

Abstract
Background
Among the pro-inflammatory lipid mediators, platelet-activating factor (PAF) is a major primary and secondary messenger that binds to the PAF-receptor (PAFR). Epidermal growth factor (EGF) is a polypeptide growth factor that binds to the EGF-receptor (EGFR). Evidence suggests that both PAF and EGF play a significant role in oncogenic transformation, tumor growth, neoangiogenesis and metastasis, including ovarian cancer. PAF has the potential to transactivate EGFR in ovarian cancer cells. This study explores the mechanisms involved in EGF-induced PAF production.
Methods
The effect of EGF on PAF production in ovarian cancer cells was observed using enzyme-linked immunosorbent assay. The receptors transactivation and the role of cytosolic phospholipase A2 (cPLA2) in modulating PAF production induced by EGF was assessed using pharmacological inhibitors, si-RNA knockdown, targeted gene overexpression and immunocytochemistry. The signaling pathways invovled in PAF production induced by EGF in ovarian cancer cells were assessed.
Results
We demonstrate that EGF increases the production of PAF in CAOV3 and SKOV3 ovarian cancer cell lines. EGF induces the transactivation of PAFR, which can be blocked by an EGFR inhibitor. Inhibition of EGFR and/or PAFR blocks PAF production in response to EGF. EGF-induced PAF production involves the phosphorylation of extracellular-regulated protein kinase (ERK) and cytosolic phospholipase A2 (cPLA2). A cPLA2 inhibitor blocks EGF-induced PAF production as well as si-cPLA2, while overexpression of cPLA2 increases PAF production.
Conclusions
These results indicate that EGF stimulates PAF production in ovarian cancer cells in a manner that requires cPLA2. We have also determined that crosstalk can occur bidirectionally between EGFR and PAFR, suggesting that EGF-induced PAF production could result in positive feedback that acts on the PAF-receptor to promote ovarian cancer progression.
Keywords: Ovarian cancer, EGF, EGF-receptor, PAF-receptor, ERK, cPLA2
Background
Chronic inflammatory microenvironments have been suggested as the major predisposing factor for ovarian and other cancers [1]. Lipid mediators such as lysophosphatidic acid (LPA) and prostaglandins (PGs), with their specific receptors and pathways, have been shown to play a critical role in cancer initiation and progression [2-4]. Platelet activating factor (PAF, 1-O-alkyl-2-acetyl-sn-glycero-3-phosphorylcholine) is also one of the most potent lipid mediators involved in many different biological pathways in inflammatory diseases and cancers [5,6]. There are two distinct pathways in which PAF can be synthesized: the de novo pathway and the remodeling pathway [7]. The de novo pathway is used to maintain PAF levels during normal cellular function, while the remodeling pathway is activated by inflammatory agents and is the primary source of PAF under pathological conditions. The initiation of the remodeling pathway requires membrane phospholipid hydrolysis by phospholipase A2 (PLA2), which supplies lyso-PAF, a precursor of PAF. Lyso-PAF acetyltransferase then converts lyso-PAF into PAF and finally, PAF activates the PAF-receptor (PAFR), a member of the superfamily of G protein-coupled receptors [8,9]. These events are thought to play an important role in the oncogenic transformation [10], proliferation [11] and metastasis [12] of several types of cancers, including ovarian cancers. However, PAF is rapidly degraded by PAF acetylhydrolases (PAF-AH), which cleaves the acetyl group at the sn-2 position to reform back to lyso-PAF [13]. Therefore, there maybe a possibility that PAF acts as an autocrine growth factor to promote ovarian cancer progression.
PLA2 is classified into three groups: group VI calcium-independent PLA2s (iPLA2s), secretory PLA2s (sPLA2s), and group IV cytosolic PLA2s (cPLA2) [14]. Group IVA cPLA2 is essential for producing PAF because PAF synthesis is significantly diminished in calcium ionophore-stimulated macrophages derived from group IVA cPLA2-deficient mice compared with those from wild-type mice [15]. However, the role of cPLA2 in growth factor-mediated PAF production in ovarian cancer cells has not been examined. The current study focuses on the role of cPLA2 in epidermal growth factor (EGF)-stimulated PAF production in ovarian cancer cells. Epidermal growth factor (EGF), a polypeptide growth factor, binds to the EGF-receptor (EGFR), which is a transmembrane protein tyrosine kinase. EGF is free of sugar groups, has excellent chemical stability and is highly prevalent in human blood. It also has been reported that EGF stimulates proliferation of ovarian cancer cells and other types of carcinoma cells [16,17].
Our earlier study demonstrated that ovarian cancer cells express high levels of PAFR as well as that PAF can stimulate transactivation of the EGFR in ovarian cancer cells [18]. PAF can activate matrix metalloproteases, which cleave pro-EGF from the membrane to release active ligands. Intracellular signaling molecules, such as phospholipase C and protein kinase A, have also been suggested as mediators of PAF-induced transactivation of receptor tyrosine kinase. Conversely, some growth factors and cytokines can activate PAF production, resulting in transactivation of the PAF receptor [19]. However, it has not yet been tested whether reverse crosstalk occurs and whether EGF can stimulate activation of PAFR.
In this study, we used CAOV3 and SKOV3 adenocarcinoma cells, two well-characterized human ovarian cancer cell lines, as models to examine the mechanisms involved in EGF-induced PAF production. Stimulation of cells with EGF increases PAF levels in the medium. This response can be inhibited by either EGFR or PAFR inhibition. EGF recruits cPLA2 by activating the ERK signaling pathway and overexpression of cPLA2 increases PAF production, while inhibition of cPLA2 blocks EGF-induced PAF production. These results show that EGF activates cPLA2, that cPLA2 is involved in the production of PAF and that bidirectional crosstalk can occur between the EGF and the PAF receptors.
Materials and methods
Cell culture and chemical reagents
The ovarian cancer cell lines CAOV3 and SKOV3 (obtained from the Cell Bank of the Chinese Academy of Science, Shanghai, China) were maintained at 37°C in a humidified 5% CO2 atmosphere in RPMI-1640 medium with 10% fetal calf serum (Gibco, Invitrogen, Carlsbad, CA), 100 IU/ml penicillin G, and 100 mg/ml streptomycin sulfate (Sigma-Aldrich, St. Louis, MO). Cells were serum starved by incubation in serum-free medium for 12–24 hours before the start of the experiments. Lipofectamine 2000 Transfection Reagent and Opti-MEM-1 Medium (Invitrogen, Carlsbad, CA) were used for plasmid and siRNA transfection. The vector encoding cPLA2 and cPLA2-targeted siRNA were synthesized by Shanghai GenePharma Co. AG1478 (EGFR inhibitor) [20] and WEB2086 (PAFR inhibitor) were purchased from Sigma-Aldrich (St Louis, MO). PD98059 (ERK inhibitor) and LY294002 (PI3K inhibitor) were obtained from Cell Signaling Technology (Boston, MA). Rabbit polyclonal antibodies that were used in this study were directed against phospho/total-EGFR, phospho/total-PLCβ, phospho/total-cPLA2, phospho/total-Akt, and phospho/total-ERK. All the antibodies were purchased from Cell Signaling Technology Co. The mouse monoclonal antibodies that were used in this study were directed against actin (Sigma, Missouri, USA).
Western blot analysis
Cellular extracts were prepared in modified radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, protease inhibitor cocktail). Protein concentrations of cellular extracts were measured using a Bio-Rad protein assay kit. Then, cellular extracts were subjected to SDS-PAGE. Proteins were transferred to PVDF membranes. After blocking for 1 h at room temperature in 5% BSA, blots were probed with the primary antibody at a 1:1000 dilution and incubated overnight at 4°C. Subsequently, blots were washed three times and incubated for 1 h at room temperature with a 1:10000 dilution of secondary peroxidase-conjugated antibodies. Following three washes, immunoreactive bands were detected using electrochemiluminescence (ECL).
Transfection with cPLA2 overexpression vector
Cells were seeded and grown to approximately 40% confluence. Cells were then transfected with the CMV-MCS-EGFP-SV40-Neomycin-cPLA2 expression vector using Lipofectamine2000. Briefly, cells were transfected for 48 hours with 1 μg of DNA and were then serum-starved for 12 hours before experimentation.
Incubation with small interference RNA for cPLA2
Cells were seeded and grown to approximately 40% confluence. Cells were incubated with 50 nM siRNA for cPLA2 or non-target siRNA, using Lipofectamine 2000 for 48 hours, based on the manufacture’s protocol. Cells were then serum starved for 12 hours before experimentation.
PAF assay
Aliquots of the supernatant from stimulated ovarian cancer cells were collected and the PAF concentrations were measured using a specific enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI). The assay was performed according to the manufacturer’s instructions. PAF production was evaluated in duplicates, and the concentrations were determined from a standard curve of PAF. The sensitivity of the assay allowed for the detection of up to 15 pg/ml. When necessary, the samples were diluted in the assay buffer.
Immunocytochemistry
After the drug treatment, the cells were fixed with 100% methanol for 6 min at −20°C, then washed with PBS and left at 4°C until use. Cells were permeabilized by incubation in PBS containing 0.3% Triton X-100 and 5% goat serum for 30 min. A polyclonal antibody against phospho-cPLA2 was used at a 1:100 dilution, and a secondary antibody FITC-conjugated goat anti-rabbit (Invitrogen, Carlsbad, CA), was used at a 1:200 dilution. The first antibody was incubated overnight at 4°C and the second antibody for 2 hours at RT. Images were captured with an Olympus DP 71 camera (Tokyo, Japan). The magnification level was 400 ×.
Statistical analysis
All experiments were performed at least three times. The data are expressed as the mean ± SD. Wherever appropriate, the data were also subjected to unpaired, two-tailed Student’s t-tests. Differences were considered significant when P < 0.05.
Results
Effects of EGF on PAF production in ovarian cancer cells
As shown in Figure 1, in CAOV3 ovarian cancer cell lines, extracellular EGF caused a significant rise in the PAF released from 0.5 ng/ml to 100 ng/ml; while in SKOV3 ovarian cancer cell lines, ext racellular EGF caused a significant rise in the PAF release from 1 ng/ml to 100 ng/ml. The maximum effect was reached with 25 ng/ml of EGF (1.952 ± 0.9-fold) in CAOV3 (Figure 1A) cells and 10 ng/ml EGF (1.414 ± 0.3-fold) in SKOV3 cells (Figure 1B). Both in CAOV3 and SKOV3 cells, PAF production increased after 20 min of stimulation with EGF (10 ng/ml) and continued to rise to a maximum after 1 h. Longer stimulation of EGF (24 h) caused no significant additional increase in PAF release over that obtained at 1 h (Figure 1C and D). Together, our data indicated that EGF stimulates PAF production in two human ovarian cancer cell lines.
Figure 1.

Effects of epidermal growth factor (EGF) on platelet-activating factor (PAF) production in ovarian cancer cells. (A and B) Release of PAF in response to increasing concentrations of EGF for 1 h. CAOV3 and SKOV3 cells were serum starved and then stimulated with indicated concentrations (0.5 to 100 ng/ml) of EGF. (C and D) Time course of PAF increase in response to 10 ng/ml EGF. CAOV3 and SKOV3 cells were treated with EGF for the indicated time (20 min to 24 h) while measuring PAF production. Bars represent the average of triplicates ± S.D.; “*” (p < 0.05) and “**” (p < 0.01) indicate a statistically significant difference compared to the untreated control.
EGFR transactivates PAFR followed by EGF stimulation
Our previous data has shown that PAF can transactivate the EGFR, and EGF and PAF are shown to activate many of the same intracellular signaling pathways. Conversely, to test whether a growth factor might transactivate PAFR, we first stimulated CAOV3 and SKOV3 cells with 10 ng/ml of EGF for varying times (5 min to 120 min) to observe the change in phosphorylation of EGFR and phosphoinositide-specific phospholipase C-β (PLCβ). PLCβ has been shown to lie downstream of the activated PAFR and, therefore, the phosphorylation of PLCβ indicates the PAFR activation [21-23]. As shown in Figure 2A and B, stimulation with EGF (10 ng/ml) evoked EGFR and PLCβ phosphorylation in a time-dependent manner in CAOV3 and SKOV3 cells. Phosphorylation of EGFR reached maximum activation at 5 min, followed by a subsequent reduction to the baseline by 120 min. Meanwhile, the phosphorylation of PLCβ increased gradually and reached a maximum activation at 120 min. We also observed that in both the cells, the phosphorylation of PLCβ was somewhat slower than the phosphorylation of EGFR after stimulation with EGF, suggesting the mechanism of EGF-induced transactivation of PAFR.
Figure 2.

PAFR-coupled PLCβ phosphorylation induced by EGF in ovarian cancer cells. CAOV3 (A) and SKOV3 (B) cells were treated with a constant dose (10 ng/ml) of EGF for different time intervals, as indicated. Total protein was extracted and analyzed for phospho-EGFR/total-EGFR and phospho-PLCβ/total-PLCβ and was examined by immunoblot analysis. β-actin was used as an internal control. CAOV3 (C) and SKOV3 (D) cells were pretreated with 10 μM AG1478 for 1 h before exposure to 10 ng/ml EGF for 10 min. Total protein was extracted and analyzed for phospho-EGFR/total-EGFR and phospho-PLCβ/total-PLCβ and was examined by immunoblot analysis. β-actin was used as the internal control. Bars represent the average of the triplicates ± S.D.; “*” (p < 0.05) and “**” (p < 0.01) indicate a statistically significant difference compared to the untreated control.
We next investigated whether EGF-induced PAFR transactivation is EGFR-dependent. Serum-starved CAOV3 and SKOV3 cells were treated with AG1478 (10 μM), an EGFR-specific tyrosine kinase inhibitor, for 1 h before exposing them to EGF (10 ng/ml) for 10 min. As shown in Figure 2C and D, AG1478 inhibited the phosphorylation of EGFR and PLCβ, in both cells, with or without EGF stimulation. These results suggest that EGF can induce PAFR transactivation and that PAFR transactivation is EGFR-dependent.
Effects of EGFR and PAFR inhibition on PAF production in ovarian cancer cells
EGF-induced PAF production is presumably mediated through EGFR-mediated activation of phospholipases. To address the potential for crosstalk between EGFR and PAFR, we tested whether EGFR and PAFR activation correlated with increased PAF production. AG1478 (an EGFR-specific tyrosine kinase inhibitor) and WEB2086 (a small molecular inhibitor of PAFR) were used to block EGFR and PAFR activation. CAOV3 and SKOV3 cells were pretreated with 10 μM of AG1478 or 50 μM of WEB2086 or a combination of AG1478 and WEB2086 for 30 min. The cells were then incubated with 10 ng/ml of EGF for 30 min, and PAF production was measured using an ELISA assay. As shown in Figure 3, AG1478 and WEB2086 significantly reduced the EGF-induced increase in PAF levels in both ovarian cancer cell lines. Additive PAF-production-inhibiting effects were observed on the combined inhibition of both receptors. Taken together, these results suggest that both EGFR and PAFR are involved in EGF-induced PAF production.
Figure 3.
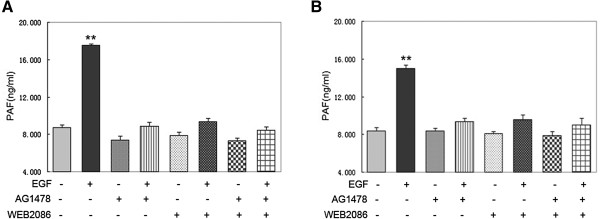
Effects of an EGFR inhibitor and PAFR inhibitor on EGF-induced PAF production. CAOV3 (A) and SKOV3 (B) cells were serum starved and then pretreated with 10 μM AG1478 or 50 μM WEB2086 or a combination of AG1478 and WEB2086 for 30 min. Cells were then stimulated with 10 ng/ml EGF for 30 min. Medium was harvested, and the amount of PAF was measured. In A and B, bars represent the average of triplicates ± S.D.; “*” (p < 0.05) and “**” (p < 0.01) indicate a statistically significant difference compared to the untreated control.
Akt and ERK lie downstream of activated EGFR and PAFR, and ERK is required for activation of cPLA2
We next investigated the signaling pathway downstream of activated EGFR and PAFR in ovarian cancer cells to elucidate the mechanisms involved in EGF-induced PAF production. Western blots using an antibody that specifically recognized the phosphorylated forms of Akt and ERK were used. As shown in Figure 4A and B, exposure to 10 ng/ml of EGF caused the rapid phosphorylation of Akt and ERK in CAOV3 and SKOV3 cells. We then investigated the effects of the EGFR inhibitor, AG1478, and the PAFR inhibitor, WEB2086, to determine whether both EGFR and PAFR were involved in Akt and ERK activation using EGF. Preincubation with 10 μM of AG1478 and/or 50 μM of WEB2086 for 1 h totally prevented the activation of Akt and ERK, following stimulation using 10 ng/ml of EGF for 10 min. These data suggest that EGF activates downstream Akt and ERK signaling via EGFR and PAFR.
Figure 4.

Akt and ERK phosphorylation induced by EGF in ovarian cancer cells and ERK is required for activation of cPLA2. CAOV3 (A) and SKOV3 (B) cells were pretreated with AG1478 (10 μM) and/or WEB2086 (50 μM) for 1 h before exposure to EGF (10 ng/ml) for 10 min. Total protein was extracted and analyzed for phospho-Akt/total-Akt and phospho-ERK/total-ERK and was examined by immunoblot analysis. β-actin was used as a loading control. CAOV3 (C) and SKOV3 (D) cells were pretreated with the ERK inhibitor PD98059 (10 μM) and the Akt inhibitor LY294002 (10 μM) before exposure to EGF (10 ng/ml) for 10 min. Total protein was extracted and analyzed, and phospho-cPLA2/total-cPLA2 was examined by immunoblot analysis. β-actin was used as a loading control. For the immunofluorescence staining of phosphorylated cPLA2 in CAOV3 (E) and SKOV3 (F) cells, after 10 min of incubation without any drug or with 10 ng/ml of EGF or with 10 ng/ml of EGF plus 10 μM of PD98059, cells were labeled with polyclonal antibody to phosphorylated cPLA2 overnight, and then cells were incubated with fluoresent secondary antibody to phospho- cPLA2 for 1 h and stained with DAPI for 10 min.
To further analyze the mechanisms of EGF-induced PAF production, we examined what is required for activation of cPLA2 among the activated downstream signaling pathways. As shown in Figure 4C and D, exposure to EGF caused the significant phosphorylation of cPLA2 in CAOV3 and SKOV3 cells. The activation of cPLA2 was treated using 10 ng/ml of EGF for 10 min. The phosphorylation of cPLA2, stimulated by EGF, could be blocked using by 10 μM of PD98059 (an ERK inhibitor), while no inhibition was observed with LY294002 (10 μM), which binds to the site of PI3K, thereby preventing the activation of target enzymes, such as Akt. Furthermore, the staining intensity of phosphorylated of cPLA2, following 10 min of 10 ng/ml EGF treatment, was much higher in CAOV3 and SKOV3 cells than in control cells, and when cells were pretreated with the ERK inhibitor PD98059, the staining intensity of phosphorylated of cPLA2 was reduced (Figure 4E and F). These data indicate that ERK is required for the phosphorylation of cPLA2 in ovarian cancer cells.
Effects of cPLA2 on EGF-induced PAF production
To determine whether cPLA2 is essential for producing PAF stimulated by EGF in ovarian cancer cells, we first tested for the effects of arachidonyl trifluoromethyl ketone (AACOCF3), a small-molecular inhibitor of cPLA2[24,25] on PAF production. As shown in Figure 5A and B, CAOV3 and SKOV3 cells were pretreated with AACOCF3 (10 μM) for 30 min before exposing the cells to EGF (10 ng/ml) for 30 min. cPLA2 inhibition by AACOCF3 restained PAF production induced by EGF in both cells. To further analyze the effect of cPLA2 inhibition on PAF production, we suppressed cPLA2, through RNA interference in CAOV3 and SKOV3 cells before the stimulation with EGF. Our data suggest that cPLA2-targeted siRNA was able to effectively silence the expression and that the suppression of cPLA2 led to the inhibition of PAF production when cells were exposed to EGF compared to EGF alone (Figure 5C and D). Furthermore, we overexpressed cPLA2 in CAOV3 and SKOV3 cells using an expression vector encoding cPLA2 to determine whether cPLA2 is involved with EGF-induced PAF production. As shown in Figure 5E and F, the vector encoding cPLA2 significantly increased the expression in CAOV3 and SKOV3 cells, and the overexpression of cPLA2 enhanced the PAF production over control cells. However, EGF did not further increase the PAF production in cells overexpressing cPLA2, which may reflect a limit to the amount of PAF that can be produced. Together, our data suggest a role for cPLA2 in EGF-induced production in ovarian cancer cells.
Figure 5.

Role of cytosolic phospholipase A2 (cPLA2) in EGF-induced PAF production in ovarian cancer cells. CAOV3 (A) and SKOV3 (B) cells were serum starved and then pretreated with the cPLA2 inhibitor AACOCF3 (10 μM) for 30 min. Cells were then stimulated with 10 ng/ml EGF for 30 min. Medium was harvested, and the amount of PAF was measured. (C and D) CAOV3 and SKOV3 cells were transfected with a negative-control (NC) or cPLA2-targeted siRNAs. Following 48 h of transfection, cells were treated with EGF (10 ng/ml) for 30 min. Medium was harvested, and the amount of PAF was measured. (E and F) CAOV3 and SKOV3 cells were transfected with the cPLA2 overexpression vector for 48 h. Cells were then treated with EGF (10 ng/ml) for 30 min. Medium was harvested, and the amount of PAF was measured. Bars represent the average of triplicates ± S.D.; “*” (p < 0.05) and “**” (p < 0.01) indicate a statistically significant difference compared to the untreated control.
Discussion
The results of this study demonstrate that EGF stimulates the release of PAF from human ovarian cancer cells by acting on the EGF-receptor and transactivating the PAF-receptor. Stimulation of EGFR and PAFR led to the activation of Akt and ERK, but only the phosphorylation of ERK could stimulate the cPLA2 enzyme, resulting in the production of PAF (Figure 6).
Figure 6.
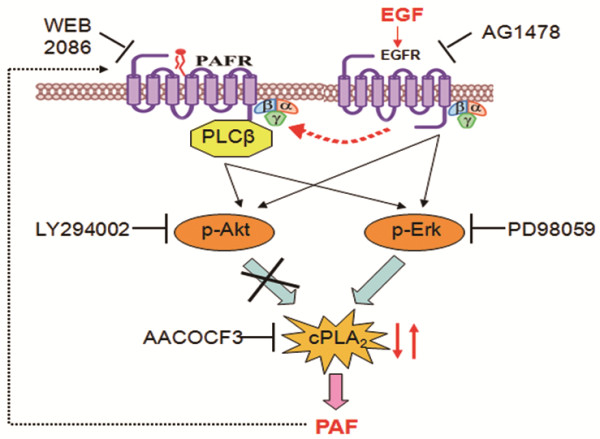
Proposed cascade of the mechanisms involved in PAF production by extracellular EGF in ovarian cancer cells. Extracellular EGF binds to the EGF-receptor (EGFR) and transactivates the PAFR. Activation of EGFR and PAFR stimulates the phosphorylation of Akt and ERK. Only the ERK pathway induces the phosphorylation of cPLA2, the latter increases the production of PAF.
This is the first study to examine the role of EGF, a mitogenic agonist that binds to EGFR, a tyrosine kinase receptor, on PAF production in ovarian cancer cells. Overexpression of EGFR is common in cancers, including 35-70% of ovarian cancers [26-28]. The release of PAF stimulated by EGF can be blocked by AG1478, an inhibitor of EGFR, as expected. Interestingly, WEB2086, an inhibitor of PAFR, also blocks EGF-stimulated PAF production, suggesting that crosstalk between the receptors (EGFR and PAFR) is required. We have previously observed that the tyrosine phospho-activation of protein targets, including EGFR, were significantly increased after PAF treatment, and that phosphorylation was blocked or inhibited by the PAFR antagonist Ginkgolide B using phospho-antibody microarray technologies [18]. Phosphoinositide-specific phospholipase C (PLC) plays a significant role in transmembrane signaling and the subfamily of PLCβ are activated by the G-proteins. Our previous data revealed that PAF can activate PLCβ-dependent PKC and Ca2+ pathways via PAFR to promote ovarian cancer progression. In this study, we demonstrate that EGF stimulates the phosphorylation of PLCβ, which can be blocked by the EGFR inhibitor AG1478, suggesting that the crosstalk occurs bidirectionally between EGFR and PAFR in ovarian cancer cell lines.
PAFR expression is elevated in non-mucinous types of ovarian cancer tissues and cells, suggesting its role in the pathogenesis and progression of ovarian cancer. PAF, the sole ligand of PAFR, is secreted by many different cell types, including endothelial, stromal and inflammatory cells, as well as many different tumor cells [29-31], thus indicating an important role of PAF/PAFR signaling in ovarian cancer progression. In addition, it has been showen that PAF/PAFR significantly promotes ovarian cancer proliferation and invasion [32,33]. The mechanisms, however, in which PAF accumulates in the extracellular space to activate PAFR is still unknown. PAF may be produced within the cell membrane and then exported out of the cell, or it may be synthetized extracellularly. In the present study, we observed that EGF treatment led to an increased production of PAF. As we have demonstrated that PAF-induced ovarian cancer cell proliferation and invasion is dependent on PAFR, it can be assumed that PAF is an autocrine growth factor for ovarian cancer.
The current study demonstrates that EGF stimulates the phosphorylation of Akt and ERK, which can be blocked by either AG1478, an inhibitor of EGFR, or WEB2086, an inhibitor of PAFR. This suggests that EGFR and PAFR, stimulated by EGF, can potentially activate common downstream intracellular signaling pathways. ERK inhibition with PD98059 completely abolishes the phosphorylation of cPLA2, while the antagonist of Akt had no effect on the activation of cPLA2, suggesting that the phosphorylation of cPLA2 induced by EGF is ERK-dependent. In rat articular chondrocytes, the phosphorylation of ERK and p38 MAPKs activated cPLA2 and increased PGE2 production, which is another type of lipid mediator, similar to PAF [34]. Phosphorylated ERK in dorsal root ganglion neurons, caused by spinal cord injury, can induce increased levels of PGE2 [35]. Whether EGF could affect the expression of cPLA2 and whether cPLA2 could affect the production of PAF requires further research in ovarian cancer cells.
cPLA2 can be activated by small GTPases, receptor tyrosine kinases, and phosphatidylinositides [36,37]. In this study, we have shown that the phosphorylation of cPLA2 is stimulated by EGF in ovarian cancer cells. Further, we have shown that cPLA2 is likely to be involved in PAF production, as both of the specific cPLA2 inhibitors, AACOCF3 and cPLA2-targeted siRNA, block PAF production, while exogenously added cPLA2 promotes PAF production. The role of cPLA2 in smooth muscle cell spreading and/or migration has also been well documented [38]. The results concerning the role of cPLA2 in EGF-induced PAF production, along with the convergence of signaling molecules on cPLA2, suggest that cPLA2 may be a potential therapeutic target in ovarian cancer.
Conclusions
Taken together, our results identify mechanisms leading to PAF production and reveal a novel autocrine loop in ovarian cancer cells. Extracellular EGF could stimulate the release of PAF, and this signaling pathway depends on the transactivation between EGFR and PAFR. This requires the phosphorylation of ERK and cPLA2, though the activation of Akt is not involved in this pathway.
Abbreviations
PAF: Platelet-activating factor; PAFR: PAF-receptor; EGF: Epidermal growth factor; EGFR: EGF-receptor; ERK: Extracellular-regulated protein kinase; cPLA2: cytosolic phospholipase A2; siRNA: Short interfering RNA; ELISA: Enzyme-linked immunosorbent assay; PLCβ: Phospholipase C-β; AG1478: An EGFR-specific tyrosine kinase inhibitor; WEB2086: A small molecular inhibitor of PAFR; AACOCF3: Arachidonyl trifluoromethyl ketone; PD98059: ERK inhibitor; LY294002: PI3K inhibitor.
Competing interest
The authors indicated that they have no conflicts of interests with regard to the content of this paper.
Authors’ contributions
YY performed the experiments and drafted the manuscript. XYZ and SSH participated in the design of this study. MXZ and QQC participated in the experiments. WJ and CJX contributed to the design of this study, final data analysis and edited the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yi Yu, Email: icefishyuyi@163.com.
Xiaoyan Zhang, Email: xyzhang80@gmail.com.
Shanshan Hong, Email: shanjenqing@aliyun.com.
Mingxing Zhang, Email: zhangmingxing@163.com.
Qingqing Cai, Email: caiqingqing85@gmail.com.
Wei Jiang, Email: jw52317@126.com.
Congjian Xu, Email: xucongjian@gmail.com.
Acknowledgements
This work was supported by National Natural Sciences Foundation of China (81202052) awarded to WJ, and the Medical Pilot Project of the Shanghai Municipal Science and Technology Commission (114119a2300) awarded to WJ.
References
- Quirk JT, Kupinski JM. Chronic infection, inflammation, and epithelial ovarian cancer. Med Hypotheses. 2001;57:426–428. doi: 10.1054/mehy.2001.1326. [DOI] [PubMed] [Google Scholar]
- Willier S, Butt E, Grunewald TGP. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: A focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biol Cell. 2013;105:317–333. doi: 10.1111/boc.201300011. [DOI] [PubMed] [Google Scholar]
- Cai H, Xu Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun Signal. 2013;11(1):31. doi: 10.1186/1478-811X-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima H, Oshima M. The role of PGE (2)-associated inflammatory responses in gastric cancer development. Semin Immunopathol. 2013;35:139–150. doi: 10.1007/s00281-012-0353-5. [DOI] [PubMed] [Google Scholar]
- Stafforini DM, McIntyre TM, Zimmerman GA, Prescott SM. Platelet-activating factor, a pleiotrophic mediator of physiological and pathological processes. Crit Rev Clin Lab Sci. 2003;40:643–672. doi: 10.1080/714037693. [DOI] [PubMed] [Google Scholar]
- Boccellino M, Camussi G, Giovane A, Ferro L, Calderaro V, Balestrieri C, Quagliuolo L. Platelet-activating factor regulates cadherin-catenin adhesion system expression and beta-catenin phosphorylation during Kaposi’s sarcoma cell motility. Am J Pathol. 2005;166:1515–1522. doi: 10.1016/S0002-9440(10)62367-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio G, Lai Y, Granata F, Mounier CM, Nallan L, Oslund R, Leslie CC, Marone G, Lambeau G, Gelb MH, Triggiani M. Expression of phospholipases A (2) in primary human lung macrophages Role of cytolsolic phospholipase A (2)-alpha in arachidonic acid release and platelet activating factor synthesis. Biochim Biophys Acta Mol Cell Biol Lipids. 2009;1791:92–102. doi: 10.1016/j.bbalip.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebmann C. G protein-coupled receptors and their signaling pathways: Classical therapeutical targets susceptible to novel therapeutic concepts. Curr Pharm Des. 2004;10:1937–1958. doi: 10.2174/1381612043384367. [DOI] [PubMed] [Google Scholar]
- Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- Kume K, Shimizu T. Platelet-activating factor (PAF) induces growth stimulation, inhibition, and suppression of oncogenic transformation in NRK cells overexpressing the PAF receptor. J Biol Chem. 1997;272:22898–22904. doi: 10.1074/jbc.272.36.22898. [DOI] [PubMed] [Google Scholar]
- Ibe BO, Portugal AMD, Raj JU. Platelet activating factor (PAF) and ovine fetal pulmonary vascular smooth muscle cell proliferation. FASEB J. 2005;19:A1089–A1089. [Google Scholar]
- Melnikova VO, Villares GJ, Zeidan AA, Wang H, Bar-Eli M. PAR-1 and PAF receptor regulate MCAM/MUC18 expression and contribute to melanoma metastasis. Proc Am Assoc Cancer Res Annu Meet. 2008;49:68–68. [Google Scholar]
- Stafforini DM. Biology of Platelet-activating Factor Acetylhydrolase (PAF-AH, Lipoprotein Associated Phospholipase A (2)) Cardiovasc Drugs Ther. 2009;23:73–83. doi: 10.1007/s10557-008-6133-8. [DOI] [PubMed] [Google Scholar]
- Scott KF, Sajinovic M, Hein J, Nixdorf S, Galettis P, Liauw W, de Souza P, Dong Q, Graham GG, Russell PJ. Emerging roles for phospholipase A (2) enzymes in cancer. Biochimie. 2010;92:601–610. doi: 10.1016/j.biochi.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Role of cytosolic phospholipase A (2) in allergic response and parturition. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- Feng Y, Dai X, Li X, Wang H, Liu J, Zhang J, Du Y, Xia L. EGF signalling pathway regulates colon cancer stem cell proliferation and apoptosis. Cell Prolif. 2012;45:413–419. doi: 10.1111/j.1365-2184.2012.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer D, Steppan I, Wiedemair A, Fleischer M, Marth C, Zeimet AG. The role of E2F3 isoforms in EGF-mediated proliferation in ovarian cancer. Geburtshilfe Frauenheilkd. 2007;67:529–529. [Google Scholar]
- Aponte M, Jiang W, Lakkis M, Li M-J, Edwards D, Albitar L, Vitonis A, Mok SC, Cramer DW, Ye B. Activation of platelet-activating factor receptor and pleiotropic effects on tyrosine phospho-EGFR/Src/FAK/paxillin in ovarian cancer. Cancer Res. 2008;68:5839–5848. doi: 10.1158/0008-5472.CAN-07-5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachogianni IC, Nomikos T, Fragopoulou E, Stamatakis GM, Karantonis HC, Antonopoulou S, Demopoulos CA. Interleukin-1beta stimulates platelet-activating factor production in U-937 cells modulating both its biosynthetic and catabolic enzymes. Cytokine. 2013;63:97–104. doi: 10.1016/j.cyto.2013.04.024. [DOI] [PubMed] [Google Scholar]
- Hsieh CY, Tsai PC, Tseng CH, Chen YL, Chang LS, Lin SR. Inhibition of EGF/EGFR activation with naphtho 1, 2-b furan-4,5-dione blocks. Toxicol In Vitro. 2013;27(1):1–10. doi: 10.1016/j.tiv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Ali H, Fisher I, Haribabu B, Richardson RM, Snyderman R. Role of phospholipase C beta 3 phosphorylation in the desensitization of cellular responses to platelet-activating factor. J Biol Chem. 1997;272:11706–11709. doi: 10.1074/jbc.272.18.11706. [DOI] [PubMed] [Google Scholar]
- Kajiwara N, Sasaki T, Bradding P, Cruse G, Sagara H, Ohmori K, Saito H, Ra C, Okayama Y. Activation of human mast cells through the platelet-activating factor receptor. J Allergy Clin Immunol. 2010;125:1137–1145. doi: 10.1016/j.jaci.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Richardson RM, Ali H, Pridgen BC, Haribabu B, Snyderman R. Multiple signaling pathways of human interleukin-8 receptor A - Independent regulation by phosphorylation. J Biol Chem. 1998;273:10690–10695. doi: 10.1074/jbc.273.17.10690. [DOI] [PubMed] [Google Scholar]
- Chang JY, Tsai P-F. IL-6 release from mouse glia caused by MeHg requires cytosolic phospholipase A (2) activation. Neurosci Lett. 2009;461:85–89. doi: 10.1016/j.neulet.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Berrios S, Gonzalez-Velazquez W, Perez-Sanchez L, Gonzalez-Mendez R, Rodriguez-del Valle N. Cytosolic phospholipase A (2): a member of the signalling pathway of a new G protein alpha subunit in Sporothrix schenckii. BMC Microbiol. 2009;9:100. doi: 10.1186/1471-2180-9-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003;21:2787–2799. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- Nicholson RI, Gee JMW, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37:S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- FischerColbrie J, Witt A, Heinzl H, Speiser P, Czerwenka K, Sevelda P, Zeillinger R. EGFR and steroid receptors in ovarian carcinoma: Comparison with prognostic parameters and outcome of patients. Anticancer Res. 1997;17:613–619. [PubMed] [Google Scholar]
- Braeuer RR, Zigler M, Villares GJ, Dobroff AS, Bar-Eli M. Transcriptional control of melanoma metastasis: The importance of the tumor microenvironment. Semin Cancer Biol. 2011;21:83–88. doi: 10.1016/j.semcancer.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camussi G, Montrucchio G, Lupia E, Soldi R, Comoglio PM, Bussolino F. Angiogenesis induced in vivo by hepatocyte growth factor is mediated by platelet-activating factor synthesis from macrophages. J Immunol. 1997;158:1302–1309. [PubMed] [Google Scholar]
- Li T, Southall MD, Yi QF, Pei Y, Lewis D, Al-Hassani M, Spandau D, Travers JB. The epidermal platelet-activating factor receptor augments chemotherapy-induced apoptosis in human carcinoma cell lines. J Biol Chem. 2003;278:16614–16621. doi: 10.1074/jbc.M211287200. [DOI] [PubMed] [Google Scholar]
- Jiang W, Wang Y-S, Cong Q, Li M-J, Ye B, Xu C-J. Effects and mechanisms of platelet-activating factor on the invasiveness of ovarian cancer cells in vitro. Zhonghua Fu Chan Ke Za Zhi. 2011;46:931–935. [PubMed] [Google Scholar]
- Zhang L, Wang D, Jiang W, Edwards D, Qiu W, Barroilhet LM, Rho J-H, Jin L, Seethappan V, Vitonis A, Wang J, Mok SC, Crum C, Cramer DW, Ye B. Activated networking of platelet activating factor receptor and FAK/STAT1 induces malignant potential in BRCA1-mutant at-risk ovarian epithelium. Reprod Biol Endocrinol. 2010;8:74. doi: 10.1186/1477-7827-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Zhu Y. Signaling Pathways of ATP-Induced PGE2 Release in Spinal Cord Astrocytes are EGFR Transactivation-Dependent. Glia. 2011;59:664–674. doi: 10.1002/glia.21138. [DOI] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E-2 contributes to pain after spinal cord injury. J Neurosci. 2007;27:2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guild SB. Effects of phospholipase A (2) activating peptides upon GTP-binding protein-evoked adrenocorticotrophin secretion. Eur J Pharmacol. 2001;424:163–171. doi: 10.1016/S0014-2999(01)01149-9. [DOI] [PubMed] [Google Scholar]
- Tong LJ, Dong LW, Liu MS. GTP-binding protein mediated phospholipase A (2) activation in rat liver during the progression of sepsis. Mol Cell Biochem. 1998;189:55–61. doi: 10.1023/A:1006804429027. [DOI] [PubMed] [Google Scholar]
- Gluck N, Schwob O, Krimsky M, Yedgar S. Activation of cytosolic phospholipase A (2) and fatty acid transacylase is essential but not sufficient for thrombin-induced smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2008;294:C1597–C1603. doi: 10.1152/ajpcell.00206.2007. [DOI] [PubMed] [Google Scholar]