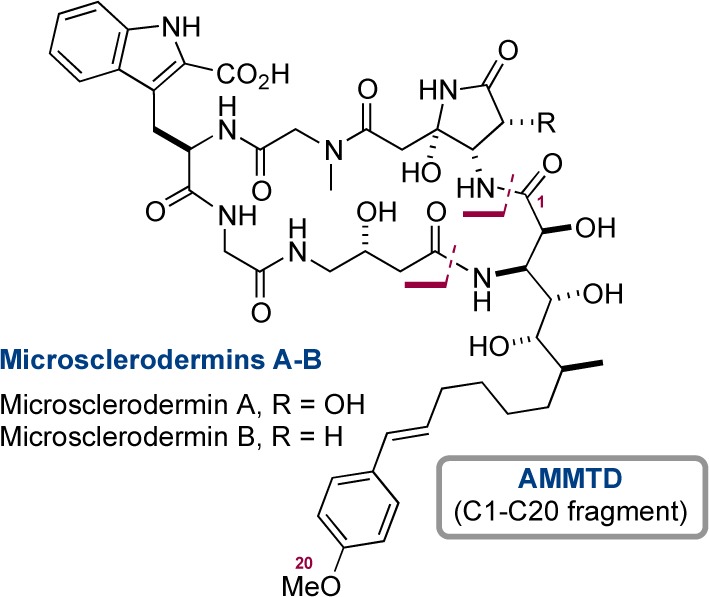
Microsclerodermins A and B.
Microsclerodermins (A–K) are a family of cyclic hexapeptides isolated from the lithistid marine sponges Microscleroderma sp., Theonella sp., and Microscleroderma herdmani,8,9 which demonstrate antifungal activity and cytotoxicity against an HCT-116 cell line. The most complex residue is a β-amino acid with either four or five contiguous stereocenters and variation in the degree of unsaturation in the side-chain tail. Early studies on the microsclerodermins by Shioiri10 were followed by Ma who reported the only total synthesis of any microsclerodermin (E, containing a β-amino acid with four contiguous stereocenters).11 Shioiri12 and Chandrasekhar13 reported the synthesis of protected variants of the β-amino acid in microsclerodermins A and B (AMMTD), while the simpler β-amino acid of microsclerodermins C and D (APTO) and E (AETD) have received the most attention.14
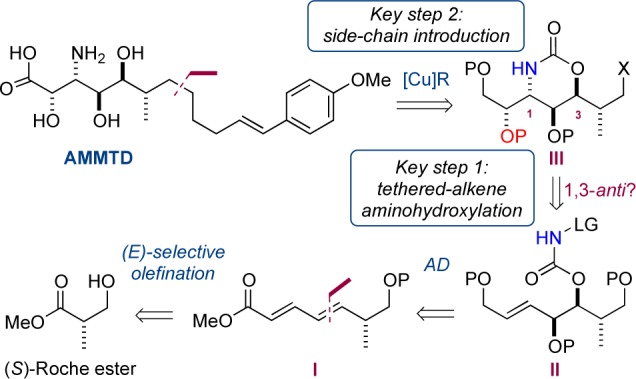
It was essential that our approach to AMMTD (Scheme 1) produced the target in a form that would permit conjugation to other amino acids, ultimately allowing a total synthesis of the microsclereodermins. Another fundamental part of our design was late-stage introduction of the aliphatic tail, to allow access to multiple members of the family. We envisaged side-chain installation via organocuprate addition to functionalized electrophile III. The key aminohydroxyl unit at C2,C3 could be installed via a regio- and stereoselective aminohydroxylation of an olefin via TA reaction of II. Precursor II could arise from manipulation of a diol, generated by dihydroxylation (AD) of α,β,γ,δ-unsaturated ester I. In turn diene I should be readily prepared as a single enantiomer from (S)-Roche ester.
Scheme 1. Retrosynthetic Analysis of AMMTD.
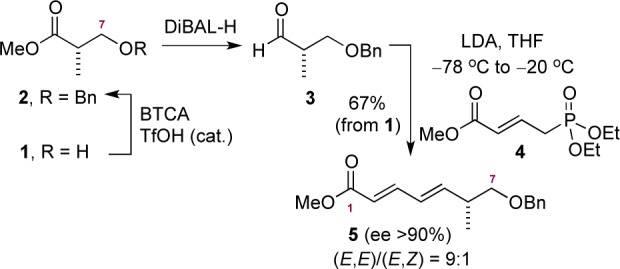
The first step involved formation of benzyl ether 2 by reaction of 1 with benzyl 2,2,2-trichloroacetimidate, followed by reduction to aldehyde 3 (Scheme 2).15 The crude aldehyde was transfomed into (E,E)-diene 5 through (E)-selective Horner–Wadsworth–Emmons reaction with phosphonate 4. We found that the desired olefination proceeded efficiently utilizing 2 equiv of phosphonate and LDA at −20 °C. This allowed for the formation of diene 5 in good yield (67%, three steps) as an (inseparable) (E,E)/(E,Z) 9:1 mixture and as a single enantiomer (ee >90%, by Mosher’s ester analysis).
Scheme 2. Construction of Diene 5.
Installation of the C4,C5 diol proceeded via regio- and stereoselective dihydroxylation of diene 5 under Sharpless AD conditions, exploiting preferential reaction at the more electron rich alkene (Scheme 3).16 Reaction of 5 with AD mix-α gave diol 6 as a single diastereoisomer17 (stereoselectivity assigned by the Sharpless mnemonic18). The preferential rate of dihydroxylation of (E)-1,2- over (Z)-1,2-disubstituted olefins19 meant that the minor unreacted (E,Z)-diene isomer could be easily removed.
Scheme 3. Formation of TA Precursor 9.
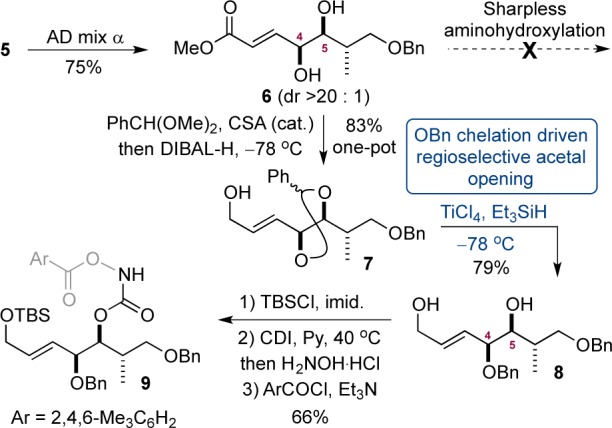
The TA sequence required differentiation between the two alcohols in 6. This was accomplished by formation of cyclic acetal 7, using benzaldehyde dimethyl acetal with one-pot ester reduction using DIBAL-H (83%).20 We envisaged that the benzyl ether of 7 might coordinate to a Lewis acid and encourage regioselective opening of the acetal. Treatment of 7 with TiCl4 and triethylsilane generated monoprotected benzyl ether 8 in 79% yield (regiochemistry confirmed by HMBC analysis).21 The TA precursor was then prepared by selective protection of the primary alcohol as a TBS ether, allowing the secondary alcohol to be transformed via treatment with CDI and hydroxylamine hydrochloride, followed by mesitoylation to afford intermediate 9.
The key transformation (the TA) was now addressed (Scheme 4). Treatment of 9 with 1 or 2 mol % potassium osmate hardly converted the substrate, presumably because of steric hindrance. An increase in catalyst loading to 5 mol % proved beneficial, and the desired aminohydroxylated product 10 was isolated in a yield of 76% at 40 °C. Notably, and consistent with previous observations, compound 10 was isolated as a single diastereoisomer, via a 1,3-anti TA reaction.5 The structure was confirmed by transformation into a solid, 12, and X-ray diffraction studies.22 At this stage we had prepared a compound (10) with the five contiguous stereocenters of AMMTD in place over 10 steps in 17% overall yield.
Scheme 4. Tethered Aminohydroxylation (TA) of 9.
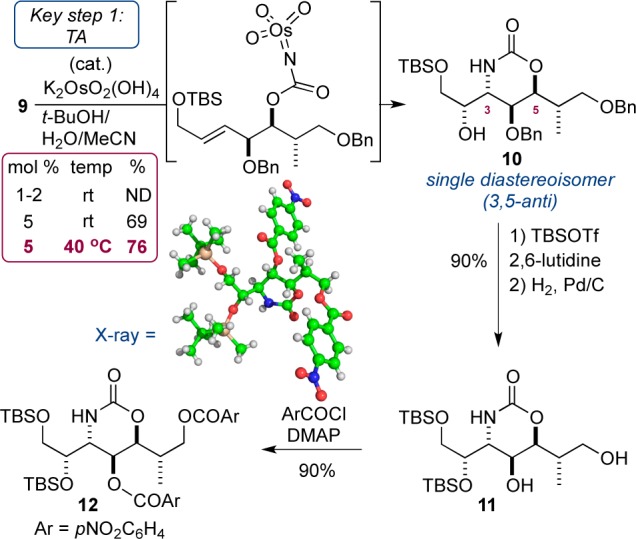
In order to append the aliphatic side chain (via addition of an organocuprate), diol 11 was converted to primary iodide 14, via selective formation of the iodide at C7 (84%) and protection of the C4 hydroxyl (91%), again as a TBS ether (Scheme 5). As part of our overall strategy we elected to prepare an organocuprate from 5-iodo-1-pentene (a side chain that bore a terminal olefin for flexibility later on). The generation of an organometallic reagent from one primary iodide, and subsequent displacement within a second primary iodide (which itself contained an acidic proton) proved to be extremely challenging. Extensive experimentation allowed us to make the following observations: (i) preparation of the organolithium from pentenyl iodide was best accomplished with t-BuLi; (ii) organolithium formation required ether solvent, while cuprate formation and subsequent displacement of 14 required THF solvent; (iii) CuCN was the best source of copper;23 (iv) addition of LiCl increased the solubility of the Lipshutz cuprate providing improved reproducibility; (v) temperature control during the reaction was essential, with organolithium formation being best at −78 °C with warming to room temperature, while transmetalation and displacement were optimal at −40 °C (Table 1).
Scheme 5. Synthesis of Iodide 14 and Side Chain Installation.
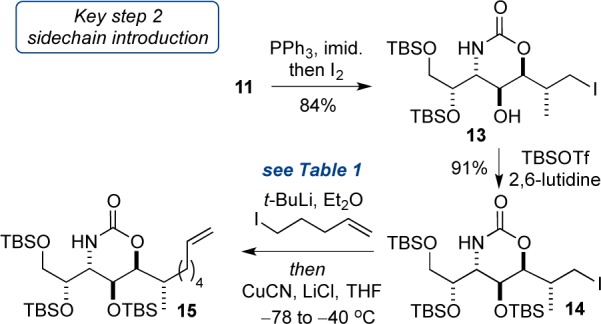
Table 1. Optimization of Organocuprate Addition to 14.
| entry | formation of pentenyl lithiuma |
additiveb | temp (°C)c |
15, yield % |
|---|---|---|---|---|
| 1 | Li metald | CuCN | –40 | 50 |
| 2 | t-BuLi | CuCN | –40 | 77 |
| 3 | t-BuLi | CuCN | –20 | 30 |
| 4 | t-BuLid | CuCN | –40 | 0e |
| 5 | t-BuLi | CuCN/LiCl | –40 | 80–97 |
4-Pentenyl iodide used as anion precursor in Et2O.
Anion transferred to the copper source in THF.
Temperature for anion formation −78 °C then warm to rt for 1 h.
4-Pentenyl bromide used as anion precursor in ether.
Iodide 14 was recovered.
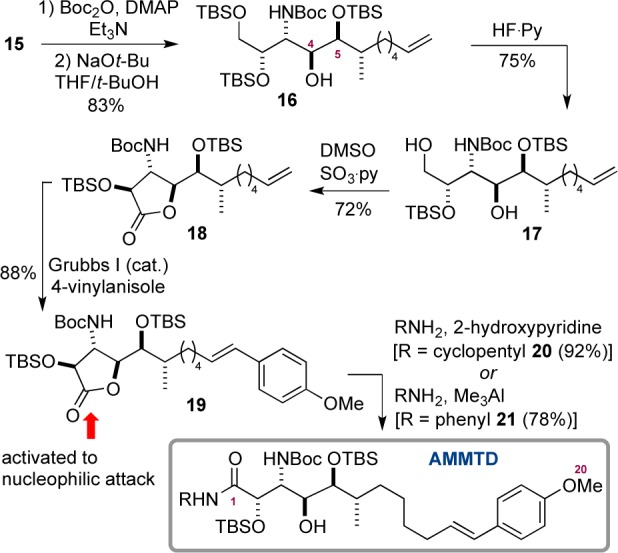
In order to deliver the amino acid in a form suitable for a later total synthesis, we cleaved the carbamate generated in the TA reaction (Scheme 6). This was accomplished via initial activation of 15 as its N-Boc derivative (Boc2O/DMAP, 95%). We found sodium tert-butoxide in t-BuOH/THF then brought about the desired cleavage in high yield (87%). Interestingly, the product 16 derived from C4-to-C5 migration of the TBS group (9:1 mixture) was isolated from the cleavage reaction. We were unable to reprotect the regioisomeric mixture as TBS ethers (to generate a single compound) and so continued by selective deprotection of the primary TBS ether, using HF/pyridine (75%).24 At this stage the regioisomers were separable, allowing 17 to be isolated as a single compound. We now sought to oxidize the C1 hydroxyl and found the most reliable procedure involved oxidation of 17 under Parikh–Doering conditions, which cleanly gave lactone 18. To prepare AMMTD, we required transformation of the terminal olefin into an (E)-p-methoxy styrene via cross-metathesis.25 Therefore, treatment of terminal alkene 18 and 4-vinylanisole (5 equiv) with sequential additions of Grubbs I catalyst (3 × 5 mol %) afforded (E)-styrene 19 in 88% yield, thus completing the synthesis of the fully protected β-amino acid AMMTD. Finally, we decided to test the effectiveness of the lactone as an activated carbonyl for amide formation. Treatment of 19 with a branched primary amine (cyclopentylamine, chosen as a model for the pyrrolidinone amino acid in the microsclerodermins), in the presence of 2-hydroxypyridine,26 generated amide 20 in 92% yield. In addition, reaction of aniline (selected since aniline amides have been shown to be excellent precursors to carboxylic acids via hydrolysis under mild conditions27) and trimethylaluminium also opened the lactone to deliver amide 21 in 78% yield.
Scheme 6. Completion of the Synthesis of Protected AMMTD.
In conclusion, we have demonstrated the synthetic utility of the tethered aminohydroxylation via the regio- and stereoselective functionalization of a complex homoallylic alcohol, serving as a key step in the synthesis of β-amino acid AMMTD. Moreover, we have designed our route to incorporate a late-stage introduction of the aliphatic side chain, a tactic that should allow access to other β-amino acid members of the microsclerodermins.
Acknowledgments
We thank the EPSRC (R.D.C.P.), the Hill Foundation (E.Y.M.), and the Deutscher Akademischer Austauschdienst (DAAD) (C.W.) for financial support. Dr. D. Klauber (University of Oxford) is thanked for some preliminary experiments.
Supporting Information Available
Experimental details and NMR spectra for all new compounds; X-ray crystallographic data for 12. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- For reviews, see:; a Bodkin J. A.; McLeod M. D. J. Chem. Soc., Perkin Trans. 1 2002, 2733. [Google Scholar]; b Kolb H. C.; Sharpless K. B. In Transition Metals for Organic Synthesis, Vol. 1, 2nd ed.; Beller C. B. M., Ed.; Wiley-VCH Verlag GmbH: 2008; p 309. [Google Scholar]; c Nilov D.; Reiser O. Adv. Synth. Catal. 2002, 344, 1169. [Google Scholar]
- For a recent review, see:; a Donohoe T. J.; Callens C. K. A.; Flores A.; Lacy A. R.; Rathi A. H. Chem.—Eur. J. 2011, 17, 58. [DOI] [PubMed] [Google Scholar]; For an example of asymmetric aminohydroxylation, see:; b Laxma Reddy K.; Sharpless K. B. J. Am. Chem. Soc. 1998, 120, 1207. [Google Scholar]
- Donohoe T. J.; Johnson P. D.; Cowley A.; Keenan M. J. Am. Chem. Soc. 2002, 124, 12934. [DOI] [PubMed] [Google Scholar]
- Donohoe T. J.; Chughtai M J.; Klauber D. J.; Griffin D.; Campbell A. D. J. Am. Chem. Soc. 2006, 128, 2514. [DOI] [PubMed] [Google Scholar]
- Donohoe T. J.; Bataille J. R.; Gattrell W.; Kloesges J.; Rossignol E. Org. Lett. 2007, 9, 1725. [DOI] [PubMed] [Google Scholar]
- Donohoe T. J.; Callens C. K. A.; Thompson A. L. Org. Lett. 2009, 11, 2305. [DOI] [PubMed] [Google Scholar]
- Donohoe T. J.; Pullin R. D. C. Chem. Commun. 2012, 48, 11924. [DOI] [PubMed] [Google Scholar]
- a Bewley C. A.; Debitus C.; Faulkner D. J. J. Am. Chem. Soc. 1994, 116, 7631. [Google Scholar]; b Qureshi A.; Colin P. L.; Faulkner D. J. Tetrahedron 2000, 56, 3679. [Google Scholar]; c Schmidt E. W.; Faulkner D. J. Tetrahedron 1998, 54, 3043. [Google Scholar]
- Zhang X.; Jacob M. R.; Rao R. R.; Wang Y. H.; Agarwal A. K.; Newman D. J.; Khan I. A.; Clark A. M.; Li X. C. Research Reports in Medicinal Chemistry 2012, 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shioiri T.; Sasaki S.; Hamada Y. ARKIVOC 2003, 103. [Google Scholar]; b Sasaki S.; Hamada Y.; Shioiri T. Synlett 1999, 453. [Google Scholar]
- Zhu J.; Ma D. Angew. Chem., Int. Ed. 2003, 42, 5348. [DOI] [PubMed] [Google Scholar]
- a Sasaki S.; Hamada Y.; Shioiri T. Tetrahedron Lett. 1999, 40, 3187. [Google Scholar]; b Sasaki S.; Hamada Y.; Shioiri T. Tetrahedron Lett. 1997, 38, 3013. [Google Scholar]
- Chandrasekhar S.; Sultana S. S. Tetrahedron Lett. 2006, 47, 7255. [Google Scholar]
- a Davies S. G.; Fletcher A. M.; Foster E. M.; Lee J. A.; Roberts P. M.; Thomson J. E. J. Org. Chem. 2013, 78, 2500. [DOI] [PubMed] [Google Scholar]; b Hjelmgaard T.; Faure S.; Lemoine P.; Viossat B.; Aitken D. J. Org. Lett. 2008, 10, 841. [DOI] [PubMed] [Google Scholar]; c Shuter E. C.; Duong H.; Hutton C. A.; McLeod M. D. Org. Biomol. Chem. 2007, 5, 3183. [DOI] [PubMed] [Google Scholar]; d Tarrade-Matha A.; Valle M. S.; Tercinier P.; Dauban P.; Dodd R. H. Eur. J. Org. Chem. 2009, 673. [Google Scholar]
- An alternative route that involved over-reduction of 2 to the primary alcohol (LiBH4) and oxidation under Parikh–Doering conditions formed 5 in 75% yield from 1; see Supporting Information.
- Xu D.; Crispino G. A.; Sharpless K. B. J. Am. Chem. Soc. 1992, 114, 7570. [Google Scholar]
- For comparison we also dihydroxylated the diene using the same conditions but with diastereomeric ligand (DHQD)2PHAL (AD mix-β) to give the diastereomeric diol, again as a single diastereomer (>20:1).
- For a review, see:Kolb H. C.; VanNieuwenhze M. S.; Sharpless K. B. Chem. Rev. 1994, 94, 2483. [Google Scholar]
- Andersson P. G.; Sharpless K. B. J. Am. Chem. Soc. 1993, 115, 7047. [Google Scholar]
- The ester group was not compatible with the TA reaction since the hydroxycarbamate nitrogen underwent conjugate addition to the alkene rather than participate in aminohydroxylation.
- Kotsuki H.; Nishikawa H.; Mori Y.; Ochi M. J. Org. Chem. 1992, 57, 5036. [Google Scholar]
- Low temperature, single crystal diffraction data for 12 were collected on I19 (EH1) at the Diamond Light Source, Harwell.; a Nowell H.; Batnett S. A.; Christensen K. E.; Teat S. J.; Allan D. R. J. Synch. Rad. 2012, 19, 435. [DOI] [PubMed] [Google Scholar]; Data were reduced using CrysAlisPro (Agilent), solved using SuperFlip:; b Palatinus L.; Chapuis G. J. Appl. Crystallogr. 2007, 40, 786. [Google Scholar]; Data refined within the CRYSTALS suite:; c Betteridge P. W.; Carruthers J. R.; Cooper R. I.; Prout K.; Watkin D. J. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar]; d Cooper R. I.; Thompson A. L.; Watkin D. J. J. Appl. Crystallogr. 2010, 43, 1100. [Google Scholar]; e Thompson A. L.; Watkin D. J. J. Appl. Crystallogr. 2011, 44, 1017–1022 as per the Supporting Information (CIF). Full refinement details are given in the Supporting Information (CIF); Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre (CCDC 947576) and can be obtained via www. ccdc.cam.ac.uk/data_request/cif. [Google Scholar]
- Lipshutz B. H.; Wilhelm R. S.; Floyd D. M. J. Am. Chem. Soc. 1981, 103, 7672. [Google Scholar]
- Crouch R. D. Tetrahedron 2004, 60, 5833. [Google Scholar]
- Chatterjee A. K.; Choi T. L.; Sanders D. P.; Grubbs R. H. J. Am. Chem. Soc. 2003, 125, 11360. [DOI] [PubMed] [Google Scholar]
- Wangsell F.; Russo F.; Savmarker J.; Rosenquist A.; Samuelsson B.; Larhed M. Bioorg. Med. Chem. Lett. 2009, 19, 4711. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Rajapakse H.; Chiu A.; Stenkamp D. Angew. Chem., Int. Ed. 2002, 43, 4573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.