


Abstract
Nucleosomes contain ∼146 bp of DNA wrapped around a histone protein octamer that controls DNA accessibility to transcription and repair complexes. Posttranslational modification (PTM) of histone proteins regulates nucleosome function. To date, only modest changes in nucleosome structure have been directly attributed to histone PTMs. Histone residue H3(T118) is located near the nucleosome dyad and can be phosphorylated. This PTM destabilizes nucleosomes and is implicated in the regulation of transcription and repair. Here, we report gel electrophoretic mobility, sucrose gradient sedimentation, thermal disassembly, micrococcal nuclease digestion and atomic force microscopy measurements of two DNA–histone complexes that are structurally distinct from nucleosomes. We find that H3(T118ph) facilitates the formation of a nucleosome duplex with two DNA molecules wrapped around two histone octamers, and an altosome complex that contains one DNA molecule wrapped around two histone octamers. The nucleosome duplex complex forms within short ∼150 bp DNA molecules, whereas altosomes require at least ∼250 bp of DNA and form repeatedly along 3000 bp DNA molecules. These results are the first report of a histone PTM significantly altering the nucleosome structure.
INTRODUCTION
Eukaryotic DNA is organized into chromatin, which consists of nucleosomes containing ∼146 bp wrapped ∼1.65 times around H2A, H2B, H3 and H4 histone protein octamers (1). This structure helps organize eukaryotic genomes within the cell nucleus and controls DNA–protein interactions to regulate DNA processing such as transcription, replication and repair. Nucleosome structure and function are regulated by a number of factors including histone posttranslational modifications (PTMs) (2), chromatin remodeling complexes (3,4) and histone chaperones (5,6).
Over 100 histone PTMs have been reported (7). A majority of histone PTMs are located on disordered N-terminal histone tails, which appear to function by providing binding sites for additional protein complexes (8) and by impacting higher order chromatin compaction (9,10). A number of histone PTMs have been identified within the structured nucleosome core (11), including 10–20 histone PTMs that have been identified within the DNA–histone interface (12–15). These are poised to directly alter DNA–histone interactions. Many of these modifications impact nucleosome stability, mobility and unwrapping (16–20), demonstrating that histone PTMs can modulate nucleosome physical properties. Furthermore, lysine acetylation in the nucleosome dyad is reported to facilitate transcription in human cells by destabilizing nucleosomes (21). These studies support an additional model of histone PTM function, in which modifications regulate transcription and DNA repair by directly controlling nucleosome stability and/or dynamics (22).
There have been a number of nucleosome crystal structures solved with histone PTMs (23) and histone mutants, including H3(T118A), H3(T118I) and H3(T118H) (24,25), but none of these studies have demonstrated a significant impact of a single histone residue on the overall nucleosome structure. While these changes appear to influence the free energy of DNA–histone interactions, they have not been reported to alter the overall structure of the nucleosome.
Threonine 118 of histone H3 is located in the DNA–histone interface near the nucleosome dyad symmetry axis (Figure 1A) where significant DNA–histone interactions occur, and was determined by mass spectrometry to be a site of phosphorylation [H3(T118ph)] (12). Genetic studies found that either a glutamic acid or alanine substitution for this residue is lethal in haploid yeast, while low-level expression of H3(T118E) and H3(T118A) resulted in defects in transcriptional silencing (27). In addition, H3(T118I) is a SWI/SNF INdependent (SIN) mutation, where this substitution relieves the requirement of the SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeling complex for mating type switching in budding yeast (28), and either mutation H3(T118I) (29) or H3(T118H) (30) lowers the transcriptional barrier for Pol II. Our recent studies of single nucleosomes containing H3(T118ph) found that this phosphorylation dramatically enhanced nucleosome mobility, reduced DNA–histone binding and facilitated nucleosome disassembly by SWI/SNF (20). This suggested that phosphorylation within the DNA–histone interface significantly impacts nucleosome stability. In addition, we noted that an additional DNA–histone complex was detected by electrophoretic mobility gel shift assay (EMSA) following nucleosome reconstitution; in our previous studies, we purified these away by sucrose gradient centrifugation (20).
Figure 1.

Phosphorylated H3(T118) forms altered DNA–histone complexes. (A) Nucleosome crystal structure (26) with histone H3 in blue and H3(T118ph) in red. (B) Integrated fluorescence intensity of each fraction for sucrose gradient purification of mp2-247 DNA alone (gray), mp2-247 reconstituted with unmodified histone octamer (HO) (red) and mp2-247 reconstituted with HO containing H3(T118ph) (blue). Fraction numbers in (B) correspond to the fractions resolved by EMSA in Supplementary Figure S1C for the H3(T118ph) sample. (C) EMSA of NPS-192 DNAs reconstituted with either unmodified or H3(T118ph) histone octamer. (D) EMSA of DNA–histone complexes with NPS-147 DNAs containing either the mp2 or L. variegatus 5S NPS reconstituted with unmodified, H3(T118E) or H3(T118ph) HO. The gray sphere superimposed on the DNA pictogram indicates a nucleosome and its position on the DNA. (E) EMSA of NPS-187 DNA reconstituted with unmodified, H3(T118E) or H3(T118ph) HO. (F) EMSA of NPS-247 DNAs reconstituted with unmodified HO or HO containing H3(T118ph).
Here, we report EMSA, sucrose gradient sedimentation, thermal disassembly, micrococcal nuclease (MNase) digestion and atomic force microscopy (AFM) measurements of these additional DNA–histone complexes that contain H3(T118ph). These measurements support a model in which phosphorylation of H3(T118) induces the formation of two alternate DNA–histone complexes that involve DNA wrapping around two complete histone octamers arranged edge-to-edge. We observe a ‘nucleosome duplex’ complex that involves two short DNA molecules wrapped around two histone octamers, and an ‘altosome’ complex that involves one DNA molecule wrapping around two histone octamers. We refer to these complexes as nucleosome duplexes and altosomes because of their similarity to structures formed by SWI/SNF chromatin remodeling (31–33). We find that nucleosome duplexes form with short DNA molecules (147, 187 and 247 bp), whereas altosomes form only with DNA molecules longer than 247 bp and repeatedly form on 3 kb DNA molecules. These results suggest that phosphorylation within the nucleosome dyad can significantly impact nucleosome structure, which could play a role in the regulation of RNA transcription and DNA repair.
MATERIALS AND METHODS
DNA constructs
The mp2-147, mp2-187, mp2-192 and mp2-247 DNA molecules were prepared as previously described (34) from the pMP2 plasmid (35). The 5S-147, 5S-187, 5S-192 and 5S-247 molecules were prepared by polymerase chain reaction from the plasmid p5S, in which the mp2 positioning sequence in pMP2 was replaced with the Lytechinus variegatus 5S RNA positioning sequence (36). The 5SX-192, Pho5-N1, Pho5-N2 and Gal4-192 molecules were prepared from plasmids containing the Xenopus borealis 5S RNA positioning sequence (37), the Pho5 promoter containing the first (N1) and second (N2) nucleosome positions of the upstream activator sequence (UASg) (38), and the Gal1-Gal10 UASg containing the nucleosome positioning sequence over the Gal4 binding site (39), respectively. The 601-17mer array was prepared as previously described (16).
Preparation of DNA–histone complexes
Nucleosomes, nucleosome duplexes and altosomes were prepared by salt double dialysis (34). Purified histone octamer and DNA were mixed at a molar ratio of 1:1.25 for single nucleosome positioning sequence (NPS) templates, and at a mass ratio of 1:1.25 and 1:1.33 for 601-2mer and 601-17mer templates, respectively, in 2 M NaCl, 5 mM Tris, pH 8.0, 0.5 mM ethylenediaminetetraacetic acid (EDTA) and 1 mM benzamidine. Samples were dialyzed into 5 mM Tris, pH 8.0, 1 mM EDTA and 1 mM benzamidine and purified by sucrose gradient centrifugation (34). Addition of 0.5 mM MgCl2 was required to maintain stability during the purification of H3(T118ph) nucleosomes. H3(T118ph) was prepared by expressed protein ligation as previously described (20,40). Wild-type histones, H3(T118E), H2A(C0) and H4(C0), were prepared by recombinant expression in Escherichia coli and purified as previously described (41).
Phosphatase treatment
Dependence of nucleosome duplex and altosome formation on H3(T118) phosphorylation was determined by treating unmodified or H3(T118ph)-containing HO with Antarctic phosphatase (AP; New England Biolabs). Octamers at 1 mg/ml were incubated with 4 U/μl AP in 25 mM Tris–HCl, 1 mM MgCl2, 0.1 mM ZnCl2, 1 M NaCl, pH 8.0, at 37°C for 30 min. Reactions were quenched by addition of EDTA to 2.5 mM final concentration. Treated samples were desalted by ZipTipC18 (EMD Millipore) before assay by MALDI-TOF MS (Bruker Daltonics Microflex). Spectra were processed with Savitzky–Golay smoothing and peaks were picked using the Centroid algorithm in Bruker FlexAnalysis software; unprocessed data are shown in Figure 2. Treated samples were used for nucleosome reconstitution with mp2-187 and mp2-247 DNA according to above protocols.
Figure 2.

Formation of nucleosome duplexes and altosomes are H3(T118ph)-dependent. (A) (Top, unmod − AP): mass spectrum of unmodified HO (H3 [M+2H]2+ m/z expected: 7637, observed: 7637), (Middle, T118ph − AP): H3(T118ph) HO before AP treatment (H3(T118ph) [M+2H]2+ m/z expected: 7677, observed 7676) and (Bottom, T118ph + AP) H3(T118ph) HO after AP treatment (H3(T118): ([M+2H]2+ m/z expected: 7637, observed: 7634). (B and C) EMSA of mp2-187 and mp2-247 DNA’s, respectively, reconstituted with unmodified HO without AP treatment (lane 1), H3(T118ph) HO without AP (lane 2), unmodified HO after AP (lane 3) and H3(T118ph) HO after AP (lane 4).
Fluorescence measures of relative histone and DNA content
The ratio of H2A/H2B heterodimer to DNA or H3/H4 tetramer to DNA of unmodified nucleosomes compared with H3(T118ph)-containing nucleosomes, nucleosome duplexes and altosomes were quantified using Cy5-labeled H2A, Alexa488-labeled H4 and Cy3-labeled DNA as follows. H2A(C0-Cy5) and H4(C0-Alexa488) were labeled before refolding into the histone octamer as previously reported (17) (Supplementary Figure S8A). Labeling efficiency was 50–60% as determined by ultraviolet-visible absorbance and MALDI-TOF MS (data not shown); fluorescent histones from the same preparation were used to refold both H3(T118ph)-containing and unmodified octamer to control for labeling efficiency. Fluorescent octamer was reconstituted onto Cy3-end-labeled mp2-147, mp2-187 and mp2-247, resolved by 5% native polyacrylamide gel electrophoresis (PAGE) in 0.3× Tris-borate-EDTA (TBE) at 20 V/cm, imaged on a Typhoon 8600 variable mode imager (GE Healthcare) and quantified using ImageQuant. Alexa488 fluorescence is resolved by 488 nm laser excitation and imaging with a 520 ± 20 nm bandpass emission filter; Cy3 by 532 nm excitation laser and 610 nm ± 20 nm emission filter; Cy5 by 633 nm excitation laser and 670 ± 20 nm emission filter. Reconstituted samples were verified for absence of fluorescence resonance energy transfer between the three fluorescent tags on a Fluoromax 3 spectrofluorometer (Horiba) (Supplementary Figure S8C). To validate that the three fluorescent dyes are spectrally separable by the Typhoon imager, three different DNA molecules were end-labeled with Alexa488, Cy3 and Cy5 and resolved by native PAGE gel before imaging (Supplementary Figure S8B).
Thermal disassembly
Purified nucleosome duplexes and altosomes containing mp2-187 or mp2-247 DNA were diluted to 50 nM in 20 mM Tris (pH 8.0) and heated at 53°C for 0 and 90 min. Reactions were quenched by transfer of 1 μl of the heated nucleosomes into 6 µl of 3% sucrose in 0.2× TBE. Samples were analyzed by PAGE with 0.2× TBE. The gel was pre-run for 3 h before running the samples for 3 h at 20 V/cm at 4°C with continuous buffer recirculation. Gels were imaged with a Typhoon 8600 variable mode imager (GE Healthcare) and quantified by ImageQuant.
Nap1 assembly
Unmodified and H3(T118ph)-containing nucleosomes were assembled from DNA and purified octamer by yNap1. His6-tagged yNap1 (generous gift from Toshio Tsukiyama) was expressed and purified as previously reported (42). DNA at 12.5 ng/μl and HO at 12.5 ng/μl were incubated with 0–7.2 μM yNap1 dimer at 30°C for 60 min in 130 mM NaCl, 0.5 mM MgCl2, 7.5 mM Tris, pH 7.5, 0.25 mM EDTA, 0.25 mM dithiothreitol, 0.1 mg/ml bovine serum albumin, 2.5% glycerol, 0.01% NP40 and 0.01% Tween20. Reactions were then resolved by PAGE with 5% polyacrylamide and 0.3× TBE at 20 V/cm at 4°C with continuous buffer recirculation.
Exonuclease III mapping
The nucleosome positions within the mp2-187 and mp2-247 DNA molecules were determined with Exonuclease III (ExoIII) mapping as previously reported (34). Reactions were carried out in an initial volume of 50 μl with 10 nM nucleosomes and 50 U/ml of ExoIII (New England Biolabs) in 20 mM Tris, 0.5 mM MgCl2, pH 8.0, at 16°C.
MNase footprinting
Histone protection of the mp2-187 and mp2-247 DNA molecules was determined by MNase digestion. Reactions were carried out in an initial volume of 10 μl with 10 nM nucleosomes and 0–40 U/ml of MNase (New England Biolabs) in 20 mM Tris, pH 8.0, 0.5 mM CaCl2 at 37°C to prevent H3(T118ph) nucleosome disassembly. After 20 min, reactions were quenched with 15 mM EDTA final concentration and resolved by PAGE. Gels were stained with SYBR Gold (Invitrogen) and imaged by a Typhoon 8600 variable mode imager (GE Healthcare).
AFM imaging
For imaging of mp2-147 and mp2-247, purified nucleosomes were fixed by dilution to 0.5 nM in 0.5× Tris EDTA, 0.5 mM MgCl2 and 0.01% glutaraldehyde and incubation on ice for 30 min; for 601-2mer and 601-17mer, purified arrays were diluted to 0.5 nM in 0.5× Tris EDTA. Samples were then deposited on poly-d-lysine–treated mica surface as previously described (16). Samples were imaged with a Dimension Icon with ScanAsyst SPM (Bruker) using Peak Force Mode and ScanAsyst Air tips (Bruker) with a scan rate of 1 Hz and 0.1 pN peak force. Images were processed and analyzed with Gwyddion 2.19 open source software.
RESULTS
EMSA analysis indicates that H3(T118ph) induces the formation of nucleosome duplexes and altosomes independent of DNA sequence
During our initial studies of the impact of H3(T118ph) on nucleosome stability and dynamics, we carried out nucleosome reconstitutions by salt dialysis (41). In addition to canonical nucleosomes, we observed the formation of DNA–histone complexes with a significantly altered electrophoretic mobility, which sediment about two times further on a sucrose gradient than canonical nucleosomes (Figure 1 and Supplementary Figure S1), consistent with the formation of a DNA–histone complex twice the size of a canonical nucleosome. We used sucrose gradient centrifugation to separate and purify canonical nucleosomes and these alternate complexes, which allowed us to characterize the impact of H3(T118ph) on the stability, mobility and remodeling of canonical nucleosomes (20). Here, we have investigated the nature of these alternate mobility complexes that were consistently observed by EMSA of nucleosome reconstitutions carried out with H3(T118ph).
Alterations in electrophoretic mobility within polyacrylamide gels are sensitive to DNA length, DNA sequence and position of the histone octamer along the DNA molecule (43,44). To rule out DNA effects on the formation of these alternate mobility complexes, we first investigated the influence of DNA sequence on the electrophoretic mobility of these complexes. We reconstituted nucleosomes with either unmodified H3 or H3(T118ph) onto 192 bp DNA with six different NPS. We consistently observed low mobility bands form with each of the NPS, but only in reconstitutions carried out with H3(T118ph) (Figure 1C). This indicates that the DNA sequence is not responsible for the alteration in mobility or the formation of this low mobility complex, which we refer to as a nucleosome duplex (32).
We also investigated the influence of DNA length on the formation of nucleosome duplexes by salt dialysis with 147, 187 and 247 bp DNA molecules containing a center-positioned mp2 (Figure 1A, Supplementary Figure S1A) or 5S (data not shown) NPS. The mp2 NPS is a variant of the 601 NPS (35,45). We found that the nucleosome duplex is formed irrespective of DNA length (Figure 1D–F). In addition, we observed an additional band in EMSA of samples prepared with 247 bp DNA molecules. This band has a faster electrophoretic mobility than center-positioned nucleosomes. However, although the electrophoretic mobility is similar to that of a depositioned nucleosome, this DNA–histone complex sediments through a sucrose gradient twice as far as canonical nucleosomes, similar to the nucleosome duplex (Supplementary Figure S1). We refer to the high mobility complex as an altosome (31) based on the additional experiments discussed below.
Phosphorylation at H3(T118) is sufficient and necessary for nucleosome duplex and altosome formation
H3(T118) phosphorylation could potentially alter nucleosome formation by generating a misfolded state of the histone octamer structure. To test this model, we treated refolded histone octamer containing H3(T118ph) with AP to remove the phosphate group from T118 within the folded histone octamer. The removal of the phosphate was confirmed by mass spectrometry (Figure 2A). We find that reconstitutions with the dephosphorylated histone octamer (Figure 2B and C) or with H3(T118E) (Figure 1D and E) only formed canonical nucleosomes. This indicates that the phosphate group on H3(T118) is necessary and sufficient for the formation of nucleosome duplexes and altosomes.
The histone–DNA ratio of nucleosome duplexes and altosomes are different
Different ratios of histone proteins within the protein core or relative to DNA within the nucleosome duplex and altosome could be an explanation for the altered electrophoretic mobility and sucrose gradient sedimentation. To investigate this possibility, we used fluorescence to determine the ratio of H2A–H2B heterodimers and H32–H42 tetramers relative to DNA molecules. We labeled H2A with Cy5 and H4 with Alexa488 using cysteine residues inserted at the N-terminus by site-directed mutagenesis. Histone octamers were refolded with fluorophore-labeled H2A and H4 and either unmodified H3 or H3(T118ph) (Supplementary Figure S8A). Following gel filtration purification, nucleosomes were reconstituted with each labeled histone octamer and Cy3-labeled DNA.
The canonical nucleosomes, nucleosome duplexes and altosomes were analyzed by EMSA, and the fluorophore emissions were detected with a Typhoon scanner (Figure 3A–C). We confirmed that none of these complexes undergo fluorescence resonance energy transfer and that each fluorophore was spectrally separable (Supplementary Figure S8B and C). We found that the canonical nucleosomes and nucleosome duplexes showed the same relative emissions ratio for H2A–H2B heterodimer: H32–H42 tetramer:DNA, implying that nucleosome duplexes contain one intact histone octamer per DNA. In contrast, altosomes maintain an equivalent ratio of heterodimers to tetramer, but twice the ratio of H2A–H2B heterodimers and H3–H4 tetramers relative to DNA, equivalent to two intact histone octamers per DNA (Figure 3D).
Figure 3.

Relative DNA and histone content of nucleosome duplexes and altosomes. (A) Crystal structure (26) of nucleosome illustrating 5′ end-labeled DNA with Cy3 (green), Alexa488-labeled H4(C0) (blue) and Cy5-labeled H2A(C0) (red); H3(T118ph) in yellow. The histone tails are flexible and largely unstructured. EMSA of (B) Cy3-labeled mp2-187 and (C) Cy3-labeled mp2-247 DNAs reconstituted with fluorescent-labeled unmodified and H3(T118ph) HO. Each EMSA gel was imaged by the Cy3-DNA label (left, excitation at 532 nm, emission at 610 ± 10 nm), Alexa488-H4 label (middle, excitation at 488 nm, emission at 520 ± 10 nm) and Cy5-H2A label (right, excitation at 633 nm, emission at 670 ± 10 nm). (D) Fluorescence ratio of two fluorophore-labeled components (blue, Cy5-H2A versus Cy3-DNA; orange, Alexa488-H4 versus Cy3-DNA; pink, Cy5-H2A versus Alexa488-H4) for H3(T118ph) canonical nucleosomes (Nuc), nucleosome duplex (ND) and altosomes (Alto) species relative to the same two fluorophore-labeled components of the unmodified nucleosome species for each indicated DNA length in base pairs. Error bars are the standard deviation of three independent reconstitutions.
Nucleosome duplexes are assembled by the Nap1 histone chaperone
Salt dialysis reconstitution is the most common approach to assemble nucleosomes in vitro (41). However, it remained possible that these altered structures might be an artifact of this experimental approach. Therefore, we used the histone chaperone Nap1 to assemble DNA–histone complexes with both unmodified H3 and H3(T118ph). Nap1 can deposit both H32–H42 tetramers and H2A–H2B heterodimers to form nucleosomes in vitro (5,6). We found that deposition of unmodified histones onto mp2-187 by Nap1 results in nucleosomes that have the same electrophoretic mobility as nucleosomes formed by salt dialysis. EMSA of H3(T118ph)-containing complexes deposited by Nap1 onto mp2-187 results in bands with the same electrophoretic mobility as the canonical nucleosomes and the nucleosome duplexes formed by salt dialysis (Figure 4). This result demonstrates that this structure is not an artifact of salt dialysis assembly. We also carried out Nap1-mediated DNA–histone assemblies with mp2-247 DNA. We observe a band with the same mobility as altosomes formed by salt dialysis (Supplementary Figure S2). However, Nap1 assembled unmodified depositioned nucleosomes with similar electrophoretic mobilities to altosomes. Therefore, our data are consistent with altosome formation by Nap1, but we could not explicitly rule out that the band was a depositioned nucleosome.
Figure 4.
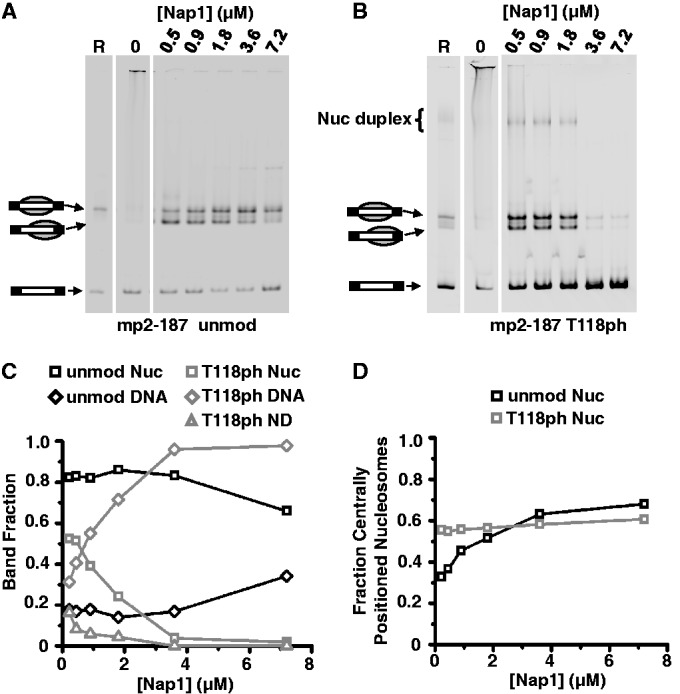
Nucleosome duplexes are assembled by yNap1. EMSA of (A) unmodified HO and (B) H3(T118ph) containing HO reconstituted on mp2-187 by salt dialysis (lane 1, ‘R’), unmodified HO mixed with DNA in the absence of yNap1 (lane 2, ‘0’) and unmodified HO assembled on DNA in the presence of increasing amounts of yNap1 as indicated for each lane. (C) Quantification of the fraction of yNap1 assembled nucleosomes (‘nuc’, squares), nucleosome duplexes (‘ND’, triangles) and remaining free DNA (‘DNA’, diamonds) as a function of [Nap1] for unmodified (black) and H3(T118ph) HO (gray) from the data in (A) and (B). (D) Fraction of centrally positioned versus depositioned nucleosomes assembled by yNap1 as a function of [Nap1] for unmodified nucleosomes (black square) and H3(T118ph) nucleosomes (gray squares).
Nucleosome assembly or disassembly is an equilibrium process that sets up a competition between histone–DNA complexes and histone–Nap1 complexes. The dependence of nucleosome duplex formation on Nap1 concentration can therefore be used as a measure of stability. We find that a significantly lower concentration of Nap1 is required to disassemble canonical nucleosomes when H3(T118ph) is present than when H3 remains unmodified. In fact, while 1.8 µM Nap1 is sufficient for the formation of positioned canonical nucleosomes both with and without H3(T118ph) (Figure 4A and B and Supplementary Figure S2A and B), an increase in Nap1 concentration to ≥3.6 µM maintains well-positioned unmodified canonical nucleosomes while H3(T118ph) structures disassemble (Figure 4C and Supplementary Figure S2C). Second, H3(T118ph)-containing canonical nucleosomes, nucleosome duplexes and altosomes all disassemble at similar concentrations of Nap1 (Figure 4C and Supplementary Figure S2C). This suggests that the nucleosome duplexes and altosomes containing H3(T118ph) have a similar stability to canonical nucleosomes containing H3(T118ph), and that the stability of these complexes is dependent on H3(T118) phosphorylation. These results are compatible with our prior studies of H3(T118ph) canonical nucleosomes (20).
Nucleosome duplexes and altosomes have different thermal stabilities and DNA footprints
We previously found that H3(T118ph) alters the thermal stability and mobility of canonical nucleosomes (20). Therefore, we investigated the stability of nucleosome duplexes to thermal disassembly by incubating them at 53°C and characterizing the products by EMSA. We find that nucleosome duplexes converted to canonical nucleosomes following 30-min incubation at 53°C irrespective of DNA length (Figure 5A and B and Supplementary Figure S3). In contrast, the electrophoretic mobility of altosomes did not change after incubation at 53°C, and we did not observe any separation of altosomes into canonical nucleosomes or free histones and naked DNA (Figure 5C and D). This suggests the altosome species is thermally stable. However, it should be noted that EMSA would not necessarily separate altosome species in which wrapping subtly changed and that the separation of one altosome into two canonical nucleosomes would require acceptor DNA.
Figure 5.

Nucleosome duplexes are decoupled into mononucleosomes. (A) EMSA of purified nucleosome duplexes containing H3(T118ph) HO and mp2-187 after heating at 53°C for the indicated amount of time. Nucleosome duplexes convert to positioned and depositioned mononucleosomes as determined by MNase and ExoIII nucleosome mapping (see Supplementary Figure S4). (B) Quantification of fraction of nucleosome duplexes (squares), nucleosome (circles) and free DNA (diamond) species for the gel in (A) versus time. Error bars are the standard deviation of three independent experiments. (C) EMSA of purified nucleosome duplexes and altosomes containing H3(T118ph) HO and mp2-247 after heating at 53°C for the indicated amount of time. Nucleosome duplexes convert in part to positioned and depositioned mononucleosomes as determined by MNase and ExoIII nucleosome mapping (see Supplementary Figure S5). (D) Quantification of the fraction of nucleosome duplexes (squares), altosomes (triangles), nucleosome (circles) and free DNA (diamond) species for the gel in (C) versus time. Error bars are the standard deviation of three independent experiments.
These thermal disassembly studies suggest that nucleosome duplexes contain miswrapped nucleosomes that convert to correctly wrapped nucleosomes upon heating to 53°C. To investigate these species, we determined the DNA footprint of nucleosome duplexes and altosomes with MNase digestion (Supplementary Figure S4) and ExoIII nucleosome mapping (Supplementary Figures S5 and S6). By each approach, we find that the nucleosome duplex containing H3(T118ph) on 187 bp DNA had a base pair footprint that is similar to the 147 bp footprint of canonical nucleosomes both before and after 30 min at 53°C. In contrast, the DNA–histone complexes formed with H3(T118ph) on 247 bp DNA molecules, which contain both nucleosome duplexes and altosomes after sucrose gradient purification, had both a 147- and 247-bp DNA footprint and increased protection from digestion when compared with canonical nucleosomes formed with unmodified H3 or H3(T118ph) on 247 bp DNA. We confirmed by EMSA that both the nucleosome duplexes and altosomes did not disassemble under the conditions of the digestions (data not shown). These results suggest that the nucleosome duplexes contain DNA that is wrapped around the histone octamer similarly to nucleosomes, while the altosomes contain a DNA organization that appears to be distinct from nucleosomal DNA organization, with a larger DNA footprint.
AFM images of the nucleosome duplexes and altosomes reveal they have twice the volume as canonical nucleosomes
The observation that both nucleosome duplexes and altosomes sediment twice as far within a sucrose gradient as canonical nucleosomes suggests the mass of the particles could be larger than a canonical nucleosome. To investigate the size of nucleosome duplexes and altosomes, we used AFM. We prepared by sucrose gradient purification unmodified nucleosomes, canonical nucleosomes containing H3(T118ph) and a mixture of both nucleosome duplexes and altosomes with mp2-247 DNA (Figure 6) or only nucleosome duplexes with mp2-147 (Supplementary Figure S7) DNA. Because nucleosome duplexes and altosomes sediment similarly through sucrose gradients, they could not be separated and were therefore imaged together by AFM.
Figure 6.

Nucleosome duplexes and altosomes are twice the size of mononucleosomes. AFM images of purified (A) mp2-247 nucleosomes with unmodified HO (247 Unmod Canonical Nucs), (B) mp2-247 nucleosomes with H3(T118ph) HO (247 T118ph Canonical Nucs) and (C) mp2-247 nucleosome duplexes with H3(T118ph) HO (247 T118ph Nuc Duplexes/altosomes). Inset is 60 nm in width. Histograms of (D) surface area and (E) height of mp2-247 containing unmodified nucleosomes (black square, n = 173), H3(T118ph) canonical nucleosomes (blue circle, n = 140) and H3(T118ph) nucleosome duplexes/altosomes (red triangle, n = 175). AFM images of 601-17mer arrays reconstituted with (F) unmodified (11.2 ± 0.5 nucleosomes per molecule, n = 27) and (G) H3(T118ph) HO (7.1 ± 0.4 discrete complexes per molecule, n = 32). Inset is 400 nm in width.
Following AFM imaging of these complexes, we quantified the average area and height of ∼200 particles of each sample type. Analysis of these images demonstrated that the height distribution of unmodified canonical nucleosomes, H3(T118ph) canonical nucleosomes, nucleosome duplexes and altosomes with either mp2-247 or mp2-147 had a maximum of ∼3 nm. This is the canonical mononucleosome height measured by AFM (46). In contrast, we found that the area distribution maximum of nucleosome duplexes and altosomes with mp2-247 was twice as large as both unmodified and H3(T118ph)-containing canonical nucleosomes with mp2-247 (Figure 6). In addition, the area distribution of the nucleosome duplexes with the mp2-147 DNA molecule had two peaks (Supplementary Figure S7). The larger area peak maximum was approximately double the area distribution maximum of canonical nucleosomes. The smaller area peak maximum was equal to the canonical nucleosome distribution maximum. We attribute this second peak to canonical nucleosomes generated by the destabilization of the nucleosome duplexes when they are diluted for AFM imaging, similar to the thermal destabilization observed by EMSA. The observation that nucleosome duplexes and altosomes are the same height and twice the area of canonical nucleosomes implies that the volume of these complexes is double that of canonical nucleosomes. We also found that the shapes of the nucleosome duplexes and altosomes were elliptical, while canonical nucleosomes containing either unmodified H3 or H3(T118ph) were circular.
These results, combined with our observation that nucleosome duplexes can be converted to canonical nucleosomes by heat and contain equal numbers of DNA molecules and histone octamers, suggest that the nucleosome duplexes contain two DNA molecules and two histone octamers, where the two DNA molecules partially wrap around each of the histone octamers. In contrast, the AFM analysis of altosomes combined with the observations that altosomes cannot be converted to canonical nucleosomes, that they have an increased DNA footprint and that they contain twice as many histone octamers as DNA molecules suggests that the altosomes contain one DNA molecule wrapped around two histone octamers.
Altosomes form on 3000 bp DNA molecules
Our observation that increasing the DNA length to 247 bp allowed for two histone octamers to form on one DNA molecule raised the question of whether a further increase in DNA length would result in larger complexes. Therefore, we reconstituted nucleosomes with unmodified H3 or H3(T118ph) on two different extended DNA constructs: either a 364 bp DNA molecule containing two 601-like NPSs (2-mer array, Supplementary Figure S7F), or a 3-kb DNA molecule that contained a tandem repeat of seventeen 601-like NPSs (16) (17-mer array, Supplementary Figure S1A). We analyzed the DNA–histone complexes by AFM. For the 2-mer arrays, we observed two nucleosomes with dimensions similar to mononucleosomes with unmodified H3, but only one DNA–histone complex that was significantly larger than a single nucleosome when H3(T118ph) was used (Supplementary Figure S7G and H). For the 17-mer arrays, we used limiting amounts of histone octamer with a ratio of 1 histone octamer to 2 NPS to prevent aggregation. As anticipated, we find that nucleosomes formed with unmodified histone octamers have dimensions similar to mononucleosomes (Figure 6F). In contrast, the 3-kb DNA molecule reconstituted with H3(T118ph)-containing histone octamer forms numerous DNA–histone complexes that are significantly larger than single nucleosomes (Figure 6G). Interestingly, compared with unmodified H3, fewer H3(T118ph) complexes formed per DNA molecule. This confirms that the altosome structures are not restricted to short DNA segments, and suggests that the altosomes that form on mp2-247 DNA molecules can also form on significantly longer DNA molecules.
DISCUSSION
We find that H3(T118ph) significantly influences DNA wrapping around the histone octamer. We observe two types of altered DNA–histone complexes: a nucleosome duplex with low electrophoretic mobility, and an altosome with a high electrophoretic mobility. However, there are key differences between the nucleosome duplex and the altosome. The nucleosome duplex has the same ratio of histone octamer to DNA, has the same DNA footprint as canonical nucleosomes and can convert to canonical nucleosome when heated. In contrast, the altosome contains two equivalents of histone octamer per DNA, has a larger DNA foot print compared with canonical nucleosomes, and is thermally stable compared with the nucleosome duplex. Both complexes have a similar height but are twice the volume relative to canonical nucleosomes, and have an elliptical shape. We conclude that the nucleosome duplex contains two histone octamers positioned side by side with two DNA molecules wrapped around the two histone octamers, and that the altosome contains one DNA molecule wrapped around two histone octamers in a similar configuration (Figure 7). While nucleosome duplexes containing H3(T118ph) convert to canonical nucleosomes, H3(T118ph) containing altosomes do not. This suggests that the altosome structure containing H3(T118ph) is energetically favorable relative to canonical nucleosomes containing H3(T118ph), or is kinetically trapped in that altosome state. This work appears to represent the first direct demonstration of a single histone PTM causing a large alteration in nucleosome structure.
Figure 7.
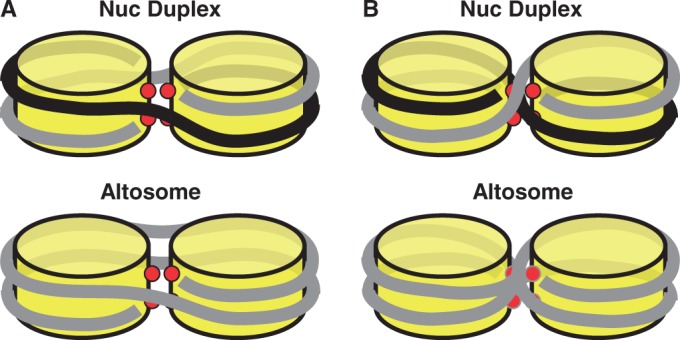
Models of nucleosome duplexes and altosomes. (A) Model of DNA-histone wrapping without DNA crossing of a nucleosome duplex containing two DNA molecules and two histone octamer components and an altosome containing one DNA molecules and two histone octamers. (B) Model of DNA–histone wrapping with DNA crossings for a nucleosome duplex and an altosome.
The DNA–histone interactions near the nucleosome dyad contribute significantly to nucleosome stability (34), with implications for nucleosome assembly and disassembly (16,21). Our results suggest that in addition to an impact on nucleosome stability, modified contacts within the dyad region can influence nucleosome structure. The DNA phosphate backbone is within 3 Å of the side chain hydroxyl of H3(T118) (26), such that the addition of a phosphate at this residue could introduce a strong electrostatic repulsion between the phosphate on the threonine and the DNA phosphate, in addition to a steric conflict between the protein and DNA phosphate moieties. The alternate structures are stabilized by submillimolar concentrations of Mg2+; while metal ions typically stabilize compacted chromatin through interactions with the phosphate backbone, it is also possible that recruitment of a metal ion by phosphothreonine could further alter local contacts in this region. In prior work, we demonstrated a dramatic destabilization of canonical nucleosomes bearing this phosphate modification (20). Notably, a Thr to Glu substitution is insufficient to replicate the effects of phosphothreonine on either canonical nucleosome destabilization in our prior studies, or the formation of nucleosome duplexes and altosomes observed here, suggesting that electrostatic effects are not sufficient and the steric effect or other specific properties of the side chain phosphorylation are necessary.
Our results suggest that in the nucleosome duplexes and altosomes, electrostatic and steric clash between the phosphothreonine side chain and the DNA is avoided by partial wrapping of the DNA around two separate histone octamers aligned edge-to-edge. For short DNA molecules, our results suggest that two DNA molecules are required to maintain DNA–histone contacts, while DNA molecules of ∼250 bp and longer can do this with one DNA molecule (Figure 7). As the DNA extends from one octamer to the other the DNA could remain on the outside of the complex (Figure 7A) or could cross between octamers (Figure 7B). The complexes that do not involve DNA crossing are not topologically constrained and reduce potential steric interactions between DNA stands. Therefore, we anticipate that complexes without DNA crossings to be energetically preferred.
Alternate large DNA–histone complexes have previously been observed in vitro. Chromatin remodeling by SWI/SNF and Remodels the Structure of Chromatin (RSC) can result in nucleosome disassembly or sliding, but also in persistent altered histone-DNA particles (31–33,47–49). Remodeling of single nucleosomes results in the formation of a nucleosome duplex that has similar characteristics to the nucleosome duplex formed with H3(T118ph) (31,33). Nucleosome arrays formed with longer DNA molecules can be converted by SWI/SNF to arrays of structures termed as altosomes that contain one DNA molecule wrapped around two histone octamers (31). AFM images of altosomes formed by chromatin remodeling appear to be similar to the structures formed with H3(T118ph) (50) such that we categorize the altered structures that we observe as altosomes, although their formation is not dependent on chromatin remodeling activity. The in vivo role of altosomes remains unclear, with competing views of these structures as kinetic intermediates (51,52) or as functional products of SWI/SNF-induced chromatin remodeling, for example, with altered DNA site exposure to transcription factor binding (32). In this context, the thermal stability of H3(T118ph) altosome structures (Figure 5) stands in contrast to the increased rate of H3(T118ph) nucleosome disassembly by SWI/SNF in our previous studies (20).
Dinucleosome-like particles have also been reported to form by manipulating the DNA sequence to overlap two strong NPS by 44 bp to simulate invasion of a neighboring nucleosome, for example, during chromatin remodeling (53). These cylindrical particles contain two H3/H4 tetramers, but only three H2A/H2B heterodimers, protect ∼250 bp of DNA and are thought to stack histone octamers face-to-face with an extended superhelical DNA wrapping to generate a cylindrical particle. However, these structures are structurally distinct from nucleosome duplex and altosomes structures formed by either chromatin remodeling or H3(T118ph).
Currently, the in vivo functions of histone PTMs near the nucleosome dyad are not well understood. Acetylation of H3(K115) and H3(K122), histone PTMs known to occur in vivo (12) near the nucleosome dyad, has been shown to destabilize nucleosomes in vitro (16,34). Following these reports, it was then found that nucleosome destabilization by H3(K122) is a significant regulator of transcription (21), demonstrating an in vivo function of nucleosome dyad modifications. Interestingly, mass spectrometry indicates that H3(T118ph) occurs with H3(K122) acetylation (12). These studies combined with our results suggest that H3(T118ph) could function with H3(K122) acetylation to destabilize the nucleosome structure and form altosomes to regulate transcription. Recent reports suggest that acetylation of the centromeric H3 homolog CENP-A at K124 in the dyad is correlated with conversion of octameric nucleosomes in the centromere to a four-histone hemisome structure, which is hypothesized to play a role in regulation of DNA replication (46). Taken in aggregate, these studies hint at major roles for the dyad region in regulating aspects of nucleosome structure. However, in vivo studies similar to the report on H3(K122) acetylation (21) will be required to determine the physiological function of H3(T118ph).
There are four additional histone residues within the DNA–histone interface that have been identified as phosphorylation sites (12–15). To date, there is little understanding of these buried phosphorylations. Future studies will be required to determine whether these additional histone PTMs impact nucleosome structure similarly to H3(T118ph) and how these modifications function in vivo. Our studies here indicate that phosphorylation in the DNA–histone interface have the potential to significantly impact nucleosome structure and DNA wrapping.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
American Heart Association Predoctoral Fellowship [0815460D to J.A.N., 10PRE3150036 to A.M.M.]; OSUCCC and the James Pelotonia Fellowship [to J.A.N.]; American Cancer Society [IRG-6700344, seed funding to J.J.O.]; National Institutes of Health (NIH) [GM083055 to M.G.P. and J.J.O.]; Career Award in the Basic Biomedical Sciences from the Burroughs Wellcome Fund [to M.G.P.]; National Science Foundation [MCB0845695 to J.J.O.]; Human Frontier Science Program [RGY0057/2009 to J.v.N.]. Funding for open access charge: NIH [R01GM083055].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Karolin Luger, Mekonnen Dechassa and members of the Ottesen and Poirier laboratories for helpful discussions.
REFERENCES
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Ann. Rev. Biochem. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- 3.Hota SK, Bartholomew B. Diversity of operation in ATP-dependent chromatin remodelers. Biochim. Biophys. Acta. 2011;1809:476–487. doi: 10.1016/j.bbagrm.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Ann. Rev. Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 5.Park YJ, Luger K. Histone chaperones in nucleosome eviction and histone exchange. Curr. Opin. Struct. Biol. 2008;18:282–289. doi: 10.1016/j.sbi.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avvakumov N, Nourani A, Cote J. Histone chaperones: modulators of chromatin marks. Mol. Cell. 2011;41:502–514. doi: 10.1016/j.molcel.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res. 2011;21:564–578. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 10.Li G, Reinberg D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011;21:175–186. doi: 10.1016/j.gde.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mersfelder EL, Parthun MR. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res. 2006;34:2653–2662. doi: 10.1093/nar/gkl338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Eugeni EE, Parthun MR, Freitas MA. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma. 2003;112:77–86. doi: 10.1007/s00412-003-0244-6. [DOI] [PubMed] [Google Scholar]
- 13.Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hurd PJ, Bannister AJ, Halls K, Dawson MA, Vermeulen M, Olsen JV, Ismail H, Somers J, Mann M, Owen-Hughes T, et al. Phosphorylation of histone H3 Thr-45 is linked to apoptosis. J. Biol. Chem. 2009;284:16575–16583. doi: 10.1074/jbc.M109.005421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tweedie-Cullen RY, Brunner AM, Grossmann J, Mohanna S, Sichau D, Nanni P, Panse C, Mansuy IM. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS One. 2012;7:e36980. doi: 10.1371/journal.pone.0036980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, Poirier MG. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl Acad. Sci. USA. 2011;108:12711–12716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J. Mol. Biol. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 20.North JA, Javaid S, Ferdinand MB, Chatterjee N, Picking JW, Shoffner M, Nakkula RJ, Bartholomew B, Ottesen JJ, Fishel R, et al. Phosphorylation of histone H3(T118) alters nucleosome dynamics and remodeling. Nucleic Acids Res. 2011;39:6465–6474. doi: 10.1093/nar/gkr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, Li G, Mittler G, Liu ET, Buhler M, et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. 2013;152:859–872. doi: 10.1016/j.cell.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 22.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004;11:1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 23.Lu X, Simon MD, Chodaparambil JV, Hansen JC, Shokat KM, Luger K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol. 2008;15:1122–1124. doi: 10.1038/nsmb.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwasaki W, Tachiwana H, Kawaguchi K, Shibata T, Kagawa W, Kurumizaka H. Comprehensive structural analysis of mutant nucleosomes containing lysine to glutamine (KQ) substitutions in the H3 and H4 histone-fold domains. Biochemistry. 2011;50:7822–7832. doi: 10.1021/bi201021h. [DOI] [PubMed] [Google Scholar]
- 25.Muthurajan UM, Bao Y, Forsberg LJ, Edayathumangalam RS, Dyer PN, White CL, Luger K. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO J. 2004;23:260–271. doi: 10.1038/sj.emboj.7600046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 27.Hyland EM, Cosgrove MS, Molina H, Wang D, Pandey A, Cottee RJ, Boeke JD. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol. Cell. Biol. 2005;25:10060–10070. doi: 10.1128/MCB.25.22.10060-10070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruger W, Peterson CL, Sil A, Coburn C, Arents G, Moudrianakis EN, Herskowitz I. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995;9:2770–2779. doi: 10.1101/gad.9.22.2770. [DOI] [PubMed] [Google Scholar]
- 29.Hsieh FK, Fisher M, Ujvari A, Studitsky VM, Luse DS. Histone Sin mutations promote nucleosome traversal and histone displacement by RNA polymerase II. EMBO Rep. 2010;11:705–710. doi: 10.1038/embor.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bintu L, Ishibashi T, Dangkulwanich M, Wu YY, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal elements that control the topography of the barrier to transcription. Cell. 2012;151:738–749. doi: 10.1016/j.cell.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulyanova NP, Schnitzler GR. Human SWI/SNF generates abundant, structurally altered dinucleosomes on polynucleosomal templates. Mol. Cell. Biol. 2005;25:11156–11170. doi: 10.1128/MCB.25.24.11156-11170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulyanova NP, Schnitzler GR. Inverted factor access and slow reversion characterize SWI/SNF-altered nucleosome dimers. J. Biol. Chem. 2007;282:1018–1028. doi: 10.1074/jbc.M609473200. [DOI] [PubMed] [Google Scholar]
- 33.Schnitzler G, Sif S, Kingston RE. Human SWI/SNF interconverts a nucleosome between its base state and a stable remodeled state. Cell. 1998;94:17–27. doi: 10.1016/s0092-8674(00)81217-9. [DOI] [PubMed] [Google Scholar]
- 34.Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. Acetylation of histone H3 at the nucleosome dyad alters DNA-histone binding. J. Biol. Chem. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poirier MG, Bussiek M, Langowski J, Widom J. Spontaneous access to DNA target sites in folded chromatin fibers. J. Mol. Biol. 2008;379:772–786. doi: 10.1016/j.jmb.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simpson RT, Stafford DW. Structural features of a phased nucleosome core particle. Proc. Natl Acad. Sci. USA. 1983;80:51–55. doi: 10.1073/pnas.80.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhodes D. Structural analysis of a triple complex between the histone octamer, a Xenopus gene for 5S RNA and transcription factor IIIA. EMBO J. 1985;4:3473–3482. doi: 10.1002/j.1460-2075.1985.tb04106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergman LW. A DNA fragment containing the upstream activator sequence determines nucleosome positioning of the transcriptionally repressed PHO5 gene of Saccharomyces cerevisiae. Mol. Cell. Biol. 1986;6:2298–2304. doi: 10.1128/mcb.6.7.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fedor MJ, Lue NF, Kornberg RD. Statistical positioning of nucleosomes by specific protein-binding to an upstream activating sequence in yeast. J. Mol. Biol. 1988;204:109–127. doi: 10.1016/0022-2836(88)90603-1. [DOI] [PubMed] [Google Scholar]
- 40.Shimko JC, Howard CJ, Poirier MG, Ottesen JJ. Preparing semisynthetic and fully synthetic histones h3 and h4 to modify the nucleosome core. Methods Mol. Biol. 2013;981:177–192. doi: 10.1007/978-1-62703-305-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 42.McBryant SJ, Park YJ, Abernathy SM, Laybourn PJ, Nyborg JK, Luger K. Preferential binding of the histone (H3-H4)2 tetramer by NAP1 is mediated by the amino-terminal histone tails. J. Biol. Chem. 2003;278:44574–44583. doi: 10.1074/jbc.M305636200. [DOI] [PubMed] [Google Scholar]
- 43.Flaus A, Owen-Hughes T. Dynamic properties of nucleosomes during thermal and ATP-driven mobilization. Mol. Cell. Biol. 2003;23:7767–7779. doi: 10.1128/MCB.23.21.7767-7779.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pennings S. Nucleoprotein gel electrophoresis for the analysis of nucleosomes and their positioning and mobility on DNA. Methods. 1997;12:20–27. doi: 10.1006/meth.1997.0443. [DOI] [PubMed] [Google Scholar]
- 45.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 46.Bui M, Dimitriadis EK, Hoischen C, An E, Quenet D, Giebe S, Nita-Lazar A, Diekmann S, Dalal Y. Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell. 2012;150:317–326. doi: 10.1016/j.cell.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe CE, Narlikar GJ. The ATP-dependent remodeler RSC transfers histone dimers and octamers through the rapid formation of an unstable encounter intermediate. Biochemistry. 2010;49:9882–9890. doi: 10.1021/bi101491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorch Y, Zhang M, Kornberg RD. RSC unravels the nucleosome. Mol. Cell. 2001;7:89–95. doi: 10.1016/s1097-2765(01)00157-5. [DOI] [PubMed] [Google Scholar]
- 49.Lorch Y, Cairns BR, Zhang M, Kornberg RD. Activated RSC-nucleosome complex and persistently altered form of the nucleosome. Cell. 1998;94:29–34. doi: 10.1016/s0092-8674(00)81218-0. [DOI] [PubMed] [Google Scholar]
- 50.Schnitzler GR, Cheung CL, Hafner JH, Saurin AJ, Kingston RE, Lieber CM. Direct imaging of human SWI/SNF-remodeled mono- and polynucleosomes by atomic force microscopy employing carbon nanotube tips. Mol. Cell. Biol. 2001;21:8504–8511. doi: 10.1128/MCB.21.24.8504-8511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lorch Y, Maier-Davis B, Kornberg RD. Chromatin remodeling by nucleosome disassembly in vitro. Proc. Natl Acad. Sci. USA. 2006;103:3090–3093. doi: 10.1073/pnas.0511050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu N, Balliano A, Hayes JJ. Mechanism(s) of SWI/SNF-induced nucleosome mobilization. Chembiochem. 2011;12:196–204. doi: 10.1002/cbic.201000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engeholm M, de Jager M, Flaus A, Brenk R, van Noort J, Owen-Hughes T. Nucleosomes can invade DNA territories occupied by their neighbors. Nat. Struct. Mol. Biol. 2009;16:151–158. doi: 10.1038/nsmb.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.