


Abstract
The robust and tissue-specific activation of the human growth hormone (hGH) gene cluster in the pituitary and placenta constitutes an informative model for analysis of gene regulation. The five-gene hGH cluster is regulated by two partially overlapping sets of DNase I hypersensitive sites (HSs) that constitute the pituitary (HSI, II, III and V) and placental (HSIII, IV, and V) locus control regions (LCRs). The single placenta-specific LCR component, HSIV, is located at −30 kb to the cluster. Here we generate a series of hGH/BAC transgenes specifically modified to identify structural features of the hGH locus required for its appropriate placental expression. We find that placental specificity is dependent on the overall multigene configuration of the cluster whereas the distance between the cluster and its LCR impacts the level of placental expression. We further observe that a major function of the placental hGH LCR is to insulate the transgene locus from site-of-integration effects. This insulation activity is linked to placenta-specific occupancy of the chromatin architectural protein, CTCF, at HSIV. These data reveal a remarkable combination of structural configurations and regulatory determinants that must work in concert to insure robust and tightly controlled expression from a complex multigene locus.
INTRODUCTION
The temporal and spatial control of gene expression in multi-cellular eukaryotes is essential for programming development and for maintenance of physiologic functions. The determinants of these controls are generally contained within non-coding regions of the genome. Genome-wide searches for cis-acting regulatory elements, including enhancers, repressors and insulators, are based on the recognition of specific epigenetic modifications and configurations, trans-acting factor occupancies, non-coding transcription units and sensitivities of chromatin to cleavage by a variety of endonucleases. Much of this information, currently centralized in the Encyclopedia of DNA Elements (ENCODE), suggests that >80% of human genome is ‘biochemically functional’ (1). The exploration of regulatory controls and their coordinating circuits constitutes a necessary foundation for interpretation of sequence variations and mutations linked to various phenotypes and disease processes (2). Defining the mechanisms by which these determinants act on target genes will demand in-depth analyses of robust and experimentally tractable model loci.
The human growth hormone (hGH) multi-gene cluster presents an informative model for the analysis of regulatory controls that impact developmental and physiologic processes. This locus encompasses five tightly packed genes that have a high level of structural similarity yet demonstrate robust and mutually exclusive gene-regulatory profiles (3). These genes are a single pituitary-specific gene (hGH-N), and four adjacent paralogs (hCS-A, hCS-B, hCS-L and hGH-V) that are expressed exclusively in the placenta (Figure 1A). Analyses of primary human pituitaries and extensive modeling in mouse transgenic lines support the conclusion that the tissue-specific activation of the pituitary gene of the hGH gene cluster is controlled by a set of distal regulatory elements located between 14.5- and 32-kb 5′ to the cluster. These determinants, marked by DNase I hypersensitivity, comprise the pituitary locus control region (LCR) (4). Transcriptional activation of hGH-N in pituitary somatotropes has been extensively explored in transgenic mouse models. This pathway is initiated by the binding of the pituitary-specific POU-homeodomain protein, Pit-1, at the LCR determinant, hypersensitive site I (HSI), located 14.5-kb 5′ to the hGH-N promoter (6). This interaction establishes a 32-kb domain of histone acetylation that encompasses the entire LCR and extends to include the hGH-N promoter (7,8). HSI activation triggers robust non-coding transcription within the LCR. This non-coding transcription is functionally linked to higher order reconfiguration of the locus (‘chromatin looping’) that juxtaposes the transcriptionally active LCR domain with the target hGH-N promoter (9). These events result in robust, somatotrope-specific transcriptional activation of hGH-N.
Figure 1.

Transgenes designed to identify structural features at the hGH locus critical to placental gene activation. (A) The hGH locus and epigenetic modifications in the placenta. The hGH gene cluster encompasses five genes: the pituitary-specific growth hormone gene (hGH-N), and four placenta-specific paralogs, hCS-L, hCS-A, hGH-V and hCS-B. The gene cluster is flanked 5′ by SCN4A and CD79b genes and 3′ by TCAM1. These flanking genes are specifically expressed in skeletal muscle, B lymphocyte and testis, respectively. Our previous studies identified two overlapping sets of DNase I HSs located 5′ to the cluster in the pituitary and placenta. HSIII and V are present in multiple tissues, including pituitaries and placentas, HSI and II are pituitary-specific and critical to hGH-N expression, and HSIV is specific to the placenta (4). Previously established patterns of histone H3 and H4 acetylation and H3K4 di- and tri-methylation at the hGH locus in the chromatin of human placental STBs are indicated (5). (B) Three hGH/BAC-derived transgenes designed to identify structural determinants of placental gene expression. The structures of four transgenes are shown. The wild-type hGH/BAC transgene (hGH/BAC) comprises a 123-kb NotI genomic fragment containing the entire hGH cluster with extensive 5′- and 3′-flanking regions, including the full set of pituitary or placental LCR determinants. The HSIII–V region (placental LCR) was selectively deleted from the hGH/BAC to generate the ΔHSIII-V/hGH/BAC transgene. A 12-kb segment between HSIII and the hGH cluster was deleted from hGH/BAC to generate the ΔSpacer/hGH/BAC transgene. In transgene LCR-CSA/BAC, the entire hGH cluster was replaced by a single placental gene repeat (PGR) unit encompassing hCS-A and its adjacent 3′-enhancer and 5′ P-element.
In contrast to this detailed understanding of hGH-N activation in the pituitary somatotropes, the corresponding pathways and determinants of the placental gene activation remain poorly understood. In vitro studies of human chorionic somatomammotropin (hCS) genes have identified several promoter-proximal transcription factor binding sites that contribute to hCS expression. These include a TATA box, Sp1-binding sites and an initiator element (InrE) site (10). Additional evidence from cell transfection studies suggests that a conserved ‘P-element’ located ∼2-kb upstream of each placental gene repeat (PGR) units and a 3′-enhancer located 2-kb downstream of each hCS gene contribute to tissue-specific activation of PGRs (11–13). These in vitro and cell-based studies, while of interest, do not appear to reflect critical determinants of expression in vivo. For example, using a series of transgenic mouse models, we have demonstrated that segments of the hGH cluster that encompass these identified elements in native contiguity with an hCS gene are insufficient for the activation of the placental hCS in the mouse placenta (4). This contrasts with the robust, placenta-specific and site-of-integration independent hCS expression from more extensive hGH/BAC and hGH/P1 transgenes encompassing the hGH cluster along with the contiguous 5′-flanking region (14,5). These studies suggest that the basis for appropriate activation of placental hCS genes in vivo is complex. This complexity may reflect the overall content and configuration of the multi-gene cluster and/or to determinants in the 5′-flanking region.
The DNase I hypersensitivity mapping of chromatin from primary human placental syncytiotrophoblasts (STBs) lining the placental villi (the site of hCS gene expression), reveals three HSs. These sites, located 28- (HSIII), 30- (HSIV) and 32-kb (HSV) 5′ to the hGH cluster, comprise the putative placental LCR (Figure 1A). Data from the ENCODE project (http://genome.ucsc.edu/ENCODE) demonstrate that HSIII and HSV are formed in multiple cell types, while our data reveal the formation of an additional HS, HSIV, is specific to the placenta (4). ChIP analyses of histone H3 and H4 acetylation along with H3K4-di- and tri-methylation in placental chromatin indicate that these ‘active’ epigenetic modifications are localized to the HSIII-V region and to the segments of the gene cluster encompassing each of the placental genes (15). Importantly, the region between HSIII and the target hCS genes lacks active histone modifications in placental chromatin (summarized in Figure 1A). Based on these data we have put forward a model in which the region encompassing HSIII-V plays a critical role in the regulation of the placental genes. This role may be mediated by direct interactions with target promoters within the cluster. A temporal analysis of epigenetic modifications during terminal differentiation of the placental STBs was additionally informative. This article suggested that the discontinuous pattern of histone modifications (H3/H4 acetylation and H3K4 methylation) within the cluster might be generated by the independent initiation of chromatin modifications at each of the four placental gene promoters and adjacent 3′-enhancer elements with subsequent extension of histone modifications to the gene bodies (5). Based on these data we have hypothesized that the hGH cluster in the placental chromatin is organized as four independently regulated PGR units with the activity of each PGR being dependent on the remote HSIII-V region of the hGH LCR. Of note, the region between HSIII and the gene cluster encompasses the pituitary-specific HSI and HSII that are essential to the expression of hGH-N (Figure 1A). Whether or not this region has additional relevance to activation of placental hCS genes, either via unexplored cis-acting elements or via critical spacing between the LCR and the target genes, remains in question.
In the current article we assemble a series of hGH transgenes designed to probe relationships between structures at the hGH locus and regulation of placental CS genes (Figure 1B). These transgenic studies directly assess the functional impact of the remote HSIII-V determinants on placental expression from the hGH cluster. The specific goals of the article are to determine whether the extensive spacing between the HSIII region and the target genes serves a purpose in gene regulation, and define whether the overall configuration of the multi-gene configuration impacts the specificity and accuracy of the activation pathway. The findings substantially identify critical parameters and determinants of hCS gene activation from the hGH multigene cluster in the placenta. These findings advance our understanding of gene regulatory interactions at this robustly expressed and tightly controlled gene locus and establish paradigms for transcriptional controls in similarly configured systems.
MATERIALS AND METHODS
Modification of hGH transgenes and generation of transgenic mouse lines
A 123-kb segment of human genomic DNA, encompassing hGH gene cluster flanked by a 55-kb 5′-region and a 20-kb 3′-region, was released by NotI digestion from a human bacteria-artificial-chromosome (BAC) genomic clone (BAC#535D15 from CITB Human B&C Library; Invitrogen Life Technologies) and used to generate a set of hGH/BAC transgenic mouse lines by standard transgenic microinjection technologies (see below) (5). The same BAC clone was used as a template for the construction of three modified hGH transgenes (Figure 1B). The first modified transgene, ΔHSIII-V/hGH/BAC, was generated by a standard recombineering approach (16) to delete a 6-kb region encompassing the placental LCR (27- to 33-kb 5′ to the hGH-N promoter). The second transgene, ΔSpacer/hGH/BAC, was constructed by the RecA-assisted restriction endonuclease (RARE) cleavage approach (17) to remove a 12-kb EcoRI-fragment (10- to 22-kb 5′ to hGH-N promoter) located between the placental LCR and hGH-N promoter. The third transgene, LCR-CSA/BAC, was generated by replacing the hGH cluster with a 7.5-kb fragment containing an isolated hCS-A gene unit, including its contiguous 5′ P-element and 3′ putative enhancer. In this construction, a 70-kb fragment of the hGH BAC clone, encompassing the hGH cluster along with a 10-kb upstream and a 10-kb downstream regions, was removed by RsrII and PacI digestion. The remaining 85-kb BAC vector was then ligated to a 7.5-kb hCS-A unit, generated by long-distance PCR with LA polymerase reagents (TaKaRa Bio Inc.) and two specific primers (5′-CGGTCCGCTGGTCCATGGTTGGCATGGTAACCCCTTAC-3′ and 5′-TTAATTAAGC ACCACAACTGCCATCTCCTTTTTCTCC-3′). The long-distance PCR was performed in a 50-μl reaction containing 100–200 ng of the BAC template, 1X LA PCR buffer, 0.4 mM concentration of dNTP mixture, 0.2 μM concentration of paired primers, 2.5 units of LA Taq and 1.3 units of Pfu Ultra DNA polymerase (Stratagene) under the following conditions: 5 min at 95°C, 35 cycles of 10 s at 98°C and 10 min at 66°C and a final extension for 10 min at 72°C (5).
Each modified-BAC construct was treated with NotI and the released inserts were isolated by pulsed-field gel electrophoresis (Bio-Rad) and gel-purified with 1 U/100 μl β-agarase I (New England Biolabs) followed by phenol–chloroform extraction. The three purified BAC fragments were adjusted to a concentration of 0.5 ng/μl in TE buffer (pH 7.5) and individually microinjected into the male pronuclei of fertilized mouse eggs. All transgenic founder mice were generated by the University of Pennsylvania Transgenic and Chimeric Mouse Core Facility (http://www.med.upenn.edu/genetics/core-facs/tcmf/index.html). Positive founders were identified by PCR analysis of tail DNA with primers specific to the BAC insertions (5′-CTGGGGCTCAGACAGACCCGGCTTCAAATT-3′ and 5′-GAATTCGGGTCAGGGCTCCTGCCTGA-3′). The integrity of each transgene was determined by Southern blot analysis of EcoRI-digested tail DNA (14) and targeted PCR analysis (Supplementary Figures S1 and S2). The transgene copy number for each line was estimated by normalizing the hybridization signal (PhosphorImager analysis of Southern blots) from transgene to the hybridization signal from a mouse endogenous reference gene, mouse ζ-globin gene (m-MX), on the same membrane (Supplementary Figure S2).
Isolation of intact nuclei from mouse placentas
Placentas from E18.5 day transgenic embryos were collected and washed in PBS (Ca and Mg free). Placental cells were dissociated in cell-free dissociation buffer (GIBCO-BRL) and filtered through a 40-µm cell strainer (Falcon) into 10% (v/v) FBS/DMEM. Cells were washed with PBS and lysed with Nonidet P-40 (NP-40) lysis buffer (10 mM Tris–HCl [pH 7.5], 10 mM NaCl, 3 mM MgCl2, 0.2% NP-40 and proteinase inhibitor cocktail (Sigma)) containing 10 mM sodium butyrate on ice. The lysate was incubated on ice for 10 min. Nuclei were collected by centrifugation. The nuclei were washed twice with RB buffer (0.1 M NaCl, 50 mM Tris–HCl [pH 8.0], 3 mM MgCl2, 0.1 mM PMSF and 5 mM sodium butyrate [pH 7.0]) and stored in glycerol storage buffer containing 20 mM sodium butyrate before analysis.
DNase I hypersensitive site mapping
Of intact nuclei, 300 μg were suspended in 1 ml RB buffer with 1 mM CaCl2. 120 U DNase I (Invitrogen) was then added into the sample and the reaction was incubated at 37°C. At different time points, 50 μg nuclei was removed and the reaction was terminated by adding EDTA to 50 mM followed by digestion of nuclei with an equal volume of solution S (1.6 M NaCl, 1% SDS and 200 μg/ml proteinase K). The samples were incubated at 55°C overnight and extracted with phenol/chloroform. The DNA was ethanol precipitated, resuspended in TE (10 mM Tris, 0.1 mM EDTA), digested by EcoRI, subjected to electrophoresis on a 0.8% agarose gel and transferred to Zetabind nylon membranes for hybridization (4,8).
RT-PCR assays
(I) Placental hGH/CS expression
Total RNAs were extracted from mouse tissues with RNA-Bee reagent (Tel-Test Inc.) according to the manufacturer’s instructions. The RNAs were treated with RNase-free DNase I (Promega) at 37°C for 1 h and purified with RNeasy column (Qiagen). The resulting RNAs were reverse transcribed with an oligo(dT) primer and Superscript III transcriptase (Invitrogen Life Technologies), and 2.5% of this RT reaction mixture was used for PCR. PCR was performed with a sense primer 32P labeled with T4 polynucleotide kinase (5′-GTCCCTGCTCCTGGCTTTTG-3′) and an antisense primer (5′-AGCAGCCCGTAGTTCTTGAG-3′). PCR was carried out for 25–30 cycles under the following conditions: 30 s at 94°C, 30 s at 57°C and 2 min at 72°C. The amplified cDNA segments were 546 bp for hGH-N, hGH-V, hCS-A and hCS-B and 492 bp for hCS-L. Digestion of the amplification products with TaqI generated an hGH-V fragment of 546 bp, an hGH-N fragment of 494 bp, an hCS-L fragment of 251 bp and hCS-A and hCS-B fragments of 305 bp (5). As an internal control, a transcript of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was amplified using a specific primer pair (5′-GCCAAAAGGGTCATCATCTC-3′ and 5′-CCTGCTTCACCACCTTCTTGA-3′). The amplified and digested products were separated on 6% polyacrylamide gels, and the signals were visualized by autoradiography or quantified by PhosphorImager (Molecular Dynamics). The ratio of placental hGH/CS to GAPDH was divided by transgene copy number to establish the value of transgene expression per copy in each case.
(II) Pituitary hGH-N expression
The pituitary mRNAs were prepared and the cDNAs were synthesized as described above. Primers (5′-GCCTGCTCTGCCTGC-3′ and 5′-GACTGGATGAGCAGCAG-3′) used for analysis of hGH-N expression corresponded to perfectly conserved regions between mouse GH (mGH) and hGH-N (4). PCR was performed under the following conditions: 30 s at 94°C, 30 s at 45°C and 1 min at 72°C. Following 28 cycles of PCR, the 3′-end-labeled products were subjected to BstNI digestion. The major band of hGH-N cDNA is 170 bp and the band for mGH is 110 bp. Fragments were separated on a 6% polyacrylamide gel, and bands were quantified by PhosphorImager analysis. The ratio of hGH-N to mGH was divided by transgene copy number to establish the transgene expression per copy in each case.
ChIP assay
The cells from placenta, kidney and liver were prepared as described above and suspended in 20 ml 10% (v/v) FBS/DMEM. The cell suspensions were fixed in 1% formaldehyde at room temperature for 10 min followed by the addition of glycine (final concentration 0.125 M) with incubation at room temperature for an additional 5 min. Cells were collected, washed with cold PBS twice and lysed in NP-40 lysis buffer containing 10 mM sodium butyrate on ice for 10 min. Nuclei were pelleted and suspended in lysis buffer (50 mM Tris–HCl [pH 8.0], 10 mM EDTA, 1% SDS, 10 mM sodium butyrate and 0.1 mM PMSF) and incubated on ice for 10 min. The lysates were then sheared by sonication (Sonic Dismembrator, Fisher Scientific) to an average size of 200–1000 bp, and the soluble chromatin was concentrated using Centricon-10 (Amicon Inc.) and taken up in IP buffer (20 mM Tris–HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 0.01% SDS, 1% Triton X-100, 10 mM sodium butyrate and 0.1 mM PMSF). An aliquot of each sample was removed as ‘input’ and used in PCR analysis. The remainder of the soluble chromatin was incubated at 4°C overnight with 20 μl of CTCF antibody (Upstate Biotech) or preimmune IgG (Upstate Biotech). Immune complexes were isolated by incubation with 60 μl of Protein A/G-agarose (Millipore) for 2 h at 4°C. The complexes were serially washed in 1 ml low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl [8.0] and 150 mM NaCl), 1 ml of the same buffer but with high salt (500 mM NaCl), 1 ml of LiCl buffer (250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA and 10 mM Tris–HCl [pH 8.0]) and twice with TE (10 mM Tris–HCl [pH 8.0] and 1 mM EDTA). The complexes were eluted with two 250 μl aliquots of elution buffer (1% SDS and 0.1 M NaHCO3) at room temperature for 15 min. DNAs were isolated by reversing the cross-links (heated at 65°C for 5 h in the presence of 0.2 M NaCl and 2 μl of 10 mg/ml RNase A and subsequently digested with 1 μl of 20 mg/ml proteinase K at 45°C for 12 h) and purified by QIAquick spin column (Qiagen). The resulting DNAs were analyzed by PCR with specified primer pairs (Table 1) and the PCR products were resolved by electrophoresis on 2% agarose gels. DNAs were visualized by fluorescence staining with SYBR Gold (Invitrogen) and quantified by PhosphorImager analysis. Band intensities were expressed relative to the signal obtained from 0.01% of input, and normalized with positive control (defined as 100) using a primer set that amplified CTCF-binding site in the Igf2/H19 imprinting-control region. All signals were demonstrated to be proportional to the amount of DNA input.
Table 1.
DNA sequences of oligonucleotide primers used in CTCF-ChIP assay
| Oligonucleotide Primers for ChIP Assay | |
|---|---|
| Primer | Sequence |
| HSV-5′ | 5′-TGCAGTCCGCGCCCTGTGAGTGGTG-3′ |
| HSV-3′ | 5′-ACACTGGGGAGGTTGGGAGCAGACC-3′ |
| HSIV-5′ | 5′-ATCCCCAGCAGCCATTGACGCAT-3′ |
| HSIV-3′ | 5′-CGCAGAGTCCACCACTTCTTCCCAC-3′ |
| HSIII-5′ | 5′-ATCAAATCCCTGCGGACACTGCGG-3′ |
| HSIII-3′ | 5′-TCCTGGGTGCTTCTGCAGTCTCTGCT-3′ |
| -25 kb-5′ | 5′-AAAATGAGACTGCTGGGCCAGGCACG-3′ |
| -25 kb-3′ | 5′-ACAAGGCAGGTTGTAGCAGCCTCGT-3′ |
| -15 kb-5′ | 5′-CCAAGCCTTTCCCAGTTATAC-3′ |
| -15 kb-3′ | 5′-CATCTTGGCCTAGGCCTCGGA-3′ |
| Promoter-5′ | 5′-TCCATTAGCACAAGCCCGTCAG-3′ |
| Promoter-3′ | 5′-TTTTTCTCTCTCCATCCCTCCAG-3′ |
| Enhancer-5′ | 5′-AGAACAATCTGCCCCTTATGGAAAGAC-3′ |
| Enhancer-3′ | 5′-CATGATGTTGCCTCACAGCAACCTCCA-3′ |
| TCAM-5′ | 5′-TGCAGCCGAATCACCAACCCGT-3′ |
| TCAM-3′ | 5′-GCATGATGTGTGATTTATCATGGC-3′ |
| (+)Control-5′ | 5′-AGCCGCTATGCCTCAGTGGTCGAT-3′ |
| (+)Control-3′ | 5′-GGGCTGTGTAGGGAATGAGTCAA-3′ |
RESULTS
Generation of transgenic mouse lines to define critical structures at the hGH gene cluster
To identify structural features of the hGH locus required for appropriate activation of placental CS genes, we parsed the hGH locus into three components: (i) the remotely situated HSIII-V (the putative placental LCR), (ii) the interval region between the placental LCR and the hGH cluster and (iii) the individual PGR units within the hGH cluster. Three corresponding transgenes were constructed; ΔHSIII-V/hGH/BAC, ΔSpacer/hGH/BAC and LCR-CSA/BAC (Figure 1B). The 123-kb hGH/BAC transgene, from which these transgenes were derived, served as the control for the studies. This hGH/BAC transgene contained the entire 48-kb hGH cluster with extensive 5′- and 3′-flanking regions encompassing the full complement of HSs that constitute the hGH LCR as well as neighboring genes (SCN4A, CD79b and TCAM1) with mutually exclusive tissue specificities (Figure 1A and B). Mice carrying this hGH/BAC are known to have robust and placenta-specific expression of the hCS genes (5).
The three modified transgenes were individually injected into fertilized mouse embryos and four transgenic lines were generated for each construct. The integrity and copy number of each transgene insertion event were verified by targeted PCR, restriction endonuclease fingerprinting and Southern blot (see Materials and methods section and Supplementary Figures). These analyses confirmed the targeted deletion of HSIII-V in ΔHSIII-V/hGH/BAC, the targeted deletion of a 12-kb segment between HSIII and hGH gene cluster in ΔSpacer/hGH/BAC, and the replacement of entire hGH cluster with a single isolated hCS-A PGR in LCR-CSA/BAC. One of the ΔSpacer/hGH/BAC lines was found to have a 3′ truncation within the hGH cluster, resulting in a loss of three placental genes (hCS-A, hGH-V and hCS-B), and was excluded from this study (Supplementary Figure S2). The impact of each of the three major alterations on placental gene expression from the hGH transgene locus was determined by comparing expression, tissue specificity and consistency of the expression to that of the wild-type hGH/BAC transgene.
Structure and expression of the intact hGH/BAC transgene locus in the mouse placenta
The components of the placental LCR are defined by a set of three DNase I HSs; HSIII, IV and V (4). To validate our transgenic model, we sought to demonstrate the formation of these sites within the intact hGH/BAC transgenic locus in the mouse placenta. Placental chromatin from E18.5 day embryos was isolated and mapped by partial DNAse I digestion and indirect end-labeling Southern analysis. A chromatin sample from the human placental choriocarcinoma cell line, JEG3, served as a positive control. The three DNase I HSs that constitute the hGH LCR in the placental chromatin were detected in the JEG3 chromatin DNA and in the hGH/BAC placental chromatin sample (Figure 2A). These data indicate that the three placental HSs were re-established in the hGH/BAC transgene in mouse genome.
Figure 2.

The hGH transgene locus is appropriately configured and expressed in the mouse placenta. (A) DNase I HS mapping of placental chromatin from transgenic mouse embryos carrying the intact hGH/BAC. Chromatin samples were exposed to DNase I for the times indicated to generate partial digestion products. The DNAs were then extracted, digested with EcoRI and analyzed by indirect-end labeling Southern blotting, as indicated in the diagram below the autoradiograph. The intact 23-kb EcoRI fragment encompassing the HSIII–V region is indicated by the gray arrowhead to the left of the autoradiograph and the sub-fragments generated by DNase I cleavage at HSIII, IV and V are indicated by black arrows and the black dots. A corresponding analysis of chromatin from a human placental choriocarcinoma line, JEG3, served as a positive control for placental HS formation. The pattern seen is consistent with that generated from normal human term placenta (4). Notably, two sub-bands (indicated by white dots) were observed in the Southern blot. These bands were detected consistently in the human placental chromatin and were previously demonstrated as non-specific bands unrelated to DNase I treatment (4,5). (B) Expression of the intact hGH/BAC transgene demonstrated appropriate tissue specificity. RT-PCR analyses of mRNAs isolated from the indicated tissues are shown. The primer set co-amplifies mRNAs generated from all five genes in the hGH cluster. GAPDH mRNA served as loading control. (C) Expression pattern from the intact hGH/BAC transgene in the mouse placenta recapitulated the expression from the native hGH locus in the human placenta. A 32P-labeled cDNA generated from co-amplification of all five of the hGH/hCS mRNAs (as in B above) was digested by TaqI to distinguish the expression of the individual mRNA species (see Materials and methods section). The products revealed robust expression of hCS-A/hCS-B mRNAs, much lower expression of hGH-V mRNA, and only trace detection of the hCS-L pseudogene transcript and the pituitary-specific hGH-N mRNA. This pattern recapitulated that observed in the human term placenta (5).
The expression of mRNAs from the intact hGH/BAC was next determined by a validated RT-PCR analysis that distinguishes the mRNAs originating from each of the five genes in the hGH cluster (5). The assay demonstrated appropriate expression from the PGRs within the hGH/BAC (Figure 2B); the two hCS genes, hCS-A and hCS-B, were robustly expressed with much lower level of hGH-V expression. In contrast, only trace of hCS-L (a pseudo gene) and hGH-N mRNAs could be detected (Figure 2C). These data are fully consistent with our prior studies of native human term placenta (5) and demonstrated that the hGH/BAC transgene contains the full set of placental LCR determinants and regulatory structures necessary for appropriate placental expression.
The HSIII-V region protects the hCS genes from site-of-integration effects
The impact of the HSIII-V determinants on hCS expression was explored by selectively deleting this region from the hGH/BAC transgene. Four ΔHSIII-V/hGH/BAC transgenic mouse lines lacking the 6-kb segment encompassing HSIII-V were generated and structurally validated (Supplementary Figures S1 and S2). Transgenic males from each line were crossed with wild-type CD-1 females and parallel crosses were carried out with lines carrying the intact hGH/BAC transgene. Transgenic E18 embryos from each mating were identified and the corresponding placentas were isolated and assessed for expression from the hGH/hCS transgene cluster. The analysis of the ΔHSIII-V/hGH/BAC lines confirmed the activation of the two hCS genes, hCS-A and hCS-B, in the placenta with significantly lower level of hGH-V mRNA, and trace levels of the hCS-L and hGH-N mRNAs (Figure 3A). This expression pattern accurately models the selective expression of the corresponding genes in human placenta and is in agreement with the analysis of the hGH/BAC lines (Figure 2C). These data demonstrate that HSIII-V function(s) is not essential for maintaining selective activation of hCS genes in the placenta.
Figure 3.

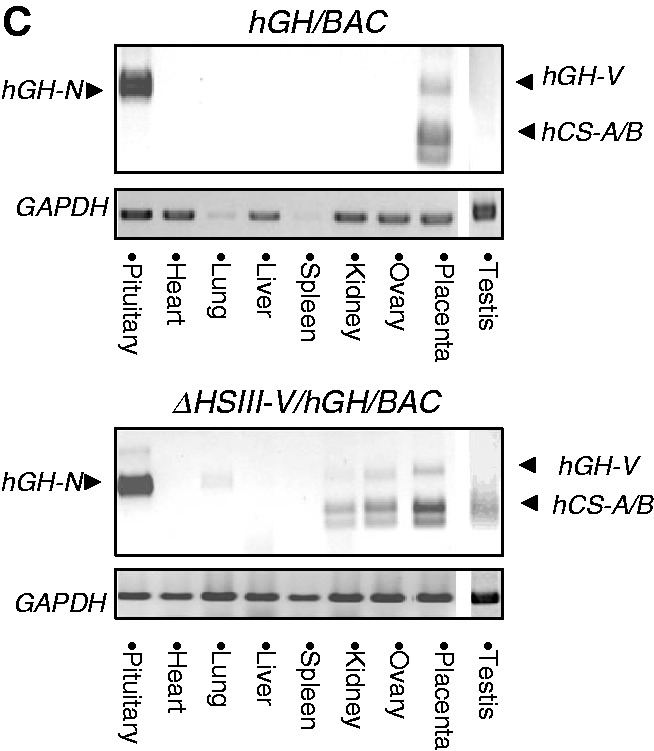
The HSIII-V region played an essential role in protecting the hGH transgene locus from site-of-integration effects in the placenta. (A) hCS mRNAs were robustly expressed from the ΔHSIII-V/hGH/BAC transgene. RT/PCR (left) and coRT/PCR-TaqI (right) analyses confirmed robust and appropriately selective expression of the hCS genes in the transgenic mouse placenta (analyses as in Figure 2B and 2C). (B) Deletion of the HSIII–V region rendered transgenic expression sensitive to site-of-integration effects in the placenta but not in the pituitary. (Top) Placental studies. Expression of hCS mRNAs in the placenta of four structurally-intact hGH/BAC lines and in four ΔHSIII-V/hGH/BAC lines was determined by RT/Q-PCR. The values were each normalized to the corresponding levels of GAPDH mRNA and then normalized to transgene copy-number (shown as mean + SD, n = 3). The copy number for each line is shown in parentheses next to the respective line designations. Regression analyses of the two sets of data, shown below the corresponding histograms, evaluated the correlation between total hCS mRNA expression (mean, n = 3) and copy number. The comparison of the linear regression r2-value for the hGH/BAC and the ΔHSIII-V/hGH/BAC lines (0.97; P-value 0.02 and 0.78; P-value 0.11) revealed a significant loss of copy-number dependence of transgene expression subsequent to deletion of the HSIII-V region. (Bottom) Pituitary studies. RT/Q-PCR analysis of pituitary hGH-N mRNA was normalized to GAPDH mRNA and transgene copy number (as in placental samples, above). The histogram represents the average of expression from triplicate assays of the indicated lines (+SD). The regression analyses of the data are shown below the histograms with high r2-values for both the intact hGH/BAC and ΔHSIII-V/hGH/BAC transgenic lines. (C) Deletion of the HSIII–V region triggered ectopic expression of hCSs. Tissue surveys from an intact hGH/BAC line (#1255B) and a ΔHSIII-V/hGH/BAC line (#01) are shown. RNAs were subjected to the coRT/PCR-TaqI analysis. These studies revealed widespread ectopic expression of hCS mRNAs from ΔHSIII-V/hGH/BAC transgene. In contrast, hGH-N mRNA expression remained tightly restricted to the pituitary in both lines.
We next assessed whether the HSIII-V region protects the hGH locus from transcriptional influences at the random transgene-insertion sites in the mouse genome. This was accomplished by determining the absolute level of hCS mRNAs normalized to the corresponding level of GAPDH in the placenta in each of the four WT/hGH/BAC lines and in each of four ΔHSIII-V/hGH/BAC lines. Line-to-line variability of expression was examined by regression analysis (Figure 3B, bottom placental panels), and mRNA expression per gene copy was calculated for each line (Figure 3B, top placental panels). In the WT/hGH/BAC mice, the expression of the placental hCS mRNAs per gene copy was maintained within a tight range, with an average value of 0.24 for the four lines (top left panel) and an r2-value of 0.95 (bottom left panel). In contrast, the expression of placental hCS mRNAs per transgene copy varied significantly among the four ΔHSIII-V/hGH/BAC lines (top right panel), varying from 0.19 (line #04) to trace (0.004 in line #06), with an r2-value of 0.78. These data revealed a marked relaxation in quantitative control of transcriptional activity of the hCS genes subsequent to deletion of the HSIII-V region.
To further assess the role of the HSIII-V region in establishing an insulated and autonomous chromatin environment, we determined the sensitivity of the ΔHSIII-V/hGH/BAC locus at each insertion site to ectopic expression in non-placental tissues. A control study with WT/hGH/BAC transgenic lines confirmed the tight control of tissue-specific regulation; hCS mRNAs were restricted to the placenta and hGH-N mRNA was restricted to the pituitary (Figure 3C, top). In contrast, the tissue-specificity of the ΔHSIII-V/hGH/BAC was clearly relaxed. In ΔHSIII-V/hGH/BAC line #01, hCS mRNAs were evident in kidneys, ovaries and testes as well as in the placentas (Figure 3C, bottom). A similar loss of tissue-specific regulation was observed in the ΔHSIII-V/hGH/BAC lines #03 and #04 (Table 2). Furthermore, in the ΔHSIII-V/hGH/BAC line #06, the hCS expression was completely inactivated in all tested tissues with only trace amount of expression detected in the E18 placenta (Figure 3B and Table 2). These data support the quantitative analyses of gene expression (Figure 3B) by demonstrating that deletion of the HSIII-V region rendered hCS expression sensitive to transcriptional influences of the host insertion site. The ectopic expression of the ΔHSIII-V/hGH/BAC transgene in non-placental tissues along with the relaxation of copy-number-dependent expression of hCS in the placenta led us to conclude that the HSIII-V region contained a boundary/insulator activity critical to the establishment of a fully autonomous hGH transgene locus.
Table 2.
Summary of hGH/CS expression in WT/hGH/BAC and ΔHSIII-V/hGH/BAC transgenic lines
| Pituitary | Placenta | Lung | Kidney | Heart | Liver | Ovary | Testis | Spleen | |
|---|---|---|---|---|---|---|---|---|---|
| WT 17 | GH-N | CSs | - | - | - | - | - | - | - |
| WT 1210B | GH-N | CSs | - | - | - | - | - | - | - |
| WT 1254D | GH-N | CSs | - | - | - | - | - | - | - |
| WT 1255B | GH-N | CSs | - | - | - | - | - | - | - |
| ΔHSIII-V 01 | GH-N | CSs | - | CSs | - | - | CSs | CSs | - |
| ΔHSIII-V 03 | GH-N | CSs | - | CSs | – | - | - | CSs | - |
| ΔHSIII-V 04 | GH-N | CSs | - | - | - | - | - | CSs | - |
| ΔHSIII-V 06 | GH-N | CSs* | - | - | - | - | - | - | - |
The tissue-specific expression of hGH-N and the hCSs was tightly controlled in all of the intact hGH/BAC transgenic lines whereas the placental genes demonstrated ectopic expression in three of the ΔHSIII-V/hGH/BAC lines. In a fourth line (#06) the expression of the placental genes was completely lost in all tested tissues, including placenta, but hGH-N remained fully expressed in the pituitary (see Figure 3B as well).
*Expressed at extremely low level.
The boundary/insulator activity mediated by the HSIII-V region is placenta-specific
The HSIII and HSV exist in both pituitary and placental chromatin. As these sites are shared between the two tissues and mark the 5′-boundary of the continuous 32-kb domain of histone acetylation established in pituitary somatotrope chromatin (13), we had previously suggested that they might serve a boundary and/or insulator function for the hGH cluster in both tissues (4). Consistent with this function, we observed that deletion of the HSIII-V region resulted in loss of site-of-integration protection for the placental genes (Figure 3B and C). To further test this model, we analyzed the impact of this deletion on hGH-N gene expression in the pituitary. Remarkably, all the lines, both WT/hGH/BAC and ΔHSIII-V/hGH/BAC, demonstrated stringently consistent level of pituitary expression (Figure 3B pituitary panels); the level of hGH-N expression in the pituitary retained tight copy-number dependence (r2-value of 0.97 in the ΔHSIII-V/hGH/BAC lines. Most strikingly, we observed robust and copy-number-dependent expression of hGH-N in the pituitary of ΔHSIII-V/hGH/BAC line #06, despite the almost complete silencing of expression of hCSs from this line in the placenta. A tissue survey of the ΔHSIII-V/hGH/BAC lines (Table 2) further confirmed that the deletion of the HSIII-V region had no adverse effect on the pituitary-restriction of hGH-N expression. These data demonstrated that the HSIII-V region is essential to the formation of an autonomous chromatin locus in the placenta; remarkably this effect is tissue specific as it does not play a corresponding boundary/insulator role for hGH-N expression in the pituitary (see Discussion).
The spacing between the hGH LCR and the hGH gene cluster has a quantitative impact on gene expression in the placenta
The preceding analyses of the ΔHSIII-V/hGH/BAC transgene revealed that the region encompassing HSIII-V serves an essential role in protecting the hGH/BAC transgene from site-of-integration effects on hCS expression in the placenta. This region is separated from the hGH cluster by 28 kb. This intervening region contains the two pituitary-specific LCR determinants, HSI and HSII. Of note, and consistent with DNase-seq data from ENCODE, we did not detect any HS in the spacing region in placental chromatin (4). It remains possible, however, that this region contains functional determinants that are unmarked by DNase I HSs. Alternatively, it is possible that it mediates a spatial/structural impact on hCS expression. The latter possibility is of particular interest in light of the high frequency with which regulatory elements in the mammalian genome are located at extensive distances from target promoters (1,18). To test these models, we removed a 12-kb segment between HSIII and hGH-N (Figure 1B). This modification in the ΔSpacer/hGH/BAC transgene deleted the region encompassing the pituitary-specific HSI and II, and moved the HSIII-V region closer to the hGH gene cluster.
The expression pattern of the genes within the ΔSpacer/hGH/BAC transgene was assessed in E18 placentas. Of the four lines established, one was eliminated from analysis due to a deletion of the placental gene segment of the cluster (Supplementary Figure S2). In all three remaining transgenic lines, we observed an appropriate pattern of expression; hCS-A and hCS-B were robustly expressed with only trace expression from the remaining three genes in the cluster (Figure 4A). This result demonstrated that the sequence between the placental LCR and the gene cluster does not contain any cis-acting element essential for the selective expression of the hCS genes in the placenta.
Figure 4.

The spacing between the HSIII–V region and the hGH gene cluster had a quantitative impact on hCS expression. (A) The native pattern of the placental gene expression from the hGH locus was fully maintained in the ΔSpacer/hGH/BAC transgene. mRNA expression from an intact hGH/BAC transgene (line #17) and a line carrying the ΔSpacer/hGH/BAC transgene (line #7) were analyzed by coRT/PCR-TaqI analysis (as in Figure 2C). (B) Gene expression from the ΔSpacer/hGH/BAC transgene was maintained in strict copy-number dependence. The RT/Q-PCR data compared copy-number expression from three hGH/BAC lines and three ΔSpacer/hGH/BAC lines. The regression analyses confirmed strict copy-number dependence in both sets of data. Remarkably, the expression per transgene copy from ΔSpacer/hGH/BAC lines was consistently higher than from the hGH/BAC lines (average of 0.51 and 0.30, respectively). (C) The placenta-specific expression of hCS mRNAs was fully maintained in the ΔSpacer/hGH/BAC lines. Analyses of nine tested tissues from the intact hGH/BAC (line #17) and the ΔSpacer/hGH/BAC (line #7) revealed that the activation of placental CS genes is restricted to the E18 placenta in both sets of transgenes. The hGH-N expression in the pituitary was selectively inactivated in the ΔSpacer/hGH/BAC transgene due to the deletion of the pituitary-specific HSI and II (located within the 12-kb deleted region).
Expression from the ΔSpacer/hGH/BAC lines was next assessed by RT/Q-PCR. The hCS mRNAs were robustly expressed in all three lines and the levels of expression were tightly correlated with copy number of transgene (r2 = 0.98; Figure 4B). Furthermore, the analysis of RNAs extracted from different mouse tissues demonstrated that the hCS genes remained exclusively placenta-specific (Figure 4C). Of note, however, the Q-PCR analysis revealed that the average level of expression per transgene copy from three ΔSpacer/hGH/BAC lines was significantly higher than that from the intact hGH/BAC transgenic lines (t-test, P-value = 0.016). This increased mRNA output from the ΔSpacer/hGH/BAC transgene suggested that moving the LCR into closer proximity to its target gene leads to an enhancement of expression.
The multi-gene structure of the hGH locus is critical to transcriptional control of the placental CS genes
The five hGH/hCS paralogs are derived from a single ancestral hGH gene via segmental duplications (19). Reflecting this evolutionary event, their gene structures and proximal regulatory elements are highly conserved (3,15). In addition to their parallel structural features, the histone modifications of regions encompassing each PGR unit, and the corresponding temporal dynamics of their establishment during STB differentiation (see Introduction section) are identical. These data suggested a model in which the hCS genes are individually controlled and activated, independent of the multigene structure of the hGH locus (5,15). To test this model, we generated a transgenic construct, in which the multi-gene cluster was replaced by a single hCS-A placental gene repeat (hCS-A PGR). The 7-kb hCS-A PGR encompasses a hCS-A gene along with its contiguous P-element and 3′-enhancer (11–13). The placental LCR as well as the remainder of hGH locus remain intact in this LCR-CSA/BAC transgene (Figure 1B). Importantly, the replacement of the gene cluster entailed a 3′-truncation of the B-cell-expressed CD79b gene, leaving the CD79b promoter and first two exons intact. As a result of this configuration, any transcription initiated at the CD79b promoter might extend into the CS-A gene. Therefore the RT/Q-PCR assay of hCS mRNA expression was specifically designed to quantify expression originating from the hCS-A promoter (Figure 5A).
Figure 5.

The multigene structure of the hGH locus was critical for the appropriate activation of the placental hCS genes. (A) Replacement of the hGH gene cluster with a single PGR unit (LCR-CSA/BAC transgene). Replacing the hGH cluster with a single hCS-A PGR unit involved truncation of the 3′ terminus of the B-cell specific CD79b gene. Thus, any transcription initiated from the B-cell promoter of the CD79b gene in contaminating B cells would have the possibility of extending into the CS-A locus (‘mRNA1’). To specifically detect transcripts originating from the hCS promoter (‘mRNA2’ originating at the hCS promoter) we used the RT/PCR primer set shown in the diagram. (B) hCS-A was robustly expressed from the LCR-CSA/BAC transgene but lacked copy-number dependence. CoRT/PCR-TaqI analysis (as in Figure 2C) confirmed robust hCS-A mRNA expression from the LCR-CSA/BAC transgene (left panel). However the levels of the expression per transgene copy demonstrated a marked line-to-line variation (r2 = 0.59) (right panels). (C) Expression of hCS from the LCR-CSA/BAC transgene demonstrated a dramatic loss of tissue specificity. Tissue survey of expression from the indicated hGH transgene loci was analyzed by RT/PCR.
Analysis of expression from the LCR-CSA/BAC transgene in the placenta revealed robust expression of hCS-A mRNA (Figure 5B, left). However a quantitative analysis of hCS-A mRNA in each of the four independent LCR-CSA/BAC lines revealed a wide variation in the copy-number-dependent transgenic expression (Figure 5B, right top), with an r2-value of 0.59 (Figure 5B, right bottom). In addition, the tightly controlled placental restriction of hCS expression, observed in WT/hGH/BAC lines (Figure 5C, top), was severely disrupted in the LCR-CSA/BAC lines with ectopic hCS-A expression in all tissues tested (Figure 5C, bottom). These data demonstrated that an isolated hCS-A gene, removed from the context of the multigene cluster, could be robustly activated but had substantially lost placental-specificity and consistency of gene expression.
Placenta-specific CTCF occupancy maps to HSIV
CTCF (or CCCTC-binding factor) has been shown to mediate multiple transcriptional controls (20,21). Prominent among proposed functions of CTCF is that of transcriptional insulation (21–23). The comparisons of expression profiles from the WT/hGH/BAC and ΔHSIII-V/hGH/BAC transgenes described above point to an essential function of the HSIII-V region in insulating the hGH cluster from site-of-integration effects at the placental chromatin locus. To explore the possible role of CTCF in this activity, the CTCF occupancy in the placental chromatin was investigated. We first scanned the hGH locus for the CTCF-binding consensus sequence (24) and located three predominant binding motifs co-mapping with HSV, IV and III (coordinates 32-, 30- and 28-kb 5′ of the hGH-N promoter). We next reviewed the distribution of CTCF occupancy in various tissues from the ENCODE data-base (25). Remarkably, the ChIP-seq data-base revealed enrichment for CTCF occupancy at HSIII and HSV, but clearly excluded HSIV (Figure 6A, top). Of note, however, the CTCF-binding data in the ENCODE data-base for primary placental cells was absent. Thus, to extend the CTCF analysis to the placental chromatin, we performed a CTCF ChIP on the E18 mouse placenta and non-placental-tissue controls (liver and kidney) from mice carrying the WT/hGH/BAC transgene. The results from analysis of the WT/hGH/BAC transgene locus were consistent with our analyses using chromatin isolated from two human placenta cell-lines, BeWo and JEG3. These studies revealed robust CTCF occupancy at sites corresponding to HSIII, IV and V in the placental tissue (Figure 6B). In contrast, and consistent with the ENCODE data-base, CTCF enrichment in the liver and kidney was limited to HSIII and HSV, and was clearly absent at HSIV. These data revealed placenta-specific CTCF recruitment at HSIV.
Figure 6.

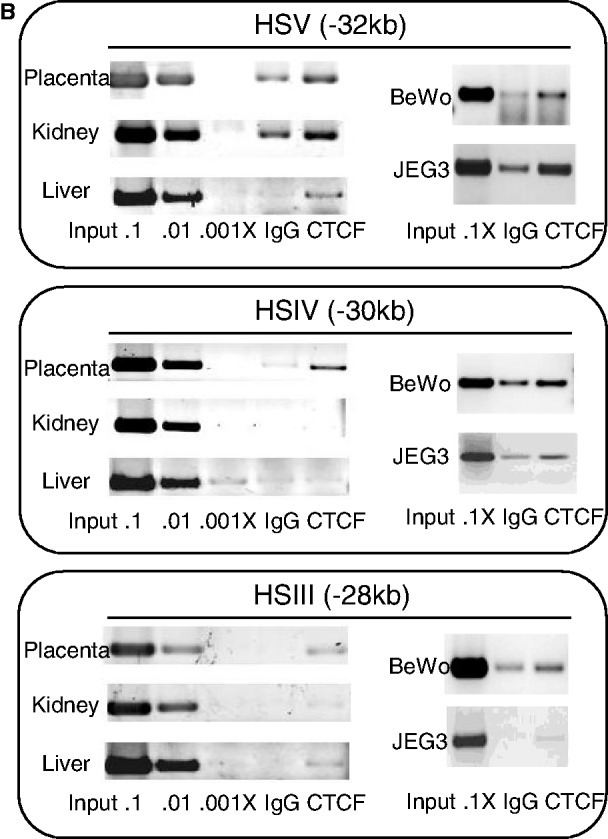
Placenta-specific occupancy of CTCF at HSIV of the hGH LCR. (A) Placental specific occupancy of CTCF at HSIV. The positions of the previously reported DNase I HSs at the hGH locus (4) and the predicted CTCF-binding site (CTCFBS) across the locus based on the presence of consensus sequences (24) are indicated (downward arrows and boxes, respectively). In vivo CTCF occupancy across the hGH locus in five primary human tissues are shown below the locus diagram (ENCODE data base). The predicted and documented CTCF-binding sites were highly correlated and indicated that the great majority of CTCF sites in this region are constitutive. The clear outlier was the predicted CTCF-binding site co-mapping with HSIV (–30 kb); this predicted CTCF site was unique in this region in that it lacks in vivo occupancy in the multiple tissues sampled. The enrichment of CTCFs in the placental LCR of hGH locus was investigated by ChIP. CTCF-bound chromatin fragments were immunoprecipitated from E18 placenta, kidney and liver of an hGH/BAC transgenic mouse. A serial dilution of input DNA was used to confirm that the PCR amplification was in linear range and an IgG antibody was used for the IP negative control. DNAs isolated from the IP pellets were assessed by PCR using three primer sets that tested the predicted CTCF-binding sites at HSIII, HSIV and HSV (shown in Figure 6B). Enrichment values were quantified and normalized with the positive control (see Materials and methods section, defined as 100). The columns in the histogram represent the average of duplicated experiments with standard deviations (n = 2). The CTCF distribution across the hGH locus showed that the CTCFs were enriched at all three HS sites in placenta (black column), but were limited to the HSIII and HSV in kidney (dark gray column) and liver (light gray column). The enrichment in the region between LCR and hGH cluster was detected at a lower level (∼30%) at coordinate –25 kb in placenta and liver, and at background level at coordinate –15 kb site in all tested tissues. The enrichment was also observed at promoters of hGH/CS genes and 3′-enhancers of hCS genes in the gene cluster (a single PGR represents all placental units in the analysis due to the highly conserved sequence in these regions). (B) CTCF binding at HSIII, IV and V in primary tissues and in two placental cell lines. CTCF ChIP was carried out on chromatin isolated from the indicated primary tissues of hGH/BAC mice and from two human choriocarcinoma cell lines, BeWo and JEG3. The assay confirmed CTCF occupancy at HSIV in these placental cell lines.
The CTCF distribution across the remainder of the locus was then investigated. The intensity of enrichment at each site was quantified after normalization to a positive control (Figure 6A, bottom). The DNA sequences corresponding to the promoters and 3′-enhancers of the hGH/hCS paralogs are highly conserved (3) and the primers for this assay included all of the individual PGR units as shown in the Figure 6A. The ChIP study failed to reveal any significant difference in CTCF occupancy between placenta and the two non-placental controls in these regions. This constitutive pattern of CTCF occupancy at all sites within the cluster was in agreement with the ENCODE studies. The mapping of a placenta-specific insulator activity within the HSIII-V region, the unique formation of DNase HSIV in the placenta chromatin, and the placenta-specific occupancy of CTCF at HSIV were consistent with the conclusion that HSIV represents a placenta-specific boundary/insulator determinant.
DISCUSSION
The eukaryotic genome exists in a tightly packed chromatin structure. To be expressed, the regulatory elements must be reconfigured and exposed to facilitate efficient interactions with trans-acting factors and RNA polymerase. These activated sites can be identified and mapped within a chromatin locus by their hypersensitivity to DNase (26,27). In prior studies we identified two overlapping sets of DNase I HSs, located between 32-and 14.5-kb 5′ to the hGH cluster in pituitary and placental chromatin (Figure 1A). HSI and II specifically assemble in pituitary chromatin and have been shown in a series of in vivo studies to serve functions that are both essential and sufficient to activation of somatotrope-specific hGH-N gene expression (4,9,28,29). The more distal HSIII and HSV assemble in the chromatin of both pituitary and placenta, as well as in multiple additional tissues, and as such they are likely to mediate constitutive functions. A single remaining site, HSIV, is remarkable in that it assembles selectively in the chromatin of placental villous STBs (our data (4) and the ENCODE data base), the site of hCS expression (30,31). Based on these data, along with the corresponding studies of histone modifications at the hGH locus (5,15), we have proposed that determinants marked by HSIII, HSIV and V constitute a placental LCR and serve regulatory roles specific to placental expression from the hGH cluster.
To test its putative regulatory activities (32), we have deleted the placental LCR region (HSIII-V) from the hGH/BAC and assessed the expression of the resultant ΔHSIII-V/hGH/BAC transgene in E18.5 embryonic placentas. The data revealed a clear relaxation of copy-number-dependent transgenic expression (Figure 3B) and an accompanying relaxation in tissue-specific expression (Figure 3C and Table 2). This impact on gene expression most likely reflected a loss of protection of the transgene from insertion-site effects. The most extreme example of this site-of-integration effects was the complete repression of hCS expression in placentas of embryos in line #06 (Figure 3B). These observations suggested that the HSIII-V region, while not essential to the robust activation of hCS genes in the placenta (Figure 3A), plays an important role(s) in the LCR boundary/insulator function.
Genome-wide analyses have mapped CTCF-binding sites to the boundaries of a number of active chromatin regions (33,34). Studies in a diverse set of experimental models support a role of CTCF in locus insulation (22,35,36). To examine whether the HSIII-V boundary activity identified at the hGH transgene locus in the placenta was related to the CTCF actions, we assessed CTCF occupancy across the intact hGH/BAC locus. The results of this article, combined with data from the ENCODE, revealed that CTCF occupancies at HSIII and HSV are ubiquitous while occupancy at HSIV is specific to the placental locus (Figure 6A). Quite remarkably, the deletion of HSIII-V region (ΔHSIII-V/hGH/BAC transgene) had no appreciable impact on the expression of hGH-N in pituitaries (Figure 3B and Table 2). The selective impact of the HSIII-V deletion on expression from the placental chromatin locus was highlighted by the observation that hGH-N expression of ΔHSIII-V/hGH/BAC line #6 in the pituitary is robust and copy-number dependent (Figure 3B). Given that HSIII and V are formed at the chromatin locus in both placentas and pituitaries while HSIV is placenta-specific, these observations support a model in which CTCF recruitment at HSIV plays an essential role in insulating the hGH transgene locus from site-of-integration effects in placental chromatin.
Regulatory functions of CTCF include its ability to organize chromatin into higher order structures (23,37–39). Genome-wide surveys of CTCF occupancy have demonstrated that the vast majority of CTCF-binding sites are constitutive and overlap extensively among different cell types (24,34). This suggested a model in which CTCFs cooperate with other proteins, such as cohesins or mediators, to generate thousands of isolated chromosomal loops throughout the genome. While many of the CTCF-mediated chromosomal structures are constitutive, a subset possesses tissue and/or developmental specificity (38,40,41). These particular structures are comprised of cell-type-specific CTCF-bindings that are often located at the boundaries of active chromatin domains (33,34). Our analysis of the placental boundary of hGH locus supported this model and suggested that the placenta-specific occupancy of CTCF at HSIV may involve the generation of a placenta-specific higher order chromatin structure critical to the establishment of a functionally autonomous hGH transgene cluster at the placental chromatin locus.
In addition to the LCR determinants 5′ to the hGH cluster, a structural feature of potential relevance to the control of placental expression from the hGH locus is its internal repeat structure. The five gene paralogs are evenly spaced within the cluster and each of the four placentally transcribed genes is flanked by a set of highly conserved proximal regulatory elements. This organization along with the patterns of epigenetic histone modifications within the cluster suggested that each of the four PGR units may be independently activated. Here we have tested this model by collapsing the multi-gene cluster to a single intact PGR unit (hCS–A PGR). This LCR-CSA/BAC transgene fully preserved the structure of the LCR as well as extensive flanking regions. Surprisingly, this conversion of a multi-gene cluster to a single gene locus resulted in a radical alteration in gene expression with a dramatic loss in tissue specificity as well as copy-number dependency (Figure 5B and C). These data argued against a model in which PGR units are individually activated and supported the conclusion that the overall structure of the hGH cluster is critical for appropriate activation of the individual placental units. This may reflect the assembly of a higher order regulatory configuration within the cluster. The occupancies by CTCFs at several sites within the hGH cluster (Figure 6A) may organize this configuration and bring the hCS genes into close proximity with the distal LCR elements. CTCF binding at HSIV might specifically modify this organization to facilitate expression of the hCS genes in the placenta. This revised model is comparable with the structure defined at the β-globin multi-gene locus where the spatial organization of the locus describes a 3D, cell-type-specific assembly of a multipartite ‘chromatin hub’ that juxtaposes the erythroid regulatory elements with their target globin genes (38,40–42). This chromatin hub is established by the interactions between CTCF-bound cis-regulatory elements (40). The conditional deletion of CTCF, or targeted disruption of the CTCF-binding site at the 3′ HSI of the globin locus, destabilizes the long-range interactions that comprise the chromatin hub and leads to a local loss of histone modifications (43). Although the chromatin conformation of the hGH cluster needs to be directly assessed in subsequent studies, it is clear from the current data that interruption of the repeat structure of the native hGH cluster, or deletion of placental LCR led to inappropriate patterns of gene regulation.
The current investigations, using a series of specifically modified hGH locus transgenes, suggested that the appropriate expression of the placental CS genes depended on the boundary actions of the HSIII-V LCR (ΔHSIII-V/hGH/BAC) and the overall gene–repeat configuration of the cluster (LCR-CSA/BAC). Both the repeated structure of the locus and the structure of the HSIII-V region were maintained in the third experimental transgene in which the spacing between the placental LCR and the gene cluster was selectively reduced (ΔSpacer/hGH/BAC). In this structural setting the hCS genes were properly regulated and retained copy-number dependency (Figure 4B and C). Remarkably, however, the absolute expression of hCS mRNAs from this transgene was significantly enhanced relative to the intact hGH/BAC locus (Figure 4B). This enhancement of hCS expression was reminiscent of, but less dramatic than, the enhancement of hGH-N expression in the pituitary when it was directly juxtaposed to the HSI and II determinants (4). In these two settings, the shortening of the distances between LCR determinants and corresponding target genes may reflect an increase in the frequency and/or productivity of the chromatin looping interactions (40,44). The dynamics of looping, as suggested by these data, may allow for flexibility in gene regulation that may be of particular importance to genes that have a highly dynamic range of gene activation, and/or genes that need to rapidly alter levels of enhancer activity in developmental or physiologic settings. The greater distance between HSIII-V to the placental genes (36 kb) as compared to that between HSI and II to the hGH-N promoter (15 kb) may necessitate selective stabilization of a putative looping structure by the HSIV-occupied CTCF complex. Subsequent studies defining the overall 3D structure of the hGH locus will be needed to further extend and validate these models.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank the University of Pennsylvania Transgenic and Chimeric Mouse Facility (supported by National Institutes of Health [P30’s DK019525, DK050306, CA016520]) for generation of the transgenic mice for our studies.
FUNDING
National Institutes of Health (NIH) [R01 HD25147 and R01 HD046737 to N.E.C and S.A.L.]. Funding for open access charge: NIH [R01 HD25147 and R01 HD046737].
Conflict of interest statement. None declared.
REFERENCES
- 1.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Portela A, Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 3.Chen EY, Liao YC, Smith DH, Barrera-Saldaña HA, Gelinas RE, Seeburg PH. The human growth hormone locus: Nucleotide sequence, biology, and evolution. Genomics. 1989;4:479–497. doi: 10.1016/0888-7543(89)90271-1. [DOI] [PubMed] [Google Scholar]
- 4.Jones BM, Monks BR, Liebhaber SA, Cooke NE. The human growth hormone gene is regulated by a multicomponent locus control region. Mol. Cell. Biol. 1995;15:7010–7021. doi: 10.1128/mcb.15.12.7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimura AP, Sizova D, Handwerger S, Cooke NE, Liebhaber SA. Epigenetic activation of the human growth hormone gene cluster during placental cytotrophoblast differentiation. Mol. Cell Biol. 2007;27:6555–6568. doi: 10.1128/MCB.00273-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shewchuk BM, Asa SL, Cooke NE, Liebhaber SA. Pit-1 binding sites at the somatotrope-specific DNase I hypersensitive sites I, II of the human growth hormone locus control region are essential for in vivo hGH-N gene activation. J. Biol. Chem. 1999;274:35725–35733. doi: 10.1074/jbc.274.50.35725. [DOI] [PubMed] [Google Scholar]
- 7.Elefant F, Cooke NE, Liebhaber SA. Targeted recruitment of histone acetyltransferase activity to a locus control region. J. Biol. Chem. 2000;275:13827–13834. doi: 10.1074/jbc.275.18.13827. [DOI] [PubMed] [Google Scholar]
- 8.Ho Y, Elefant F, Cooke NE, Liebhaber SA. A defined locus control region determinant links chromatin domain acetylation with long-range gene activation. Mol. Cell. 2002;9:291–302. doi: 10.1016/s1097-2765(02)00447-1. [DOI] [PubMed] [Google Scholar]
- 9.Ho Y, Tadevosyan A, Liebhaber SA, Cooke NE. The juxtaposition of a promoter with a locus control region transcriptional domain activates gene expression. EMBO Rep. 2008;9:891–898. doi: 10.1038/embor.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang SW, Shepard AR, Eberhardt NL. An initiator element is required for maximal human chorionic somatomammotropin gene promoter and enhancer function. J. Biol. Chem. 1995;270:3683–3692. doi: 10.1074/jbc.270.8.3683. [DOI] [PubMed] [Google Scholar]
- 11.Nachtigal MW, Nickel BE, Cattini PA. Pituitary-specific repression of placental members of the human growth hormone gene family. J. Biol. Chem. 1993;286:8473–8479. [PubMed] [Google Scholar]
- 12.Jacquemin P, Martial JA, Davidson I. Human TEF-5 is preferentially expressed in placenta and binds to multiple functional elements of the human chorionic somatomammotropin-B gene enhancer. J. Biol. Chem. 1997;272:12928–12937. doi: 10.1074/jbc.272.20.12928. [DOI] [PubMed] [Google Scholar]
- 13.Elefant F, Su Y, Liebhaber SA, Cooke NE. Patterns of histone acetylation suggest dual pathways for gene activation by a bifunctional locus control region. EMBO. 2000;19:6814–6822. doi: 10.1093/emboj/19.24.6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su Y, Liebhaber SA, Cooke NE. The human growth hormone gene cluster locus control region supports position-independent pituitary- and placenta-specific expression in the transgenic mouse. J. Biol. Chem. 2000;275:7902–7909. doi: 10.1074/jbc.275.11.7902. [DOI] [PubMed] [Google Scholar]
- 15.Kimura A, Liebhaber SA, Cooke NE. Epigenetic modifications at the human growth hormone locus predict distinct roles for histone acetylation and methylation in placental gene activation. Mol. Endocrinol. 2004;18:1018–1032. doi: 10.1210/me.2003-0468. [DOI] [PubMed] [Google Scholar]
- 16.Gong S, Yang XW, Li C, Heintz N. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6K gamma origin of replication. Genome Res. 2002;12:1992–1998. doi: 10.1101/gr.476202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrin LJ, Camerini-Otero RD. Selective cleavage of human DNA: RecA-assisted restriction endonuclease (RARE) cleavage. Science. 1991;254:1494–1497. doi: 10.1126/science.1962209. [DOI] [PubMed] [Google Scholar]
- 18.West AG, Fraser P. Remote control of gene transcription. Hum. Mol. Genet. 2005;14:R101–R111. doi: 10.1093/hmg/ddi104. [DOI] [PubMed] [Google Scholar]
- 19.Barsh GS, Seeburg PH, Gelinas RE. The human growth hormone gene family: structure and evolution of the chromosomal locus. Nucleic Acids Res. 1983;11:3939–3958. doi: 10.1093/nar/11.12.3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallace JA, Felsenfeld G. We gather together: insulators and genome organization. Curr. Opin. Genet. Dev. 2007;17:400–407. doi: 10.1016/j.gde.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 23.Ghirlando R, Giles K, Gowher H, Xiao T, Xu Z, Yao H, Felsenfeld G. Chromatin domains, insulators, and the regulation of gene expression. Biochim. Biophys. Acta. 2012;1819:644–651. doi: 10.1016/j.bbagrm.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.ENCODE Project Consortium. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010 doi: 10.1101/pdb.prot5384. doi:10.1101/pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zentner GE, Henikoff S. Surveying the epigenomic landscape, one base at a time. Genome Biol. 2012;13:250. doi: 10.1186/gb-2012-13-10-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fleetwood MR, Ho Y, Cooke NE, Liebhaber SA. DNase I hypersensitive site II of the human growth hormone locus control region mediates an essential and distinct long-range enhancer function. J. Biol. Chem. 2012;287:25454–25465. doi: 10.1074/jbc.M112.365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunsaker TL, Jefferson HS, Morrison JK, Franklin AJ, Shewchuk BM. POU1F1-mediated activation of hGH-N by deoxyribonuclease I hypersensitive site II of the human growth hormone locus control region. J. Mol. Biol. 2012;415:29–45. doi: 10.1016/j.jmb.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Walker WH, Fitzpatrick SL, Barrera-Saldaña HA, Resendez-Perez D, Saunders GF. The human placental lactogen genes: structure, function, evolution and transcriptional regulation. Endocrine Rev. 1991;12:316–328. doi: 10.1210/edrv-12-4-316. [DOI] [PubMed] [Google Scholar]
- 31.Liebhaber SA, Urbanek M, Ray J, Tuan RS, Cooke NE. Characterization and histological localization of human growth hormone-variant gene expression in the placenta. J. Clin. Invest. 1989;83:1985–1991. doi: 10.1172/JCI114108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Q, Peterson KR, Fang X, Stamatoyannopoulos G. Locus control regions. Blood. 2002;100:3077–3086. doi: 10.1182/blood-2002-04-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 34.Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yusufzai TM, Tagami H, Nakatani Y, Felsenfeld G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol. Cell. 2004;13:291–298. doi: 10.1016/s1097-2765(04)00029-2. [DOI] [PubMed] [Google Scholar]
- 36.Xiao T, Wallace J, Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol. Cell Biol. 2011;31:2174–2183. doi: 10.1128/MCB.05093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. Architectural Protein Subclasses Shape 3D Organization of Genomes during Lineage Commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Junier I, Dale RK, Hou C, Képès F, Dean A. CTCF-mediated transcriptional regulation through cell type-specific chromosome organization in the β-globin locus. Nucleic Acids Res. 2012;40:7718–7727. doi: 10.1093/nar/gks536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Merkenschlager M, Odom DT. CTCF and cohesin: linking gene regulatory elements with their targets. Cell. 2013;152:1285–1297. doi: 10.1016/j.cell.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 40.Palstra RJ, de Laat W, Grosveld F. Beta-globin regulation and long-range interactions. Adv. Genet. 2008;61:107–142. doi: 10.1016/S0065-2660(07)00004-1. [DOI] [PubMed] [Google Scholar]
- 41.Kim A, Dean A. Chromatin loop formation in the β-globin locus and its role in globin gene transcription. Mol. Cells. 2012;34:1–5. doi: 10.1007/s10059-012-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patrinos GP, de Krom M, de Boer E, Langeveld A, Imam AM, Strouboulis J, de Laat W, Grosveld FG. Multiple interactions between regulatory regions are required to stabilize an active chromatin hub. Genes Dev. 2004;18:1495–1509. doi: 10.1101/gad.289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W. CTCF mediates long-range chromatin looping and local histone modification in the betaglobin locus. Genes Dev. 2006;20:2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krivega I, Dean A. Enhancer and promoter interactions-long distance calls. Curr. Opin. Genet. Dev. 2012;22:79–85. doi: 10.1016/j.gde.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.