

Abstract
The current standard method to culture human limbal stem/progenitor cells (LSCs) in vitro is to culture limbal epithelial cells directly on a layer of murine 3T3 feeder cells (standard method). The direct contact between human cells and murine feeder cells poses the potential risk of incomplete removal of feeder cells after culture and cross-contamination in clinical applications. We present here a novel three-dimensional (3D) sandwich method in which LSCs and feeder cells were separately cultured on opposite sides of a porous membrane. Limbal epithelial cells in the form of single-cell suspensions, cell clusters, and tissue explants were subjected to standard culture or to a 3D sandwich culture method. The 3D sandwich method consistently yielded LSCs derived from cell clusters and tissue explants. The expanded LSCs exhibited a small, compact, cuboidal stem-cell morphology and stem cell phenotypes comparable to those of LSCs derived from the standard culture method. Limbal epithelial cell clusters cultured with the sandwich method had a significantly higher proliferation rate than did those cultured with the standard method. The 3D sandwich method did not favor the propagation of single LSCs. In summary, the 3D sandwich method permits complete separation between cultured cells and feeder cells, while providing an even and maximal proximity between them. This alternative method permits culturing of LSCs without the risk of feeder cell contamination.
Introduction
Human corneal epithelial stem cells reside at the basal layer of the limbal epithelium; therefore, these cells are referred to as limbal stem/progenitor cells (LSCs).1–5 Limbal stem cell deficiency causes inflammation, vascularization, scarring, pain, and ultimately blindness.6–8 Transplantation of ex vivo expanded LSCs has successfully restored vision.9,10 Currently, the standard method to propagate LSCs in vitro is to culture the LSCs directly on growth-arrested mouse fibroblast 3T3 feeder cells. Cultured stem/progenitor cells form two-dimensional (2D) colonies that expand and push away the feeder cells.
Several issues are associated with the 2D culture method. One is the varying distances between the cultured cells and the feeder cells. Feeder cells support the ex vivo expansion of LSCs by secreting soluble niche molecules, including growth factors and cytokines, and probably also by signaling through cell–cell contact.11 Because of the distance between the center of colonies and the feeder cells, gradients of nutrients form. Putative stem cell markers, including N-cadherin, p63, and ABCG2, are expressed at higher levels at the edge of the colonies, while the expression of the differentiation marker K12 is greatest near the center of the colonies12,13; this indicates that the close proximity to feeder cells helps in maintaining the less differentiated state of LSCs.
A second shortcoming of the standard 2D culture method is the competition between the stem cells and the feeder cells for the growth surface. As the epithelial colonies grow, they push away the feeder cells; this might result in a progressive decrease in the number of feeder cells in culture, which may lead to an insufficient supply of nutrients for the LSCs.
The third issue of the standard 2D culture method is the possible contamination by murine feeder cells. Because of the direct contact between the LSCs and the feeder cells, it is possible that not all feeder cells are removed from the LSC population after harvest. Thus, feeder cells are a potential cross-contamination risk in clinical applications.
To mimic the in vivo environment of LSCs and to improve the current 2D culture method, various three-dimensional (3D) methods to culture LSCs in vitro have been examined. LSCs are presumed to be in close proximity with their niche cells. LSCs and their subjacent mesenchymal niche cells have been isolated by collagenase treatment and cocultured in a 3D Matrigel to form cell spheres.14 However, the cell proliferation rate was not optimal and the percentage of epithelial cells in the cell spheres after culture was not known. Efforts have also been made to culture in vitro propagated limbal epithelial cells on top of the corneal stromal cells embedded either in collagen or in a fibrin matrix.15,16 Again, the expansion rate and epithelial stem cell phenotypes after this type of 3D culture are unknown.
A culture system that can address the above-mentioned shortcomings of the current culture methods is desirable. In the current study, we evaluated a novel 3D culture method in which LSCs are separated from feeder cells, while maximal contact between them is maintained. This novel 3D sandwich culture method appears to be efficient in expanding LSCs for use in transplantation.
Materials and Methods
Human sclerocorneal tissue
Human sclerocorneal tissue was obtained from the Illinois Eye Bank (Watson Gailey, Bloomington, IL) and the Lions Eye Institute for Transplant and Research (Tampa, FL). Tissue donors ranged in age from 20 to 65 years. Experimentation on human tissue adhered to the tenets of the Declaration of Helsinki. The experimental protocol was evaluated and exempted by the University of California, Los Angeles Institutional Review Boards.
The tissues were preserved in Optisol (Chiron Ophthalmics, Inc., Irvine, CA), and the death-to-preservation time was less than 8 h.
Preparation of limbal epithelial cell culture
Limbal epithelial cells were isolated from corneoscleral rims as previously described.17 In brief, the residual blood vessels, iris, endothelium, Tenon's capsules, and conjunctiva were removed from the rim. The rim was digested in 2.4 U/mL Dispase II (Roche, Indianapolis, IN) in a supplemented hormonal epithelial medium containing the DMEM/F12 medium (Gibco, Grand Island, NY) supplemented with N-2 (Gibco), 2 ng/mL epidermal growth factor (Gibco), 8.4 ng/mL cholera toxin (Sigma-Aldrich, St. Louis, MO), 0.5 μg/mL hydrocortisone (Sigma-Aldrich), 0.5% dimethyl sulfoxide (Sigma-Aldrich), 5% fetal bovine serum (Invitrogen, Carlsbad, CA), penicillin/streptomycin (Invitrogen), and gentamicin/amphotericin B (Invitrogen) for 2 h at 37°C. Sheets of limbal epithelium were scraped from the limbus and pipetted up and down to break the cell sheets into smaller clusters and more evenly distribute the clusters in the medium. Some clusters were further treated with 0.25% trypsin and 1 mM EDTA (Gibco) for 10–15 min at 37°C to obtain single-cell suspensions. For the in vitro propagation of LSCs, epithelial cells, either in cell clusters or in single-cell suspension, were seeded at a density of 300 cells/cm2. The cell proliferation rate was evaluated as the total number of epithelial cells recovered from the culture after moving the feeder cells. The absolute number of cells from each culture method was defined as the total number of cells that were produced per limbal epithelial cells seeded.
To prepare tissue explants for culture, corneoscleral rims were separated from residual blood vessels, iris, endothelium, Tenon's capsules, and conjunctiva and then cut into pieces that were approximately 2×2 mm. The explant pieces were placed on the growth surface with the epithelium side facing up. Only one explant piece was cultured per well or per insert.
Standard and sandwich culture methods
Subconfluent murine 3T3-J2 cells (from Howard Green, Harvard Medical School, Boston, MA) were treated with 4 μg/mL of mitomycin C (Sigma-Aldrich) for 2 h, and plates were seeded with 3×104 cells/cm2. For the standard culture method, suspensions of single limbal epithelial cells, cell clusters, or explants were seeded directly onto the 3T3 feeder cells (Fig. 1A). Standard culture, in which single limbal epithelial cells were seeded directly on 3T3 feeder cells, was used as control. Alternatively, six-well inserts with polyethylene terephthalate (PET) membrane with a pore size of 1 μm (Millipore, Billerica, MA) were placed upside down into six-well plates, and 1 mL of medium containing the 3T3 feeder cells was carefully loaded onto the bottom side of each PET membrane (Fig. 1A, B). The feeder cells were incubated at 37°C for 3h to overnight to allow cell attachment. The inserts were placed upright into the six-well plates, and the limbal epithelial cells or explants were seeded onto the inner side of the membrane. The PET membrane with 1 μm pore size was selected because it has been reported that only the 1 μm pore can effectively minimize the migration of mouse embryonic fibroblast feeder cells to zero during culture, while the 3 and 8 μm pores cannot.18 This method of 3D culture is called the “sandwich method” in the rest of the article. Limbal epithelial cells in single-cell suspension were cultured for 14–21 days and collected before confluence. Limbal epithelial cell clusters and explants were cultured for 14 days and collected before confluence. The medium was replaced every 2–3 days.
FIG. 1.
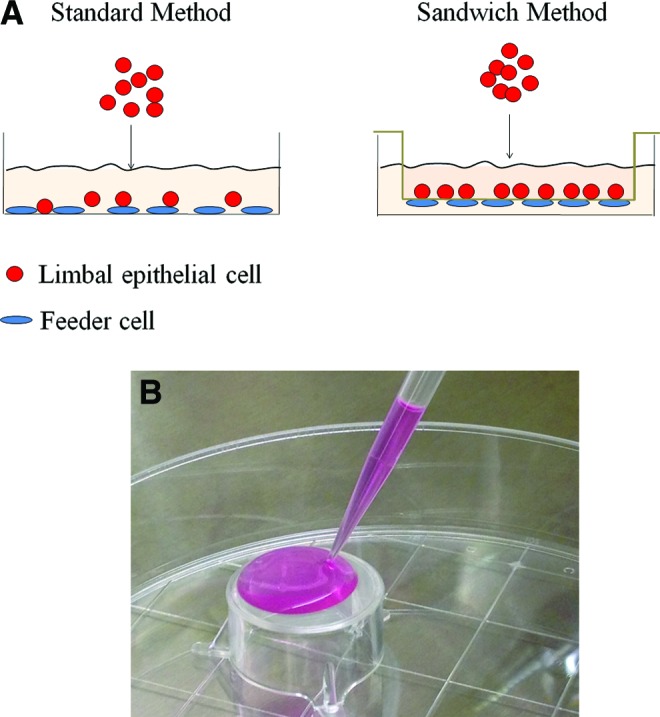
The standard and sandwich culture methods. (A) Diagram of the standard and sandwich culture methods. In the standard culture method, limbal epithelial cells were cultured directly on feeder cells. In the sandwich culture method, feeder cells attached to the bottom of the polyethylene terephthalate (PET) membrane, and the cultured cells were seeded onto the inner side of the membrane. (B) Loading of the 3T3 feeder cells on the inverted PET membrane. Color images available online at www.liebertpub.com/tec
RNA isolation, reverse transcription, and quantitative real-time polymerase chain reaction
After culture, epithelial cells were collected, and RNA was extracted (RNeasy Mini Kit; Qiagen, Valencia, CA), treated with DNase (DNA-free kit; Ambion, Austin, TX), and reverse-transcribed into cDNA (SuperScript II; Invitrogen) according to the manufacturer's instructions. Transcripts were detected by using the Kapa Sybr Fast qPCR kit (Kapa Biosystems, Woburn, MA). Cycle conditions were as follows: the reactant was denatured for 20 s at 95°C; amplified for 40 cycles (temperatures in each cycle were 95°C for 3 s, 60°C for 20 s, and 72°C for 8 s); and subjected to a melting curve program to obtain the dissociation curve. The primers used in quantitative real-time–polymerase chain reaction are listed in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/tec).
Immunocytochemistry and quantitation
Expanded epithelial cells were cytospined on slides by a cytocentrifuge (Cytofuge; Fisher Scientific, Hampton, NH) and stored at −20°C until use. Cytospin slides were fixed with 4% paraformaldehyde at room temperature for 10 min and washed three times with phosphate-buffered saline (PBS) containing 0.3% Triton X-100 (Sigma-Aldrich). PBS with 10% donkey serum was used for 30 min at room temperature to block the sections. Sections were incubated with one or more primary antibodies diluted in PBS with 1% bovine serum albumin (BSA) overnight at 4°C in a moisture chamber. Sections were washed 3 times with PBS with 1% BSA, incubated with one or more secondary antibodies diluted in PBS with 1% BSA at room temperature for 1 h, and washed with PBS containing 1% BSA and 0.3% Triton X-100. Nuclei were labeled with Hoechst 33342 (4 μg/mL; Invitrogen) at room temperature for 15 min, washed five times with PBS, and mounted in the Fluoromount medium (Sigma-Aldrich). The primary and secondary antibodies and their dilution ratios are listed in Supplementary Table S2.
Images were taken by a confocal microscope (Confocal Laser Scanning Microscopy; Olympus, San Jose, CA) and an image capture system (Fluoview FV10-ASW 3.1 Viewer; Olympus). The nuclear intensity of p63α was determined by the Definiens Tissue Studio software (Larchmont, NY).
Calculation of cell population doubling
Cell proliferation capacity was presented as the number of cell population doubling (PD), which was calculated as log2 (no. of cells harvested/no. of cells seeded).
Confocal and high-resolution light microscopy
Cultured limbal epithelial cells using the sandwich method formed cell colonies on the PET membrane. The membrane was cut off and fixed for confocal or high-resolution light microscopy. For confocal microscopy, the membrane was fixed with 4% paraformaldehyde, washed with PBS containing 0.3% Triton X-100, and stained with phalloidin (Santa Cruz Biotechnology, Santa Cruz, CA) following the manufacturer's protocol. The membrane was mounted in the Fluoromount medium (Sigma-Aldrich), scanned using a confocal microscope (Confocal Laser Scanning Microscopy; Olympus), and processed by an image capture system (Fluoview FV10-ASW 3.1 Viewer; Olympus). The serial images were composed to form a 3-dimensional model.
For high-resolution light microscopy, the membrane was fixed with 2% glutaraldehyde (Electron microscope Sciences, Hatfield, PA) and 2% paraformaldehyde (Electron microscope Sciences) in a 0.1 M cacodylate buffer, washed with the 0.1 M cacodylate buffer, osmicated for 1 h, washed extensively, and embedded in Epon resin (Momentive Specialty Chemicals, Houston, TX). Tissue was sectioned at 700 nm for light microscopy by Confocal Laser Scanning Microscopy (Olympus).
Statistical analysis
The Student's t-test was performed to analyze the data. Each error bar represents the standard error of the mean from at least three experiments. p-Values≤0.05 were considered to indicate statistical significance.
Results
LSC cultures generated from single-cell suspensions
LSCs generated from single-cell suspension cultured by the standard or sandwich method showed similar compact, cuboidal epithelial morphology (Fig. 2A); however, cells obtained with the sandwich method had a PD that was 37% lower compared with the standard method (Fig. 2B). There was a 30% chance that no epithelial growth was observed using the sandwich method. LSCs cultured by using the sandwich method had better stem cell phenotypes than did those cultured by using the standard method: the expression of ABCG2 was 2.8-fold greater in LSCs grown with the sandwich method (p<0.05), and the expression levels of other markers were comparable between LSCs obtained by either method (Fig. 2C). However, because of poor and inconsistent proliferation, the use of the sandwich method to culture single-cell suspensions was abandoned.
FIG. 2.
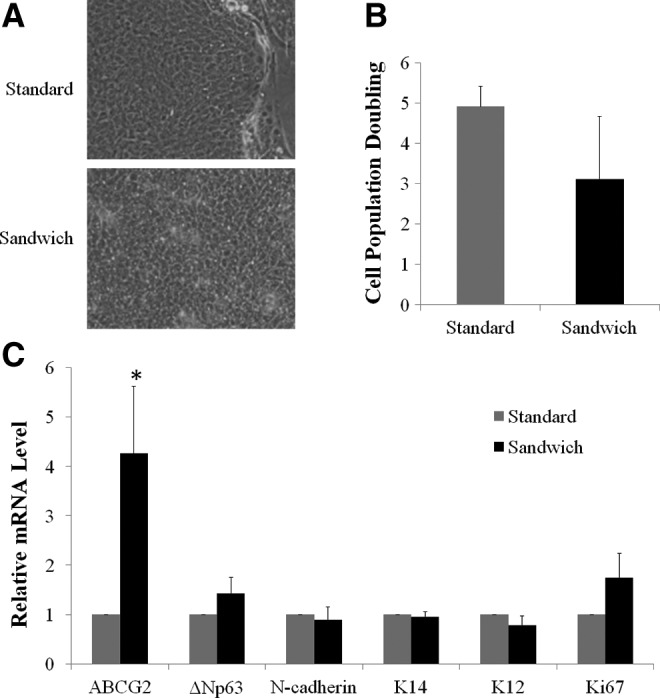
Limbal stem/progenitor cells (LSCs) cultured in single-cell suspension. (A) Morphology of LSC colonies. (B) Cell population doubling (PD) of limbal epithelial cells. (C) Relative mRNA levels of putative stem cell markers and maturation markers as evaluated by quantitative real-time–polymerase chain reaction (qRT-PCR). The expression of markers from single-cell culture using the standard method served as a control and was designated a value of 1. Asterisks indicate p<0.05 in comparison with results for the standard method.
LSC cultures generated from cell clusters
Cells derived from cluster cultures using standard (cluster standard method) or sandwich methods (cluster sandwich method) were compact and displayed cuboidal, epithelial stem-cell morphology (Fig. 3A). The PD obtained from cell clusters in the sandwich cultures was greater compared with the cell clusters in the regular method and the control (i.e., a single-cell suspension cultured directly on feeder cells). The PDs in control, cluster standard, and cluster sandwich methods were 4.9, 5.8, and 7, respectively (Fig. 3B).
FIG. 3.
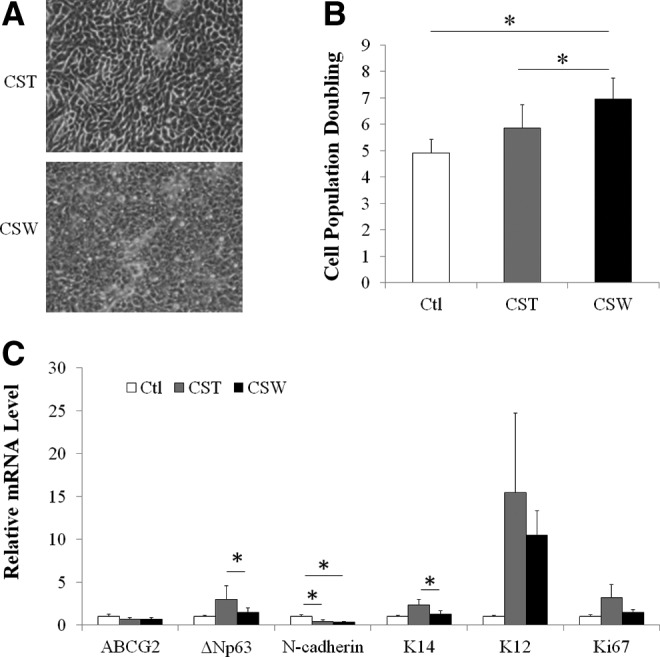
Comparison of LSCs derived from cell cluster standard and sandwich cultures. (A) Morphology of colonies. (B) Cell population doubling of limbal epithelial cells. (C) Relative mRNA levels of the putative stem cell markers and maturation marker as evaluated by qRT-PCR. The expression of markers by control single-cell standard culture served as a control and was designated a value of 1. Asterisks indicate p<0.05 in comparison with results for the cluster standard method or the control method. Ctl, control; CST, cluster standard culture method; CSW, cluster sandwich culture method.
Limbal epithelial cells from the control culture expressed a higher level of N-cadherin mRNA than did cells derived from cluster standard culture (1.3-fold higher) and cluster sandwich culture (1.8-fold higher; p<0.05 for both comparisons). Lower mRNA levels of putative LSC markers, ΔNp63 and K14, were seen in cells derived from the cluster sandwich method and cluster standard method (lower by 34% and 43%, respectively; p<0.05 both comparisons with the control) (Fig. 3C). The percentage of p63α-bright (p63αbr) cells in the cultured LSCs is a prognostic factor of clinical success after transplantation in humans.10 We then investigated the portion of p63αbr cells in our cultures. Immunocytochemistry data showed no significant differences in the percentages of p63αbr cells among the control, cluster standard method, and cluster sandwich methods (Fig. 4A). The percentage of K14+ cells was also evaluated (Fig. 4B). All three types of cultures contained 6–7% p63αbr cells and 80–90% K14+ cells. The cluster sandwich method produced slightly higher absolute numbers of p63αbr cells and K14+ cells than did the control and the standard method; however, the differences did not reach significance. The percentages and absolute numbers of K12+ differentiated corneal epithelial cells were extremely low in all three groups (0.8–2.3% and 0.1–3, respectively) (Fig. 4C).
FIG. 4.

Expression of p63α, K14, and K12 in limbal epithelial cells derived from cluster standard and sandwich methods. (A) Representative images of p63α expression, and percentage and absolute number of p63αbr cells generated from cultured limbal cell sheets. (B) Representative images of K14 expression, and percentage and absolute number of K14+ cells generated from cell cluster cultures. (C) Representative images of K12 expression, and percentage and absolute number of K12+ cells generated from cell cluster cultures. The absolute number of cells was the total number of cells obtained from the culture divided by the number of cells seeded. Scale bar represents a distance of 100 μm. Color images available online at www.liebertpub.com/tec
LSCs generated from limbal explant cultures
The 2×2 mm explants were seeded either on top of the feeder cells in a cell-culture Petri dish (standard method) or on top of a porous membrane with feeder cells seeded on the bottom side of the membrane (sandwich method). Both methods produced limbal epithelial cells with small compact cuboidal undifferentiated epithelial morphology (Fig. 5A). The proliferation rate of explant outgrowth in the sandwich culture was 6-fold greater than that in the standard culture (Fig. 5B); however, the difference did not reach significance due to big variations among tissue pieces. Explants in the sandwich culture tended to have higher levels of ΔNp63, K14, and K12 mRNA than did explants in the standard method, but none of the differences reached significance (Fig. 5C). Immunocytochemical analysis showed that in comparison with explants in the standard culture, explants in the sandwich culture generated a comparable percentage of p63αbr cells, a higher percentage of K14+ cells, and a lower percentage of K12+ cells (Fig. 6A–C). However, the differences failed to reach significance. Both samples contained a low percentage of p63αbr cells (3–5%), a high percentage of K14+ cells (80–90%), and a low percentage of K12+ cells (1.5–5.5%). The absolute numbers of p63αbr cells, K14+ cells, and K12+ cells in the sandwich culture were comparable to those generated in the standard culture.
FIG. 5.
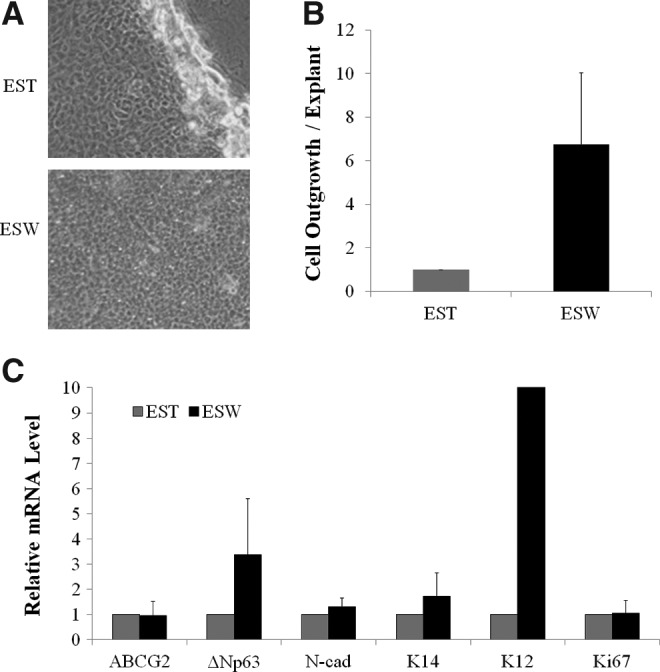
Limbal epithelial cells expanded from tissue explant cultures using the standard and sandwich methods. (A) Morphology of cells from explant outgrowth. (B) Relative rate of cell outgrowth. The cell outgrowth rate was calculated as the number of cells harvested per piece of explant. (C) The relative mRNA expression levels of putative stem cell markers and maturation marker of the outgrowth. EST, explant standard culture method; ESW, explant sandwich culture method.
FIG. 6.

Expression of p63α, K14, and K12 in cell outgrowth from limbal explants in standard and sandwich cultures. (A) Representative images of p63α expression, and percentage and absolute number of p63αbr cells in the outgrowth of explants. (B) Representative images of K14 expression, percentage and absolute number of K14+ cells in the outgrowth of explants. (C) Representative images of K12 expression, and percentage and absolute number of K12+ cells in the outgrowth of explants. The absolute number of cells was the total number of cells obtained from culture divided by the number of cells seeded. Scale bar represents a distance of 100 μm. Color images available online at www.liebertpub.com/tec
No obvious cell–cell contact was observed between cultured LSCs and feeder cells in the sandwich method
Whether there is any cell–cell contact between cultured LSCs and feeder cells in the sandwich method was investigated further. Derived 3D images showed compact cultured LSCs, with bright F-actin labeling, above the PET membrane (Fig. 7A) and the 3T3 feeder cells beneath the PET membrane (Fig. 7B). In a reconstructed image, the z-axis contact between the top cultured LSCs and the bottom feeder cells was not evident (Fig. 7C). These two layers of cells were completely separated by the PET membrane and no cells were seen within the membrane. In further analysis, we examined semithin (700-nm) transverse sections of the cells and the PET membrane. More than 50 pores were examined and none of them showed obvious cell–cell contact through the pores; representative images are shown in Figure 7D.
FIG. 7.
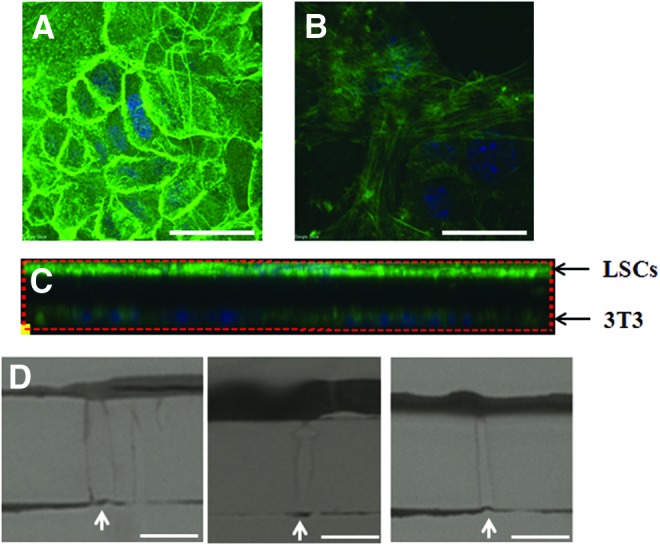
No obvious cell–cell contact was observed between cultured LSCs and feeder cells in the sandwich method. LSCs were cultured on 3T3 feeder cells using the sandwich method for 2 weeks to form colonies. The cells were stained with phalloidin and whole-mounted for a three-dimensional confocal microscopy. (A) Cultured LSCs showed compact and strong expression of F-actin on the top side of the PET membrane. (B) Feeder cells showed spread expression of F-actin underneath the PET membrane. Scale bar represents a distance of 50 μm. (C) Contact between the LSCs (upper) and the 3T3 feeder cells (lower) was not evident in transverse sections. (D) Representative images of 700-nm cross sections of pores also showed no obvious cell–cell contact between the culture LSCs and 3T3 feeder cells in high-resolution light microscopy. Pores were indicated by white arrows. Scale bar represents a distance of 5 μm. Color images available online at www.liebertpub.com/tec
Discussion
Compared with the standard 2D culture method, the 3D sandwich method presented in this article better resembles the in vivo environment of LSCs. Our results showed that no matter which form of LSCs (i.e., single-cell suspension, cell cluster, or explant) was seeded, cells derived from the sandwich culture method had phenotypes comparable to those of stem cells. Moreover, the expansion rate of epithelial cells from the cluster sandwich method was greater than that from the cluster standard method and the control method.
We propose several hypotheses to explain the increased proliferation rate and the concurrent maintenance of the stem cell phenotypes in the sandwich culture. First, the evenly close proximity of all epithelial cells to feeder cells in the sandwich culture may have provided sufficient growth support to promote a better proliferation rate, while maintaining the less differentiated stem/progenitor phenotype. Previous reports indicated that the edge of LSC colonies exhibited more stemness and more active proliferation than the cells in the center of the colonies12,13; these results suggest that close proximity to the feeder cells support the stem cell phenotype of LSCs by providing adequate nutrient levels and/or cell–cell contact. Second, the sandwich method may help to maintain the polarity of LSCs, and thus, the proliferation and maintenance of the phenotype. Limbal epithelial cells, as a type of nonkeratinized stratified epithelium, have an apical-basolateral polarity, which is crucial for their proliferation, differentiation, and proper functioning.19,20 In the standard method, feeder cells are seeded to the side of the epithelial cells, whereas in the sandwich method, feeder cells are located below the epithelial sheet. This arrangement in the sandwich culture may better maintain stem cell polarity and thus may better maintain stemness and sustain proliferation.
It is interesting that the growth rate of cells derived from the single-cell sandwich method was lower compared with cells derived from the single-cell standard method. In some instances, no growth from single-cell suspensions in sandwich culture was observed. It is possible that for survival and proliferation, a single LSC may need direct contact with feeder cells and that the PET membrane in the sandwich culture may not permit sufficient contact. Another possibility is that the PET membrane is not an ideal substrate for the initial attachment of single limbal epithelial cells for proliferation. However this difficulty was compensated by a cell cluster method.
Lee and colleagues described a similar 3D culture method.18,21 In their method of culture, human embryonic stem cells (hESCs) were placed on top of a porous PET membrane with feeder cells attached to the other side. This 3D method effectively propagated hESCs and maintained their stem cell phenotype. There is still cell–cell contact through the pores between the feeder cells and the hESCs, and maintaining the physical cell–cell contact might be important in supporting the expansion of hESCs. However, no comparison was made between this 3D method and the conventional method of culture for hESCs. Their reports focused on the benefit that this 3D method can effectively separate stem cells and feeder cells and thus can facilitate stem cell harvest and reduce xenogenic contamination by the feeder cells. This benefit also applies to our sandwich method. In the standard method of culture, LSCs can be harvested by removing the feeder cells through enzymatic or chemical dissociation. Removal of feeder cells can be incomplete, and the dissociation process can be harmful to the cultured stem/progenitor cells, which tend to locate at the edge of colonies. In the sandwich method, LSCs are separated from feeder cells by the membrane; therefore, the cultured LSCs can be easily removed by enzymatic digestion within the insert. Cell migration will not occur with a pore size less than 3.0 μm. Therefore, contamination of feeder cells in the cultured LSCs is unlikely. Our data from confocal and high-resolution light microscopy further support the complete separation between the cultured LSCs and feeder cells in the sandwich method. No direct cell–cell contact was observed in pores between the cultured LSCs and the feeder cells, thus the contamination from feeder cells appears to be minimized.
In summary, a novel 3D sandwich method described in the current study can efficiently expand LSCs without contamination by feeder cells.
Supplementary Material
Acknowledgments
This work was supported by the California Institute for Regenerative Medicine (CIRM TR2-01768), the National Eye Institute (NEI R01EY021797 and 2P30E4000331-44), and Voucher Award from the UCLA Clinical and Translational Science Institute (CTSI) in 2011.
Disclosure Statement
A patent application has been filed.
References
- 1.Ebato B., Friend J., and Thoft R.A.Comparison of central and peripheral human corneal epithelium in tissue culture. Invest Ophthalmol Vis Sci 28,1450, 1987 [PubMed] [Google Scholar]
- 2.Huang A.J., and Tseng S.C.Corneal epithelial wound healing in the absence of limbal epithelium. Invest Ophthalmol Vis Sci 32,96, 1991 [PubMed] [Google Scholar]
- 3.Tsai R.J., Sun T.T., and Tseng S.C.Comparison of limbal and conjunctival autograft transplantation in corneal surface reconstruction in rabbits. Ophthalmology 97,446, 1990 [DOI] [PubMed] [Google Scholar]
- 4.Cotsarelis G., Cheng S.Z., Dong G., Sun T.T., and Lavker R.M.Existence of slow-cycling limbal epithelial basal cells that can be preferentially stimulated to proliferate: implications on epithelial stem cells. Cell 57,201, 1989 [DOI] [PubMed] [Google Scholar]
- 5.Davanger M., and Evensen A.Role of the pericorneal papillary structure in renewal of corneal epithelium. Nature 229,560, 1971 [DOI] [PubMed] [Google Scholar]
- 6.Tseng S.C.Concept and application of limbal stem cells. Eye 3 (Pt 2),141, 1989 [DOI] [PubMed] [Google Scholar]
- 7.Sejpal K., Bakhtiari P., and Deng S.X.Presentation, diagnosis and management of limbal stem cell deficiency. Middle East Afr J Ophthalmol 20,5, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dua H.S., Saini J.S., Azuara-Blanco A., and Gupta P.Limbal stem cell deficiency: concept, aetiology, clinical presentation, diagnosis and management. Indian J Ophthalmol 48,83, 2000 [PubMed] [Google Scholar]
- 9.Pellegrini G., Traverso C.E., Franzi A.T., Zingirian M., Cancedda R., and De Luca M.Long-term restoration of damaged corneal surfaces with autologous cultivated corneal epithelium. Lancet 349,990, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Rama P., Matuska S., Paganoni G., Spinelli A., De Luca M., and Pellegrini G.Limbal stem-cell therapy and long-term corneal regeneration. N Engl J Med 363,147, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Miyashita H., Shimmura S., Higa K., Yoshida S., Kawakita T., Shimazaki J., et al. A novel NIH/3T3 duplex feeder system to engineer corneal epithelial sheets with enhanced cytokeratin 15-positive progenitor populations. Tissue Eng Part A 14,1275, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Secker G.A., and Daniels J.T.Limbal epithelial stem cells of the cornea. The Stem Cell Research Community, StemBook, 2009, doi: 10.3824/stembook.1.48.1, http://www.stembook.org [DOI] [PubMed]
- 13.Higa K., Shimmura S., Miyashita H., Kato N., Ogawa Y., Kawakita T., et al. N-cadherin in the maintenance of human corneal limbal epithelial progenitor cells in vitro. Invest Ophthalmol Vis Sci 50,4640, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Xie H.T., Chen S.Y., Li G.G., and Tseng S.C.Isolation and expansion of human limbal stromal niche cells. Invest Ophthalmol Vis Sci 53,279, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bray L.J., George K.A., Hutmacher D.W., Chirila T.V., and Harkin D.G.A dual-layer silk fibroin scaffold for reconstructing the human corneal limbus. Biomaterials 33,3529, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Papini S., Rosellini A., Nardi M., Giannarini C., and Revoltella R.P.Selective growth and expansion of human corneal epithelial basal stem cells in a three-dimensional-organ culture. Differentiation 73,61, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Truong T.T., Huynh K., Nakatsu M.N., and Deng S.X.SSEA4 is a potential negative marker for the enrichment of human corneal epithelial stem/progenitor cells. Invest Ophthalmol Vis Sci 52,6315, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S., Ahn S.E., Lee J.H., Lim D.S., Kim K.S., Chung H.M., et al A novel culture technique for human embryonic stem cells using porous membranes. Stem Cells 25,2601, 2007 [DOI] [PubMed] [Google Scholar]
- 19.St. Johnston D., and Ahringer J.Cell polarity in eggs and epithelia: parallels and diversity. Cell 141,757, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Martin-Belmonte F., and Perez-Moreno M.Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 12,23, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Hwang S.T., Kang S.W., Lee S.J., Lee T.H., Suh W., Shim S.H., et al The expansion of human ES and iPS cells on porous membranes and proliferating human adipose-derived feeder cells. Biomaterials 31,8012, 2010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.