


Abstract
Natural transformation contributes to the maintenance and to the evolution of the bacterial genomes. In Streptococcus pneumoniae, this function is reached by achieving the competence state, which is under the control of the ComD−ComE two-component system. We present the crystal and solution structures of ComE. We mimicked the active and non-active states by using the phosphorylated mimetic ComED58E and the unphosphorylatable ComED58A mutants. In the crystal, full-length ComED58A dimerizes through its canonical REC receiver domain but with an atypical mode, which is also adopted by the isolated RECD58A and RECD58E. The LytTR domain adopts a tandem arrangement consistent with the two direct repeats of its promoters. However ComED58A is monomeric in solution, as seen by SAXS, by contrast to ComED58E that dimerizes. For both, a relative mobility between the two domains is assumed. Based on these results we propose two possible ways for activation of ComE by phosphorylation.
INTRODUCTION
Bacteria sense and respond to the fluctuations of their environment by the use of two-component signaling systems (TCS). A prototypical TCS consists of a membrane-integrated histidine kinase (HK), which perceives a stimulus, and a cytoplasmic response regulator (RR), which mediates the output signal, often an alteration in gene expression (1). The HK transfers information via an auto-phosphorylation step followed by the transphosphorylation of the RR. Many 3D structures of full-length RR are available: PhoP, DrrB, MtrA that belong to the OmpR/PhoB family (2–4), RocR from Pseudomonas aeruginosa (5), that is dedicated to the cyclic di-GMP signaling and belongs to the 13% of effector domains that perform an enzymatic activity, as some examples. Several structures of RR dimers are also available (2,6–9).
RRs are composed of a canonical conserved N-terminal receiver REC domain (Pfam00072, Flavodoxin-like-fold) that is phosphorylated on an aspartate residue by the dedicated HK sensor (10). A fundamental concept of phosphorylation-mediated signaling is the precise switching between discrete functional conformations (11,12). According to the traditional view, phosphorylation stabilizes an alternate conformation. But it has been also demonstrated by dynamic NMR experiments that, before becoming phosphorylated, isolated REC domains can switch between two conformations, corresponding to the inactive and active states (13).
The second domain of the RR family is the variable C-terminal effector domain. Mostly involved in DNA-binding functions, the structural variability of the effector domains reflects the great functional diversity of the output responses controlled by the REC domain. Gao et al. showed by FRET that phosphorylation-mediated dimerization is a common mechanism for OmpR/PhoB subfamily members (14). Dimerization of the RR is usually considered necessary for transcription regulation in order to recognize their double-site promoters. For some of the RR proteins, dimerization is only induced upon highly cooperative binding to DNA. In the case of PhoP from Salmonella typhimurium, the full-length non-phosphorylated protein is a monomer in solution and binds the two-site DNA-specific box in a step-wise manner, with a second PhoP molecule binding weakly (15). A moderate increase in PhoP concentration can promote its dimerization on the DNA, but this could also be achieved by phosphorylated PhoP at much lower protein concentration. However PhoP from Mycobacterium tuberculosis, in the common active state, crystallize as a dimer via the REC domains whereas the two effector domains are not constrained and the in between linkers are flexible (2).
Natural genetic transformation is a mechanism developed by bacteria for the uptake of naked extracellular DNA and integration into their genome by homologous recombination, leading to genetic variability. A physiological state called competence has to be reached in order to perform transformation. In Streptococcus pneumoniae the induction of the competence occurs at a specific cell density during logarithmic growth, without perturbing the growth rate (16,17). It is believed to account for serotype switching, evolution of virulence factors and rapid emergence of antibiotic resistance of this major human pathogen (18,19). Competence develops in S. pneumoniae for a brief period of ∼20 min, under control of a quorum-sensing signaling pathway involving the TCS ComD−ComE (20,21) and the auto-synthesized competence-stimulating peptide CSP pheromone (22,23). The trans-membrane HK ComD senses the extracellular concentration of CSP (20) leading to autophosphorylation on H248, probably via a trans-mechanism. The phosphoryl group is subsequently transferred to D58 of the RR ComE, leading to its activation and to the induction of the early genes of the comAB, comCDE and comX operons (24–27). The promoter regions of the three operons are organized as two imperfect direct repeat motifs DR1 and DR2 of 9 base pairs each, separated by a 12-mer linker. The comAB operon encodes the machinery required for the maturation and the export of the pre-CSP, while the activation of the comCDE operon creates an auto-catalytic, rapid and synchronous activation of the competence (23). The comX gene, coding an alternative sigma-factor (24,28), is the unique link to competence-specific genes (29,30). It activates the so-called late genes required for the uptake of the external DNA (31–33) and for the integration of this DNA into the genome by homologous recombination (26,34–36). Expression and maintenance of ComD−ComE has recently been studied in S. pneumoniae (37), both under CSP-induced and under basal conditions. The basal conditions require ComD and a phosphate-accepting form of ComE but not the CSP, suggesting that ComD can phosphorylate ComE even in absence of CSP. A ComED58A mutant, that is a non-phosphorylatable form of the RR, abolishes the basal comCDE expression. A contrario a phosphorylmimetic ComED58E mutant expressed in ΔcomD S. pneumoniae cells displays full spontaneous competence (27,38). Finally contrary to ComED58A, ComED58E strongly interacts in yeast two-hybrid experiments with DprA, the transformation-dedicated loader of RecA, suggesting the involvement of DprA in the shut-off of the competence in S. pneumoniae via an interaction preferentially with the phosphorylated form of ComE, that could lead to the blockage of the early genes (38). These two mutants of ComE, ComED58A and ComED58E, even if their in vivo activity can not be directly correlated with active or inactive conformations in vitro, are the focus of the present article.
ComE belongs to the AlgR/AgrA/LytR transcription factors subfamily (LytTR domain: Pfam04397, OB-fold), with some members involved in pathogenicity (39). Its promoter DNA regions have been identified in S. pneumoniae since some years (24) or more recently in Streptococcus mutans (40). The gene control by the ComD−ComE two-component system in the natural genetic transformation regulation for competence development (30,36,41–43) has been well studied. However, only fragmented knowledge are accumulated for the molecular mechanism of ComE concerning its activation by the dimerization of the REC domains and its binding to DR1 and DR2 direct-repeat DNA sites. The only two structures available for the members of this subfamily are the X-ray structures of the AgrALytTR domain from Staphylococcus aureus bound or not to DNA (PDB ID: 3BS1 and 4G4K) (44–46) and the LytTR DNA-binding domain of a putative methyl-accepting/DNA RR from Bacillus cereus (PDB ID: 3D6W).
To get deeper insight into the molecular mechanisms responsible for the activation of ComE from S. pneumoniae, we have investigated its crystallographic and small angle X-ray scattering (SAXS) structures. We have focused on the ComED58A and ComED58E mutants that mimic the unphosphorylated and phosphorylated states, respectively. We studied both the full-length ComE and isolated REC or LytTR domains. Based on our observations that RECD58A is predominantly a monomer in solution but with a tendency to dimerize, and that RECD58E is a stable dimer, we propose a mechanistic model, in which the phosphorylation by ComD activates ComE by favoring its dimer configuration.
MATERIALS AND METHODS
Cloning, site-directed mutagenesis, expression, labeling and purification of full-length ComE, REC and LytTR isolated domains
The comE gene (NC_003098.1) was amplified by PCR using genomic DNA of S. pneumoniae strain R6 as a template. The PCR product was then cloned into a derivative of pET28 vector. Site-directed mutagenesis creating the D58A and D58E mutants was performed by GeneCust Europe. The fragments coding for the REC [1–137] and the LytTR [138–250] domains were amplified by PCR using ComED58A or ComED58E as a template and were cloned into pET28. An additional sequence coding for a 6-histidine tag was systematically introduced at the 3′-end of the genes during amplifications.
Escherichia coli Gold (DE3) strains were co-transformed by the constructs and by pG-KJE3 to co-express protein chaperones in the case of full-length ComED58A and ComED58E (47) and growth in 2xYT medium (BIO101 Inc.) complemented by kanamycin and chloramphenicol antibiotics. When the cell cultures reached an OD600 nm of 0.6, chaperone expression was induced by arabinose; then at OD600 nm = 1, ComE expression was induced with 0.5 mM IPTG (Sigma) and the cells were grown o/n at 15°C. RECD58A, RECD58E and LytTR isolated domains were expressed in E. coli Gold (DE3) without chaperones. The expression of the proteins was induced during 4 h at 37°C for LytTR and o/n at 15°C for the REC domains. Cells were harvested by centrifugation, resuspended in 40 ml of buffer A (200 mM or 500 mM NaCl, 20 mM Tris–HCl pH 7.5, 5 mM ß-mercaptoethanol, 5% (v/v) glycerol) for, respectively, ComED58A and ComED58E, and in buffer B (200 mM NaCl, 20 mM Tris–HCl pH 7.5, 5 mM ß-mercaptoethanol) for the isolated domains, and stored overnight at −20°C. Cell lyses were completed by sonication (probe-tip sonicator Branson).
The His-tagged proteins were purified on a Ni-NTA column (Qiagen Inc.), eluted with imidazole in buffer A or B; loaded onto a Heparin column (Qiagen Inc.), eluted with a gradient of 200–1500 mM NaCl; and loaded after concentration using Vivaspin 5000 nominal molecular weight limit cut-off centrifugal concentrators (Vivascience), onto a SuperdexTM75 column (Amersham Pharmacia Biotech), equilibrated against 20 mM Tris–HCl pH 7.5 or 50 mM MES pH 6.5, 5 mM ß-mercaptoethanol, 5% (v/v) glycerol, 200 mM NaCl for ComED58A and isolated LytTR, and 500 mM NaCl for ComED58E that was less stable at lower salt concentration. Se-Met-labeled ComED58A was prepared as described in Quevillon-Cheruel et al. (48) and purified as the native protein. The REC-isolated domains were purified on Ni-NTA column and SuperdexTM75 column in buffer B. Proteins were concentrated and flash frozen in liquid nitrogen, and stored at –80°C or dialyzed in a 50% (v/v) glycerol buffer for storage at −20°C.
Crystallization, data collection, model building and refinement
The first crystals of the RECD58A- and RECD58E-isolated domains were obtained in a 1:1 ratio mixture of 8.4 mg/ml and 6.4 mg/ml protein solutions, respectively, in buffer 200 mM NaCl, 20 mM Tris–HCl pH 7.5, 5 mM ß-mercaptoethanol and crystallization liquor of respectively the conditions 69 and 62 of the PEG suite (Qiagen), composed by polyethylene glycol 3350 20% (w/v) and 0.2 M sodium formate for RECD58A and 0.2 M potassium thiocyanate for RECD58E, at 18°C, in 100 nl sitting drops. Crystals appeared within 2–3 days. The RECD58A crystals were optimized by growing from a 1:1 ratio mixture of the same protein solution and crystallization liquor containing polyethylene glycol 3350 25% (w/v) and 0.2 M sodium formate. The crystals were cryo-protected by a brief soaking into the crystallization liquors supplemented with 30% (v/v) glycerol and then flash frozen in liquid nitrogen. Data were collected at 100 K on the Proxima-1 beam line at SOLEIL synchrotron (Saint-Aubin, France) and processed with the XDS package (49). A 3.2 and 2.8Å resolution datasets, respectively, for RECD58A and RECD58E were recorded, belonging to space group P63 with four molecules in both the asymmetric units (Supplementary Table S1). The structures were determined by molecular replacement with PHASER (50) using the REC domain of the following ComED58A full-length structure as a template. Initial refinement was performed using REFMAC of the CCP4 suite (51,52). Later rounds of refinement were performed by series of manual rebuilding with COOT (53) and refinement with PHENIX (54). Validation of the structures was performed using MOLPROBITY (55).
The first crystals of the SeMet-labeled ComED58A were obtained from a 1:1 ratio mixture of 2.5 mg/ml protein solution in buffer 200 mM NaCl, 20 mM Tris–HCl pH 7.5, 5 mM ß-mercaptoethanol, 5% (v/v) glycerol and crystallization liquor of the condition 68 of the JCSG+ Suite (Qiagen), composed of 2.1 M DL-malic acid pH 7.0, at 18°C, in 200 nl sitting drops. The optimized crystals were grown from a 1:1 ratio mixture of 3 mg/ml protein solution in buffer 200 mM NaCl, 20 mM Tris–HCl pH 7.5, 5 mM ß-mercaptoethanol, 5% (v/v) glycerol and crystallization liquor containing 1.8 M DL-malic acid. Crystals appeared within 1–2 days. Crystals were cryo-protected by transfer into FOMBLIN Y LVAC 14/6 oil (Sigma) and then flash-cooled in liquid nitrogen. Data were collected at 100 K on the Proxima-1 beamline at SOLEIL synchrotron (Saint-Aubin, France) and processed with the XDS package (49). A 3.4Å resolution dataset could be recorded from a SeMet crystal, belonging to space group C2221 with six molecules in the asymmetric unit (Supplementary Table S1). The structure was determined by the single-wavelength anomalous-dispersion method using the anomalous signal of selenium (Se) atoms. The program SHELXD was used to find an initial set of 34 Se sites in the 45–5.5Å resolution range (56). Refinement of the Se sites and phasing were carried out with the program SHARP using the SAD dataset (57). The final substructure model comprises 19 Se atoms. The experimental phases allowed initial manual building of α-helices and ß-strands of one molecule using COOT (53). This initial model was used to search for structural homologues in the PDB with the PDBeFold server, the closest results being the REC domain of NtrC4 (PDB ID: 1DC8) and the LytTR domain of AgrA (PDB ID: 3BS1). Molecular replacement with MOLREP (58) found four REC domains and two LytTR domains. This model was completed semi-automatically by the BUCCANEER program (59). Manual building using COOT led to a 70% completed model. This model was combined with the experimental phases using the PHASER program to improve electron density map (50), and submitted to a new series of manual rebuilding with COOT and refinement with PHENIX (54) and BUSTER (60). The program BUSTER was used for the last refinements with TLS (60). Validation of the structure was performed using MOLPROBITY (55). The statistics for data collection and refinement are summarized in Supplementary Table S1. Due to the absence of electron density, the following residues (corresponding to the six added histidine-tag for residues 251–256) were omitted from the final model: 252–256 (chain A), 252–256 (chain B), 250–256 (chain C), 251–256 (chain D), 1 and 250–256 (chain E).
Exploration of the 3D structures was performed using the following tools: Dali server (61), I-TASSER (62) and Swiss-modeling servers (63), PyMOL Molecular Graphics System (http://www.pymol.org) (64).
SAXS measurements and data analysis
The measurements for ComED58A and LytTR were made in 200 mM NaCl, 50 mM MES pH 6.5, 5 or 2% (v/v) glycerol, 5 mM ß-mercaptoethanol. The measurements for ComED58E were made in this buffer but at 500 mM NaCl. The measurements for RECD58A and RECD58E were made in 200 mM NaCl, 20 mM Tris–HCl pH 7.5, 5 mM ß-mercaptoethanol, 5% (v/v) glycerol.
SAXS experiments were carried out on the SWING beamline at the SOLEIL synchrotron radiation facility (Saint-Aubin, France), except those of domains REC that have been carried on the in-house Nanostar instrument (Bruker, Karlsruhe, Germany). The sample to detector (Aviex CCD) distance was set to 1820 mm, allowing useful data collection over the momentum transfer range 0.01Å – 1 < q < 0.5Å – 1 with q = 4πsinθ/λ where 2θ is the scattering angle and λ the wavelength of the X-rays (λ = 1.0Å). The solution was injected into the SAXS flow-through capillary cell (1.5 mm in diameter) under vacuum. In order to use mono-disperse solutions fully devoid of aggregates, SAXS data were collected directly after elution through an on-line size-exclusion high-performance liquid chromatography (SEC-HPLC) column available on SWING (65). Flow rate was 150 µl/min, frame duration was 2 s and the dead time between frames was 0.5 s. For each frame, the protein concentration (between 0.5 and 1 mg/ml at the top of elution peak) was estimated from UV absorption at 280 nm using a spectrometer located immediately upstream of the SAXS measuring cell. Selected identical frames corresponding to the main elution peak were averaged. A large number of frames were collected before the void volume and averaged to account for buffer scattering. SAXS data were normalized to the intensity of the incident beam and background (i.e. the elution buffer) subtracted using the programs FoxTrot (from Swing beamline) and Primus (66). The scattered intensities were displayed on an absolute scale using the scattering by water.
The molar mass M of the scattering objects is usually calculated from the forward (or zero-angle) scattered intensity I(0) which is proportional to M and to the concentration. In the case of ComE which does not contain any tryptophan, the determination of the concentration by UV absorption is difficult. In order to determine unambiguously the oligomeric state of the protein or complex the molar mass was obtained from the whole I(q) curve using the macromolecule volume and the method developed by Craievich’s team (67).
The program BUNCH was used to model the conformations in solution of the monomer of ComED58A by a combination of rigid-body and ab initio modelling approaches (68). The REC domain (residues 1–129) and the LytTR domain (residues 140–250) from the crystallographic structure of molecule A were taken as rigid bodies whereas the linker (residues 130–139) and the His-tag (residues 251–256) were modelled as dummy residues. The program finds the optimal positions and orientations of domains and probable conformations of the linker. The modelling was repeated 10 times. The agreement between experimental data and the scattering curve calculated on the model was the same for all runs. Another approach consists to choose a subset from a large pool of conformations using the program EOM (69). The conformer pool is constructed from the above two domains related by a flexible linker. A genetic algorithm refines the composition of the ensemble so that the average scattering pattern of the conformations within the ensemble fits the experimental curve.
For the dimer of ComED58E we used the program CORAL (EMBL Hamburg, in preparation) which combines the program BUNCH allowing to describe the linkers and the rigid-body modelling program SASREF suitable to determine the quaternary structure of a complex formed by subunits with known atomic structure. During the modeling we have imposed that the two REC domains retain the same dimerization interface as in the crystallographic structure and the LytTR domains are free to move.
In all cases the goodness of fit was characterized by the following parameter,
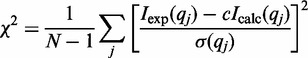 |
where N is the number of experimental points, c is a scaling factor, and Icalc(qj) and σ(qj) are the calculated intensity and the experimental error at the scattering vector qj, respectively.
RESULTS
The isolated RECD58A and RECD58E domains of ComE crystallized both as atypical similar dimers, but are respectively monomeric and dimeric in solution
We solved the X-ray structures of the RECD58A and RECD58E isolated domains at, respectively, 3.2 and 2.8Å resolution. The statistics for data collection and refinement are summarized in Supplementary Table S1. Four identical copies of the domain are present in the asymmetric units of the two crystals. They form in each case two identical dimers with a 2-fold rotational symmetry axis. The dimers are similar for the two mutants, with an RMSD of 0.61Å (Figure 1A and Supplementary Figure S1). The interface [967Å2 buried surface corresponding to 6.5% of the total surface of each REC domain according to the EBI-PISA server (70)] involves the α-helix 4 and the loop between α4 and β5 and is very different from other REC dimer interfaces (Figure 1A). Other dimerization modes for REC domains have been described. For instance the activated REC dimer of the members of the OmpR/PhoB subfamily also possesses a 2-fold rotational symmetry axis with a similar size of interface surface (974Å2/7.5% of each monomer surface) but involves the α4 -β5-α5 motif (Figure 1A) (71). Still another mode of REC domain dimerization is illustrated by the phosphorylated FixJ REC dimer that exhibits contacts only between α-helix 4 and β-strand 5 (Figure 1A; 824Å2/6% of each monomer surface) (72).
Figure 1.

Dimerization modes of homologous REC domains (A) Dimerization mode of the REC domain of full-length ComED58A, RECD58A or RECD58E domains of ComE (this work) compared to PhoB and FixJ REC domains. The secondary structures involved in dimerization are colored: α4 is in yellow, α5 is in green, β5 is in pink and the α4–β5 loop is in blue. The same color as the secondary structures code is applied. (B) Exclusion chromatography elution profile of isolated RECD58A (red line) and RECD58E (blue line) domains. Vo indicates the exclusion volume of the column. (C) Experimental scattering curves for RECD58A (red dots) and RECD58E (blue dots) compared to the calculated curves (colored lines) from crystal structures.
The structures of the RECD58A and the RECD58E monomers, as well as the spatial organization of the functionally important residues involved in the conformational changes stabilized by the phosphorylation of the strictly conserved Asp, are conserved and similar to that of the activated form of the other REC domains (Supplementary Figure S1). Indeed, the region centered on D58 is structurally much closer to the equivalent region in the activated form than in the inactivated form of PhoB (73). The side chain of F107 of RECD58A and of RECD58E adopts an orientation corresponding to those of Y102 in the activated PhoB (Supplementary Figure S1b). We assume that, due to the high protein concentrations used during crystallization of both froms of the REC domain, the active conformation was selected from the equilibrium distribution and stabilized by the crystal packing. The interface between the two subunits of REC from ComE is mostly hydrophobic and F107 sits into a pocket formed by F86, F93 and Y98 (from the other subunit in the dimer). This hydrophobic environment may stabilize the location of F107 in the active state.
This possibility of the existence of an equilibrium between the active and inactive forms was then checked by measuring the oligomeric state of RECD58A and RECD58E in solution. Size exclusion chromatography shows that RECD58E is exclusively dimeric (c ≈ 1 mg/ml in the most concentrated fractions), while the RECD58A is in a monomer/dimer equilibrium tending to favor the monomer (Figure 1B). SAXS, performed at low concentration (c ≈ 0.4 mg/ml), showed unambiguously that RECD58A is mainly monomeric while RECD58E is fully dimeric (χ ≈ 1 and 0.9 respectively; Figure 1C).
The full-length ComED58A dimer combines rotational and translational symmetries
We solved the X-ray structure of the full-length ComED58A at 3.4Å resolution. The statistics for data collection and refinement are summarized in Supplementary Table S1. The structure is composed of a REC and a LytTR domain, connected by a linker (Figure 2). Six copies (subunits A–F) of the protein are present in the asymmetric unit of the crystal. They form 3 identical dimers (AB, CD and EF), with an RMSD inferior to 2Å (Figure 2A). The total interface area excluded from solvent upon complex formation is 1780Å2 representing about 6% of the total surface of each monomer. The main dimer contacts are provided by the N-terminal REC domains (55% of the total interface area). The REC domains of ComED58A adopt the same dimerization mode as the isolated REC domains, indicating that this atypical dimerization mode is very unlikely to result from a crystal packing artifact or imposed by the LytTR domains (Figure 2B).
Figure 2.

X-ray structure of the ComED58A dimer. (A) Superimposition of the three copies of the ComED58A dimer of the asymmetric unit. (B) ComED58A crystallographic dimer. The REC domains share a 2-fold rotational symmetry axis while the LytTR domains are in tandem. The REC domains are in orange and dark blue, the LytTR domains are in yellow and cyan. (C) Color-coded representation of the B-factors of the residues. (D) Comparison of the two conformations of ComE present in the dimer. The linker between the REC and LytTR domains is in red. (E) SAXS curves of the ComE constructs. Left and middle: experimental scattering curves (black dots) with the calculated curves (continuous colored lines) derived from typical models (inserts) obtained using the rigid-body modeling programs BUNCH and CORAL for ComED58A and ComED58E, respectively. The rigid bodies are the crystal structures of the REC and LytTR domains. Right: calculated curve (blue line) from the crystal structure of the LytTR monomer compared with the experimental curve (black dots). The His-Tag has been added by using the program BUNCH.
The structure of the linker regions between the REC and LytTR domains, comprised between residues L132 and D140, is well defined (the thermal B-factors of the backbone atoms are similar as those of the REC and LytTR domains) (Figure 2C). The conformations of these linkers between residues 132 and 135 however are very different for both subunits. In one monomer L132-L133-E134 forms the C-terminal of α-helix 5 of REC, while in the other subunit it adopts an extended conformation (Figure 3A). The remainder of the linkers is superimposable. The different conformations of the linkers impose a translational symmetry to the LytTR domains, as opposed to the 2-fold rotational symmetry of the REC domains. The LytTR domains are arranged in tandem (Figure 2B). Interestingly the tandem arrangement of the LytTR domains is compatible with the binding to the tandem boxes DR1 and DR2 of their comcde, comx or comab promoters. The linkers and dimer configurations are identical for the three independent copies in the asymmetric unit, excluding particular crystal packing effects.
Figure 3.

Conformation of the linker. (A) Superimposition of the linkers of the two monomers of ComED58A. The two segments [N135-D137] are identical (in rainbow), whereas the L132-L133-E134 hinge (in red) is helical in subunit A and extended in subunit B. The V139-D140 segments (end of the rainbow) do not superimpose, directing the LytTR domains in different ways. (B) Superimposition of the isolated RECD58A (in cyan) and RECD58E (in yellow) domains with the REC domain of ComED58A (in grey). The secondary elements superimposed well except of the α-helix 2. The only large variation is for the loop between α2 and β3 which is stretched in subunit A in contrary to the other structures (in blue dotted line for RECD58A, orange for RECD58E and red for subunit A of ComED58A). (C and D) The α2–β3 loop is a hinge between monomer A and B. Same color code as in (A and B) except for the α2–β3 loop that is red. In subunit A, the link between V51 and I153 tighten the LytTR domain in one conformation whereas subunit B adopts another one.
The REC domains of ComED58A and of isolated RECD58A and RECD58E superpose very well except for the loop between α-helix 2 and β-strand 3 which is different only in the subunit A of full-length ComED58A (Figure 3B and Figure 2D). This loop adopts a different conformation in the two subunits. In one monomer its tip residue V51 stretches out to engage in a hydrophobic packing with I153 from the β-strand 7 of the LytTR (Figure 3C). In the other subunit, due different configuration of the linker, this interaction is interrupted and the loop falls back upon the core of the REC domain (Figure 3D). The α-helix 2 of subunit A is then deviated by about 20° but the β-strand 3 that carried D58 is not affected.
ComED58A crystallizes as a dimer, but is a flexible monomer in solution
We checked the oligomeric state of full-length ComED58A in solution by SAXS (Figure 2E). The characteristic parameters of the protein measured by SAXS are recapitulated in Supplementary Table S2. Size-exclusion high-performance liquid chromatography (SEC-HPLC) of ComED58A showed its elution is delayed as compared to ComED58E (Supplementary Figure S2a) suggesting their oligomeric states may be different. The determination of the molar mass of the protein using SAXS data established unequivocally that ComED58A is a monomer in solution, even at the relatively high protein concentration of 3 mg/ml used for the crystallization (Supplementary Table S1). We then compared the intensity curves calculated from each monomer (data not shown) present in the crystal dimer to the experimental curves. The calculated patterns did not provide a good fit (χ ≈ 4.0 for one monomer and 3.5 for the other) and yielded for both monomers a slightly smaller value of the radius of gyration than the experimental one. The comparison of P(r) profiles (Supplementary Figure S2b) also indicated that there are some differences between crystal and solution conformations: the shoulder visible in the 45–60Å range, which reports on the distance between LytTR and REC domains, is more pronounced in the experimental curves than in the calculated ones. This suggests that both domains are slightly more apart in solution. We then fitted the experimental scattering data by models that were created by freely moving each domain as a rigid body around the linker (residues S130–V139), which was represented as a chain of dummy residues (BUNCH program). This approach resulted in a great variety of conformations whose calculated scattering curves were in good agreement with the experimental data (χ ≈ 1.6) (Figure 2E). The monomer in solution seems to adopt an ensemble of conformations and the LytTR domain is mobile relative to the REC domain. Therefore, instead of using the scattering pattern of a single model (BUNCH approach), we fitted the data by the average scattering pattern of an ensemble of models (EOM approach). Improved adjustment (χ ≈ 1.2) demonstrates the suitability of this approach. The resulting distribution of values for the radius of gyration (Rg) for ComED58A is shown in Supplementary Figure S2d. One observes that the distribution of selected ensembles is much narrower than that of the complete random pool. This indicates that the two domains do not explore all accessible positions, which would have been the case if the linker S130–V139 had adopted a completely random conformation, and that the linker has only a restricted flexibility, in agreement with the above observations about the linkers in the crystal structure. The Rg value of the crystal structure is slightly smaller than the average value derived from selected ensembles. This is in agreement with the analysis of the P(r) profiles and indicates that the REC and LytTR domains are on average slightly more distant in solution than in the crystal structure.
The phosphorylated-mimicking ComED58E is a dimer in solution
We further checked by SAXS the oligomerization of the ComED58E mutant that mimics the phosphorylated state of the protein. In contrast to ComED58A, the data show unambiguously that ComED58E is a dimer in solution (Figure 2E), consistent with the elution shift observed during size exclusion chromatography (Supplementary Figure S2a). Indeed the experimental scattering curve I(q) and the corresponding P(r) of ComED58E were compared to the patterns calculated from the crystal dimer. As for ComED58A, the experimental and calculated scattering patterns exhibit significant differences (χ ≈ 10.1, data not shown) and the experimental P(r) is slightly higher than the calculated one beyond 50Å (Supplementary Figure S2c), showing that the protein is slightly more extended in solution that in the crystals. We have previously observed that the RECD58E domain alone forms a stable dimer and we predict that this will also be the case within the full-length ComED58E protein. Moreover, the LytTR domain alone is monomeric in solution as checked by SAXS (χ ≈ 1.1, Figure 2E). We then built a model of the ComED58E dimer (program CORAL) keeping the REC dimer fixed and allowing flexibility within the linkers between REC and LytTR (χ ≈ 1.8, Figure 2E). A great variety of positions of LytTR domains with respect to the REC dimer are compatible with the experimental data. Nevertheless, as for the monomer ComED58A an improved adjustment is obtained if we describe the ComE dimer in terms of ensemble of conformations using the program EOM (χ ≈ 1.15). Supplementary Figure S2e shows that the distribution of Rg of selected ensembles is very narrow and displaced towards the low values of Rg as compared to that of the complete random pool. This indicates that the two LytTR domains explore a restricted number of conformations.
We conclude that the ComED58E dimerizes in solution through the REC domains, probably through the same interface as the RECD58E, RECD58A and ComED58A structures, while the LytTR domains have a certain degree of mobility.
DISCUSSION
We present here the X-ray structure of a full-length DNA-binding RR dimer that informs on the domain orientations within the active dimer of an LytTR subfamily RR (Figure 2). We combine crystallographic and SAXS data to investigate the dynamic activation mechanism of ComE with respect to two mutants that mimic the phosphorylated (ComED58E, active-state mimic) and unphosphorylated (ComED58A, inactive-state mimic) states. The crystal structure of ComED58A revealed an original dimerization mode (Figure 1). The compact dimer is characterized by a 2-fold rotational symmetry between the REC domains that was also present in the dimer structures of the isolated REC mutants. The LytTR domains in the full-length dimer however do not follow this symmetry since they pack in tandem, compatible with the direct repeat arrangement of the three comcde, comab and comx promoters. The different dimer configurations for the REC and LytTR domains are correlated with the structure of the linkers, which are different in both monomers (Figure 3).
We propose that the dimer observed in the crystal structure of ComED58A corresponds to the active form of ComE bound to DNA. The fact that our three structures of ComED58A, RECD58A and RECD58E are perfectly superimposable, with the only exception of the α2–β3 loop involved in the packing of the asymmetric dimer, is not surprising. Inactive REC domains tend to crystallize as active-like dimers unless alternative dimers are preferred (a relatively rare case that occurs in E. coli PhoB for example) or stabilized by interactions with effector domains in full-length RRs. In these active-like dimer conformations, which are not stabilized by phosphoryl groups or phosphoryl analogues, switch residues have been observed in all combinations of active and inactive configurations.
We observed that ComE in solution forms an equilibrium between monomers and dimers whose relative populations depend upon the functional state of the protein. The isolated RECD58A domain is in monomer/dimer equilibrium (Figure 1B) but the RECD58E is a stable dimer. ComED58E (that mimics the active state) dimerizes, most probably via the REC domains. For both inactive and active forms, a certain degree of mobility between the REC and LytTR domains is observed in solution, likely supported by the flexibility of the linker region. The different conformations of the linker in the two subunits of the crystal dimer allows the positioning in tandem of the LytTR domains ideally positioned to interact with the two tandem operator sub-sites.
From our data, two non-exclusive mechanisms for the binding of ComE to its promoter can be proposed: (i) the phosphorylation of apo-ComE induces first its dimerization via the REC domains as proposed by SAXS measurements on ComED58E, followed by the binding to DNA via the LytTR domains or (ii) two monomers of ComE bind first independently to each of the two DR sites of comcde, followed by the phosphorylation-induced dimerization of the REC domains and the reunification of the LytTR domains in tandem as in the crystal structure of ComED58A. Because ComD is anchored to the membrane, it seems likely that the phosphorylation of ComE occurs before the binding to DNA. SAXS studies are in progress to elucidate the structure of ComE-comcde complexes.
CONCLUSIONS
In this article we demonstrate that the non-phosphorylatable ComED58A from S. pneumoniae, which in vivo abolishes the basal comCDE operon expression (27,37), is a monomer. A contrario the ComED58E mutant which mimics the phosphorylated state of ComE and renders S. pneumoniae constitutively competent (27,38), is a dimer in absence of DNA. This is in accordance with the results obtained from phosphorylatable Asp (D60A and D60E) mutants of ComE from S. mutans (74) showing that the phosphorylated state of ComE has little effect on DNA-binding affinity, but rather promotes oligomerization of the protein. We propose that the dimer interaction of SpComED58E occurs via REC–REC interactions that are reinforced by their phosphorylation (75), triggering the activation of the LytTR RR. The presence of dimers in the crystal structure of the non-phosphorylatable ComED58A mutant is probably caused by the high concentrations of ComED58A needed for the crystallogenesis. This was previously noticed in the case of the StPhoP RR for which a moderate concentration increase was sufficient to promote its dimerization on the DNA (15).
All together our results considerably improved the molecular mechanistic knowledge of the ComD–ComE two components system, which is responsible for the transcription of about hundred genes via the comx promoter activation. These late genes code for proteins involved in natural genetic transformation (DNA uptake, e.g. ComEG, ComEC, ComFA; Recombination, e.g. RecA, SsbB, DprA, CoiA). ComD–ComE also regulates the expression of virulence factors required for infection. We can expect that characterization of RR–DNA interaction at the atomic level will help to fight pathogens via development of new drugs that block protein–protein or protein–DNA interactions.
ACCESSION NUMBERS
The atomic coordinates and structure factors of full-length ComED58A and of isolated RECD58A and RECD58E domains have been deposited at the Brookhaven Protein Data Bank, respectively, under the accession numbers 4CBV, 4ML3 and 4MLD.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre National de la Recherche Scientifique; University Paris-Sud 11 [UMR8619]; French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-05-01] (PhD fellowships to M.B. and D.S.). Funding for open access charges: Centre National de la Recherche Scientifique and the University Paris-Sud 11 [UMR8619].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
X-ray and SAXS data were collected at Synchrotron SOLEIL (Saclay France) at beamlines PROXIMA-1 and SWING, respectively. We thank the beamline staff for assistance and advice during data collections. We thank Noureddine Lazar and Anthony Doizy for help with purification and crystallization, Karine Blondeau for help during labeling of ComED58A, Ines Li de la Sierra-Gallay and Sylvie Nessler for help with the resolution of the structure of the REC domains. We are grateful to Jean-Pierre Claverys and Bernard Martin for sharing purified genomic DNA from R6 S.pneumoniae strain and for discussions, and Bernard Gilquin for discussions.
REFERENCES
- 1.Jung K, Fried L, Behr S, Heermann R. Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 2012;15:118–124. doi: 10.1016/j.mib.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 2.Menon S, Wang S. Structure of the response regulator PhoP from Mycobacterium tuberculosis reveals a dimer through the receiver domain. Biochemistry. 2011;50:5948–5957. doi: 10.1021/bi2005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson VL, Wu T, Stock AM. Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J. Bacteriol. 2003;185:4186–4194. doi: 10.1128/JB.185.14.4186-4194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedland N, Mack TR, Yu M, Hung LW, Terwilliger TC, Waldo GS, Stock AM. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry. 2007;46:6733–6743. doi: 10.1021/bi602546q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen MW, Kotaka M, Vonrhein C, Bricogne G, Rao F, Chuah ML, Svergun D, Schneider G, Liang ZX, Lescar J. Structural insights into the regulatory mechanism of the response regulator RocR from Pseudomonas aeruginosa in cyclic Di-GMP signaling. J. Bacteriol. 2012;194:4837–4846. doi: 10.1128/JB.00560-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong E, Lee HM, Ko H, Kim DU, Jeon BY, Jung J, Shin J, Lee SA, Kim Y, Jeon YH, et al. Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J. Biol. Chem. 2007;282:20667–20675. doi: 10.1074/jbc.M609104200. [DOI] [PubMed] [Google Scholar]
- 7.Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science (New York, N.Y.) 2010;327:866–868. doi: 10.1126/science.1181185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonard PG, Golemi-Kotra D, Stock AM. Phosphorylation-dependent conformational changes and domain rearrangements in Staphylococcus aureus VraR activation. Proc. Natl Acad. Sci. USA. 2013;110:8525–8530. doi: 10.1073/pnas.1302819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wassmann P, Chan C, Paul R, Beck A, Heerklotz H, Jenal U, Schirmer T. Structure of BeF3- -modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure (London, England: 1993) 2007;15:915–927. doi: 10.1016/j.str.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 10.Pao GM, Saier MH., Jr Response regulators of bacterial signal transduction systems: selective domain shuffling during evolution. J. Mol. Evol. 1995;40:136–154. doi: 10.1007/BF00167109. [DOI] [PubMed] [Google Scholar]
- 11.Gao R, Stock AM. Biological insights from structures of two-component proteins. Ann. Rev. Microbiol. 2009;63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao R, Stock AM. Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 2010;13:160–167. doi: 10.1016/j.mib.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gardino AK, Kern D. Functional dynamics of response regulators using NMR relaxation techniques. Methods Enzymol. 2007;423:149–165. doi: 10.1016/S0076-6879(07)23006-X. [DOI] [PubMed] [Google Scholar]
- 14.Gao R, Tao Y, Stock AM. System-level mapping of Escherichia coli response regulator dimerization with FRET hybrids. Mol. Microbiol. 2008;69:1358–1372. doi: 10.1111/j.1365-2958.2008.06355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh V, Ekka MK, Kumaran S. Second monomer binding is the rate-limiting step in the formation of the dimeric PhoP-DNA complex. Biochemistry. 2012;51:1346–1356. doi: 10.1021/bi201257d. [DOI] [PubMed] [Google Scholar]
- 16.Morrison DA, Baker MF. Competence for genetic transformation in pneumococcus depends on synthesis of a small set of proteins. Nature. 1979;282:215–217. doi: 10.1038/282215a0. [DOI] [PubMed] [Google Scholar]
- 17.Tomasz A. Model for the mechanism controlling the expression of competent state in Pneumococcus cultures. J. Bacteriol. 1966;91:1050–1061. doi: 10.1128/jb.91.3.1050-1061.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowalko JE, Sebert ME. The Streptococcus pneumoniae competence regulatory system influences respiratory tract colonization. Infect. Immun. 2008;76:3131–3140. doi: 10.1128/IAI.01696-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson D, Karlsson S, Gustafsson E, Normark BH, Nilsson P. Modeling the regulation of the competence-evoking quorum sensing network in Streptococcus pneumoniae. Biol. Syst. 2007;90:211–223. doi: 10.1016/j.biosystems.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Havarstein LS, Gaustad P, Nes IF, Morrison DA. Identification of the streptococcal competence-pheromone receptor. Mol. Microbiol. 1996;21:863–869. doi: 10.1046/j.1365-2958.1996.521416.x. [DOI] [PubMed] [Google Scholar]
- 21.Pestova EV, Havarstein LS, Morrison DA. Regulation of competence for genetic transformation in Streptococcus pneumoniae by an auto-induced peptide pheromone and a two-component regulatory system. Mol. Microbiol. 1996;21:853–862. doi: 10.1046/j.1365-2958.1996.501417.x. [DOI] [PubMed] [Google Scholar]
- 22.Havarstein LS, Coomaraswamy G, Morrison DA. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl Acad. Sci. USA. 1995;92:11140–11144. doi: 10.1073/pnas.92.24.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hui FM, Zhou L, Morrison DA. Competence for genetic transformation in Streptococcus pneumoniae: organization of a regulatory locus with homology to two lactococcin A secretion genes. Gene. 1995;153:25–31. doi: 10.1016/0378-1119(94)00841-f. [DOI] [PubMed] [Google Scholar]
- 24.Ween O, Gaustad P, Havarstein LS. Identification of DNA binding sites for ComE, a key regulator of natural competence in Streptococcus pneumoniae. Mol. Microbiol. 1999;33:817–827. doi: 10.1046/j.1365-2958.1999.01528.x. [DOI] [PubMed] [Google Scholar]
- 25.Claverys JP, Havarstein LS. Extracellular-peptide control of competence for genetic transformation in Streptococcus pneumoniae. Front. Biosci. 2002;7:D1798–D1814. doi: 10.2741/claverys. [DOI] [PubMed] [Google Scholar]
- 26.Claverys JP, Prudhomme M, Martin B. Induction of competence regulons as a general response to stress in gram-positive bacteria. Ann. Rev. Microbiol. 2006;60:451–475. doi: 10.1146/annurev.micro.60.080805.142139. [DOI] [PubMed] [Google Scholar]
- 27.Martin B, Soulet AL, Mirouze N, Prudhomme M, Mortier-Barriere I, Granadel C, Noirot-Gros MF, Noirot P, Polard P, Claverys JP. ComE/ComE∼P interplay dictates activation or extinction status of pneumococcal X-state (competence) Mol. Microbiol. 2013;87:394–411. doi: 10.1111/mmi.12104. [DOI] [PubMed] [Google Scholar]
- 28.Lee MS, Morrison DA. Identification of a new regulator in Streptococcus pneumoniae linking quorum sensing to competence for genetic transformation. J. Bacteriol. 1999;181:5004–5016. doi: 10.1128/jb.181.16.5004-5016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrison DA, Lee MS. Regulation of competence for genetic transformation in Streptococcus pneumoniae: a link between quorum sensing and DNA processing genes. Res. Microbiol. 2000;151:445–451. doi: 10.1016/s0923-2508(00)00171-6. [DOI] [PubMed] [Google Scholar]
- 30.Luo P, Morrison DA. Transient association of an alternative sigma factor, ComX, with RNA polymerase during the period of competence for genetic transformation in Streptococcus pneumoniae. J. Bacteriol. 2003;185:349–358. doi: 10.1128/JB.185.1.349-358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo P, Li H, Morrison DA. ComX is a unique link between multiple quorum sensing outputs and competence in Streptococcus pneumoniae. Mol. Microbiol. 2003;50:623–633. doi: 10.1046/j.1365-2958.2003.03714.x. [DOI] [PubMed] [Google Scholar]
- 32.Hahn J, Maier B, Haijema BJ, Sheetz M, Dubnau D. Transformation proteins and DNA uptake localize to the cell poles in Bacillus subtilis. Cell. 2005;122:59–71. doi: 10.1016/j.cell.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claverys JP, Martin B, Polard P. The genetic transformation machinery: composition, localization, and mechanism. FEMS Microbiol. Rev. 2009;33:643–656. doi: 10.1111/j.1574-6976.2009.00164.x. [DOI] [PubMed] [Google Scholar]
- 34.Mortier-Barriere I, Velten M, Dupaigne P, Mirouze N, Pietrement O, McGovern S, Fichant G, Martin B, Noirot P, Le Cam E, et al. A key presynaptic role in transformation for a widespread bacterial protein: DprA conveys incoming ssDNA to RecA. Cell. 2007;130:824–836. doi: 10.1016/j.cell.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 35.Quevillon-Cheruel S, Campo N, Mirouze N, Mortier-Barriere I, Brooks MA, Boudes M, Durand D, Soulet AL, Lisboa J, Noirot P, et al. Structure-function analysis of pneumococcal DprA protein reveals that dimerization is crucial for loading RecA recombinase onto DNA during transformation. Proc. Natl Acad. Sci. USA. 2012;109:E2466–E2475. doi: 10.1073/pnas.1205638109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnsborg O, Havarstein LS. Regulation of natural genetic transformation and acquisition of transforming DNA in Streptococcus pneumoniae. FEMS Microbiol. Rev. 2009;33:627–642. doi: 10.1111/j.1574-6976.2009.00167.x. [DOI] [PubMed] [Google Scholar]
- 37.Martin B, Granadel C, Campo N, Henard V, Prudhomme M, Claverys JP. Expression and maintenance of ComD-ComE, the two-component signal-transduction system that controls competence of Streptococcus pneumoniae. Mol. Microbiol. 2010;75:1513–1528. doi: 10.1111/j.1365-2958.2010.07071.x. [DOI] [PubMed] [Google Scholar]
- 38.Mirouze N, Berge MA, Soulet AL, Mortier-Barriere I, Quentin Y, Fichant G, Granadel C, Noirot-Gros MF, Noirot P, Polard P, et al. Direct involvement of DprA, the transformation-dedicated RecA loader, in the shut-off of pneumococcal competence. Proc. Natl Acad. Sci. USA. 2013;110:E1035–E1044. doi: 10.1073/pnas.1219868110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikolskaya AN, Galperin MY. A novel type of conserved DNA-binding domain in the transcriptional regulators of the AlgR/AgrA/LytR family. Nucleic Acids Res. 2002;30:2453–2459. doi: 10.1093/nar/30.11.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hung DC, Downey JS, Ayala EA, Kreth J, Mair R, Senadheera DB, Qi F, Cvitkovitch DG, Shi W, Goodman SD. Characterization of DNA binding sites of the ComE response regulator from Streptococcus mutans. J. Bacteriol. 2011;193:3642–3652. doi: 10.1128/JB.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Havarstein LS. Bacterial gene transfer by natural genetic transformation. APMIS Suppl. 1998;84:43–46. doi: 10.1111/j.1600-0463.1998.tb05647.x. [DOI] [PubMed] [Google Scholar]
- 42.Martin B, Prudhomme M, Alloing G, Granadel C, Claverys JP. Cross-regulation of competence pheromone production and export in the early control of transformation in Streptococcus pneumoniae. Mol. Microbiol. 2000;38:867–878. doi: 10.1046/j.1365-2958.2000.02187.x. [DOI] [PubMed] [Google Scholar]
- 43.Peterson SN, Sung CK, Cline R, Desai BV, Snesrud EC, Luo P, Walling J, Li H, Mintz M, Tsegaye G, et al. Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol. Microbiol. 2004;51:1051–1070. doi: 10.1046/j.1365-2958.2003.03907.x. [DOI] [PubMed] [Google Scholar]
- 44.Galperin MY. Telling bacteria: do not LytTR. Structure. 2008;16:657–659. doi: 10.1016/j.str.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sidote DJ, Barbieri CM, Wu T, Stock AM. Structure of the Staphylococcus aureus AgrA LytTR domain bound to DNA reveals a beta fold with an unusual mode of binding. Structure (London, England : 1993) 2008;16:727–735. doi: 10.1016/j.str.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leonard PG, Bezar IF, Sidote DJ, Stock AM. Identification of a hydrophobic cleft in the LytTR domain of AgrA as a locus for small molecule interactions that inhibit DNA binding. Biochemistry. 2012;51:10035–10043. doi: 10.1021/bi3011785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T. Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl. Environ. Microbiol. 1998;64:1694–1699. doi: 10.1128/aem.64.5.1694-1699.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quevillon-Cheruel S, Collinet B, Tresaugues L, Minard P, Henckes G, Aufrere R, Blondeau K, Zhou CZ, Liger D, Bettache N, et al. Cloning, production, and purification of proteins for a medium-scale structural genomics project. Methods Mol. Biol. 2007;363:21–37. doi: 10.1007/978-1-59745-209-0_2. [DOI] [PubMed] [Google Scholar]
- 49.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 50.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uhlin U, Eklund H. Structure of ribonucleotide reductase protein R1. Nature. 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 52.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta. Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 55.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta. Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta. Crystallogr. D Biol. Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 57.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta. Crystallogr. D Biol. Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 58.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta. Crystallogr. D Biol. Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 59.Cowtan K. Fitting molecular fragments into electron density. Acta. Crystallogr. D Biol. Crystallogr. 2008;64:83–89. doi: 10.1107/S0907444907033938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, et al. BUSTER version 2.10.0. Cambridge, United Kingdom: Global Phasing Ltd; 2011. [Google Scholar]
- 61.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 64.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- 65.David G, Pérez J. Combined sampler robot and HPLC: a fully automated system for biological SAXS experiments at the Synchrotron SOLEIL Swing beamline. J. Appl. Cryst. 2009;42:892–900. [Google Scholar]
- 66.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Cryst. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fisher H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Cryst. 2010;43:101–109. [Google Scholar]
- 68.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 70.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 71.Bachhawat P, Swapna GV, Montelione GT, Stock AM. Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure. 2005;13:1353–1363. doi: 10.1016/j.str.2005.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Birck C, Mourey L, Gouet P, Fabry B, Schumacher J, Rousseau P, Kahn D, Samama JP. Conformational changes induced by phosphorylation of the FixJ receiver domain. Structure (London, England: 1993) 1999;7:1505–1515. doi: 10.1016/s0969-2126(00)88341-0. [DOI] [PubMed] [Google Scholar]
- 73.Sola M, Gomis-Ruth FX, Serrano L, Gonzalez A, Coll M. Three-dimensional crystal structure of the transcription factor PhoB receiver domain. J. Mol. Biol. 1999;285:675–687. doi: 10.1006/jmbi.1998.2326. [DOI] [PubMed] [Google Scholar]
- 74.Hung DC, Downey JS, Kreth J, Qi F, Shi W, Cvitkovitch DG, Goodman SD. Oligomerization of the response regulator ComE from Streptococcus mutans is affected by phosphorylation. J. Bacteriol. 2012;194:1127–1135. doi: 10.1128/JB.06565-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. Regulation of response regulator autophosphorylation through interdomain contacts. J. Biol. Chem. 2010;285:32325–32335. doi: 10.1074/jbc.M110.157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.