

Abstract
The EphB receptors have key roles in cell morphology, adhesion, migration and invasion, and their aberrant action has been linked with the development and progression of many different tumor types. Their conflicting expression patterns in cancer tissues, combined with their high sequence and structural identity, present interesting challenges to those seeking to develop selective therapeutic molecules targeting this large receptor family. Here, we present the first structure of the EphB1 tyrosine kinase domain determined by X-ray crystallography to 2.5Å. Our comparative crystalisation analysis of the human EphB family kinases has also yielded new crystal forms of the human EphB2 and EphB4 catalytic domains. Unable to crystallize the wild-type EphB3 kinase domain, we used rational engineering (based on our new structures of EphB1, EphB2, and EphB4) to identify a single point mutation which facilitated its crystallization and structure determination to 2.2 Å. This mutation also improved the soluble recombinant yield of this kinase within Escherichia coli, and increased both its intrinsic stability and catalytic turnover, without affecting its ligand-binding profile. The partial ordering of the activation loop in the EphB3 structure alludes to a potential cis-phosphorylation mechanism for the EphB kinases. With the kinase domain structures of all four catalytically competent human EphB receptors now determined, a picture begins to emerge of possible opportunities to produce EphB isozyme-selective kinase inhibitors for mechanistic studies and therapeutic applications.
Keywords: EphB1, EphB2, EphB3, EphB4, crystallography, ligand-binding sites, protein engineering, activation mechanism
Introduction
The Eph receptors (EC 2.7.10.1) are type I transmembrane proteins that interact with their membrane-bound ligands, the ephrins. These interactions facilitate cell-to-cell contacts, resulting in bidirectional intracellular signaling cascades.1 Upon ephrin stimulation and receptor dimerization, the intracellular kinase domains transphosphorylate their juxtamembrane regions and cytoplasmic tails, driving the subsequent recruitment of down-stream signaling molecules.2 These interacting proteins include Src homology 2 and 3 adapter proteins, Src family kinases, phosphatidylinositol 3-kinase, mitogen-activated protein kinases, small GTPases, guanine nucleotide exchange factors, and phosphatases, each of which contribute to the complex cell repulsion and adhesion pathways that modulate cell shape, motility and attachment.3
The complex role of the Eph receptors in a variety of tumor types has led to a number of drug-discovery programs aiming to develop a range of different molecules to modulate Eph signaling, including antibodies, peptides and small-molecule kinase inhibitors.4 While the peptides and antibodies are focused on the extracellular part of the receptors, and also mimic their interaction with the ephrin ligands, the kinase inhibitors are aimed at intercepting the signaling inside the cell. A number of groups have described the development of specific Eph kinase inhibitors, with varying degrees of potency and selectivity,5–7 while others have described small molecules designed to target other kinases that also inhibit Eph kinases.8,9 Structural biology has played an essential part in the development of many of these Eph-kinase inhibitors, and is generally accepted to be a core drug-discovery tool for optimizing the potency, selectivity, and physical properties of small-molecule kinase inhibitors.10,11
The Eph receptors have been the subject of extensive structural studies by a number of different research groups, using both X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy. Such studies have elucidated the structures of many Eph extracellular domains and their interaction with their ephrin ligands,12–15 the transmembrane helices and their homodimeric interactions,16 and the formation of intracellular SAM domain oligomers.17,18 Early structural studies with the murine EphB2 and EphA4 kinase domains revealed important mechanistic insights into the auto-inhibitory and activation mechanisms of the Eph kinases.19,20 These studies were built on the use of EphA3,21 and were expanded to include characterisation of Eph-kinase substrate specificity.22 Further, structures of EphA3, EphA7, and EphB4 have driven the development of potent inhibitory molecules targeting these kinases.5,23,24 Of the fourteen human Eph receptors, half have had their kinase domain structures defined, leaving just six to be determined: EphA1, EphA6, EphB1, EphB3, and catalytically dead kinases EphA10 and EphB6. The high degree of sequence and structural conservation between the Eph kinase domains, and confusing data around their roles in different oncological settings, means that structure-driven optimisation of Eph-kinase selectivity is likely to prove essential in the future development of Eph-kinase inhibitors for clinical and mechanistic applications.
The EphB kinases are a small subfamily of the Eph receptors, comprising just five members. EphB1 to 4 are catalytically competent, while the more divergent EphB6 is catalytically dead. Further to the data we have presented on the biophysical and biochemical characterization of the four catalytically active EphB kinases,25 this article goes on to investigate their amenability to crystallization and crystallography. Crystallization screening revealed that wild-type forms of EphB1, EphB2, and EphB4 all crystallized readily, leading to the determination of the first EphB1 kinase domain structure, the first human EphB2 kinase domain structure and the first wild-type EphB4 structure. Although the equivalent EphB3 kinase domain construct was recalcitrant to crystallization, we have used the structures of the other EphB kinases to rationally engineer a crystallizable mutant form of EphB3 that has enabled its structure solution. This mutation also gave rise to higher yields of recombinant protein in Escherichia coli, which we have attributed to elevated intrinsic stability. The EphB3 structure contains a partially ordered activation loop which adopts a catalytically competent binding mode, indicating a possible cis-phosphorylation auto-activation mechanism for the EphB kinase family. We also discuss potential opportunities for routes to obtain small molecules with Eph-kinase selectivity.
Results
We have previously described a number of key differences between the four catalytically competent human EphB kinase domains EphB1, 2, 3, and 4 in terms of their amenability to recombinant expression in E. coli, their intrinsic stability, their enzymatic parameters and, importantly, their compound inhibition profiles.25 In this study, we sought to explore how these differences, most notably their recombinant expression and intrinsic stability, correlated with their amenability to structure determination by X-ray crystallography.
Crystallization of wild-type EphB kinases
To determine their intrinsic crystallizability, samples of each of the four EphB wild-type kinase-domain proteins were subjected to crystallization screening by sitting-drop vapour diffusion against 192 separate conditions, using three different ligand and buffer formulations. Three of the four proteins (EphB1, EphB2, and EphB4) produced a number of proteinaceous crystalline hits, some of which were suitable for X-ray diffraction testing. The highest hit-rate was observed for EphB4, which produced hits in ∼6% of conditions tested, compared with EphB1 and EphB2, which had a ∼2% hit rate. No screening hits were, however, obtained for EphB3 in any of the conditions/formulations tested. The initial screening hits required one to two rounds of optimization at microliter scale to produce diffraction-quality crystals from which diffraction data-sets for EphB1, B2 and B4 could be obtained [Fig. 1(B)].
Figure 1.
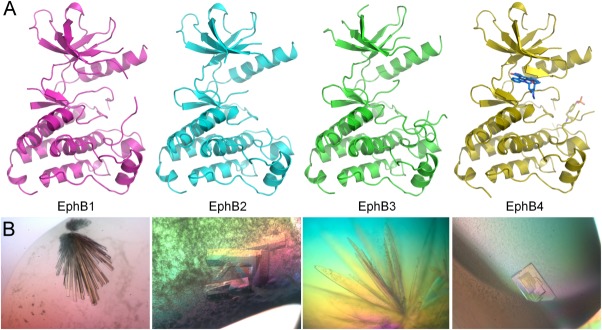
Structural gallery of the EphB kinase domains. (A) Backbone ribbon representations of the kinase domain structures of EphB1 (pink), EphB2 (cyan), EphB3 A899P (green), and EphB4 plus staurosporine (yellow). (B) Corresponding light microscope images of diffraction-quality crystals of EphB kinase domains. PyMol was used to prepare all structure images (http://www.pymol.org).
Crystals obtained for EphB2 and EphB4 were suitable for collection using in-house X-ray generators, whereas those obtained for EphB1 required a synchrotron light source to achieve the required data quality. The apoprotein structure of EphB2 and structure of EphB4 co-complexed with staurosporine, were solved by molecular replacement using previously published structures of the same proteins. The novel EphB1 structure was solved by using the published EphB2 structure, 1JPA, as a search model; refinement statistics and Protein Data Bank (PDB) accession codes for the three structures can be found in Table I.
Table I.
Summary of Data Collection and Refinement Statistics
| EphB1 | EphB2 | EphB4 + Staurosporine | EphB3 A899P | |
|---|---|---|---|---|
| PDB accession code | 3ZFX | 3ZFM | 3ZEW | 3ZFY |
| Data collection | ||||
| Space group | P32 | P1 | P21 | P1 |
| Cell dimensions | ||||
| a | 195.99 | 34.59 | 52.17 | 46.99 |
| b | 195.99 | 41.19 | 88.17 | 56.48 |
| c | 60.21 | 54.47 | 80.78 | 61.35 |
| α | 90 | 92.65 | 52.17 | 93.02 |
| β | 90 | 97.15 | 88.71 | 90.65 |
| γ | 120 | 114.79 | 80.78 | 90.03 |
| Resolution (Å) | 2.50 (2.50–2.64) | 2.27 (2.27–2.39) | 2.50 (2.50–2.59) | 2.20 (2.28–2.20) |
| Total observations | 464,293 (62,305) | 45,906 (6452) | 73,290 (9852) | 58,327 (4795) |
| Unique reflections | 89,433 (13,046) | 11,890 (1679) | 22,752 (3102) | 29,560 (2529) |
| Completeness | 100.0 (100.0) | 95.1 (92.6) | 91.3 (90.3) | 92.31 (83.0) |
| Rmerge | 0.154 (0.600) | 0.035 (0.117) | 0.111 (0.328) | 0.054 (0.144) |
| <I/σ(I)> | 8.3 (2.8) | 25.7 (10.3) | 6.3 (2.4) | 9.7 (3.8) |
| Refinement | ||||
| Resolution | 2.50 | 2.27 | 2.50 | 2.20 |
| No. reflections | 84,918 | 11,689 | 22,656 | 28,046 |
| R (%) | 19.09 | 23.7 | 24.98 | 22.82 |
| Rfree (%) | 21.80 | 26.11 | 28.03 | 25.24 |
| No. atoms | ||||
| Protein | 18,042 | 1933 | 4235 | 3989 |
| Other | 102 | 79 | 284 | 111 |
| Ramachandran (%) | ||||
| Favored | 96.5 | 96.3 | 95.8 | 94.3 |
| Allowed | 2.8 | 3.7 | 2.2 | 4.3 |
| Disallowed | 0.7 | 0.0 | 2.0 | 1.4 |
| r.m.s. deviations | ||||
| Bonds (Å) | 0.014 | 0.009 | 0.009 | 0.010 |
| Angles (degrees) | 1.373 | 1.050 | 1.040 | 1.289 |
| Mean isotropic B values | 28.31 | 28.66 | 65.75 | 33.02 |
Data in parentheses refer to the highest resolution shell.
Predictably, all EphB structures reported here adopt the traditional bi-lobed kinase fold, with a few observed differences; these are, to a large extent, the result of the hinge motion of the kinase domains upon ligand binding. As illustrated in Figure 1(A), the staurosporine-bound EphB4 adopts the most closed conformation, with the Gly-rich loop folding tightly over the ligand, while the apoprotein structures of EphB1 and EphB2 are more open, with the Gly-rich loops partly disordered. The asymmetric unit of the novel EphB1 structure contains nine molecules, with only minor differences between them. The EphB4 structure is different from the previously published EphB4 structures23,24 in that it exhibits a much more ordered activation loop with a phosphorylation on Tyr774, and slight changes in the glycine loop to accommodate the staurosporine molecule. The EphB2 structure is the first published apoprotein EphB2 structure, and is also the first wild-type human EphB2 kinase domain structure, although the two previously published EphB2 structures use a mouse sequence (UniProtKB/Swiss-Prot: P54763), which is identical to the human sequence in the kinase domain region.
EphB3 kinase domain model and mutant design
Instead of screening a greater number of conditions, ligands and alternative phosphoforms for the EphB3 kinase domain, we chose to identify a crystallizable form of the protein using rational engineering. In order to identify potential sites on EphB3 for mutational engineering, we used SWISS-MODEL (http://swissmodel.expasy.org/26), with its default settings, to construct a three-dimensional model of the EphB3 kinase domain, using the highest resolution EphB structure in the PDB, that of EphB4 in co-complex with a small-molecule kinase inhibitor (PDB: 2VWX). EphB4 shares an 86% identity with EphB3 within the chosen domain boundaries. The resultant model is shown in Supporting Information Figure 1(A). The model highlights the residues in EphB3 that are not conserved in the three other EphB kinases for which the X-ray structures have so far been solved (EphB1, EphB2, and EphB4). In total 19 residues are found only in EphB3 within our previously determined kinase domain boundaries, of which 15 are visible within the model. The majority of these unique residues are found in loop regions and as surface exposed hydrophilic residues on α-helices, together with a few conservative hydrophobic residue differences. The most striking difference, from a structural perspective, was an alanine residue at position 899. In each of the other three EphB kinases, this residue is a proline [Supporting Information Fig. 1(B)] that creates a kink in the structure between two helices just before the C-termini of the kinase domains. This kink allows a small section of α-helix to fold back and associate with the rest of the C-terminal lobe, making a series of stabilising interactions. We hypothesised that the lack of a proline in this position in EphB3 might lead to a destabilisation in this region of the C-terminal lobe, and also its recalcitrance to crystal formation and X-ray structure determination.
EphB3 A899P expression and stability profiling
A single point mutation of the EphB3 kinase domain (616–910) was created to determine whether swapping the alanine at position 899 for a proline (subsequently referred to as A899P), as found in the EphB paralogues, would lead to increased stability and potentially aid crystallogenesis. E. coli expression studies were undertaken in the presence and absence of human PTP1B, which we had previously shown to be essential for obtaining soluble, homogeneously unphosphorylated EphB3.25 We found that the A899P mutation did not have a detrimental effect on soluble recombinant expression, and, in the presence of PTP1B, the expression of A899P was enhanced by around threefold compared with the wild-type protein (Fig. 2). We also determined that both proteins were active when recombinantly expressed, as evidenced by auto-phosphorylation, and that co-expression with PTP1B enabled production of homogeneously unphosphorylated material (Fig. 2).
Figure 2.
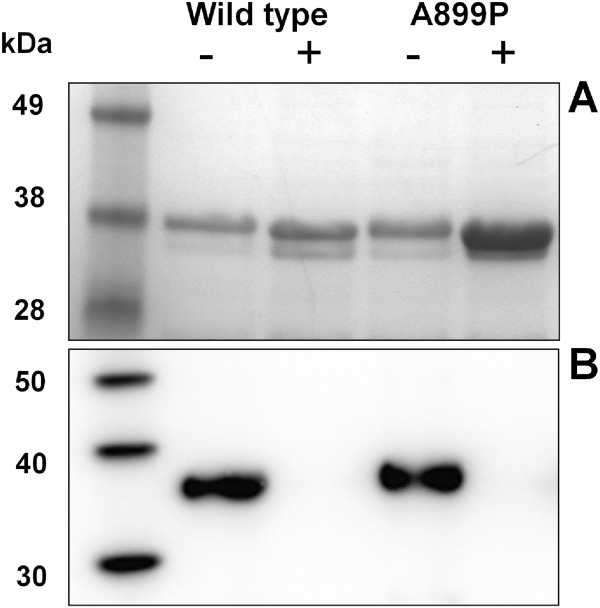
E. coli expression results of EphB3 kinase domains. (A) shows an SDS PAGE gel analysis of hexahistidine affinity-purified samples from soluble fraction of E. coli cell lysates; 0.5 ml of culture equivalent loaded per lane,-/+ refers to co-expression with PTP1B. (B) Shows an anti-phosphotyrosine western blot analysis of the same affinity purified samples; 0.5 µg of kinase loaded per lane.
Large-scale expression and purification resulted in sufficient quantities of purified material to attempt stability analysis, to determine whether this single point-mutation had enabled the production of a more thermodynamically stable enzyme. The intrinsic stability of the isolated EphB3 A899P kinase domain was investigated using two separate techniques: thermal denaturation using Circular Dicroism (CD), and chaotropic unfolding following changes in intrinsic tryptophan fluorescence. Initial wavelength scans were performed by CD in phosphate buffer at physiological pH, and demonstrated very similar secondary-structure profiles for the wild-type and mutant enzymes [Fig. 3(A)]. Clear unfolding transitions were observed for both proteins over a 20°C to 80°C range [Fig. 3(B)], which demonstrated a large increase in thermal stability for the mutant A899P versus the wild-type enzyme of +7.2°C (Table II).
Figure 3.
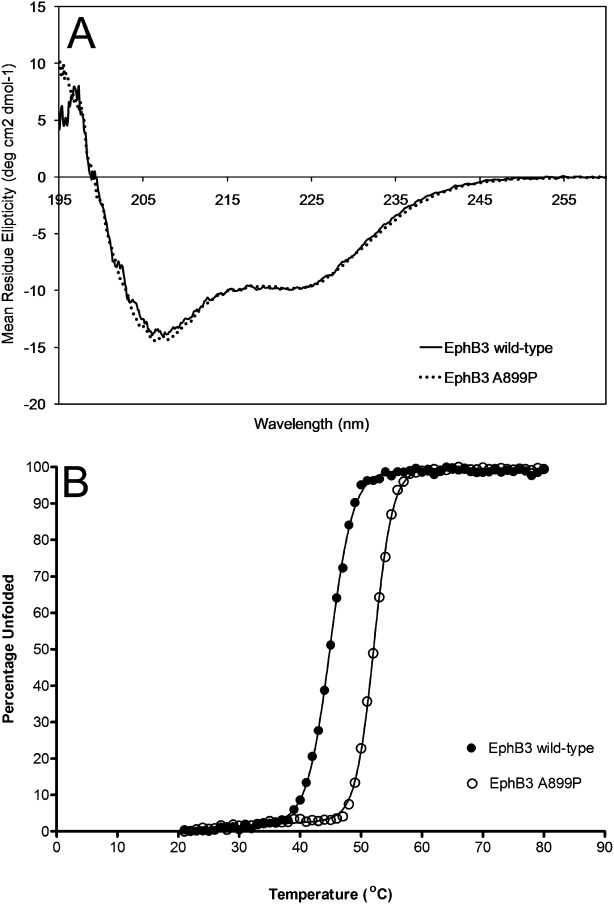
Thermal unfolding of EphB3 kinases. (A) shows a molar concentration adjusted 260 to 195 nm CD wavelength scan of each of EphB3 and EphB3 A899P kinase domains to demonstrate their secondary-structure profiles, n = 3—the web server K2d27 was used to estimate the percentages of protein secondary structure from these circular dichroism spectra; EphB3 wild-type: 32% alpha helical, 15% beta strand, 52% random coil, EphB3 A899P: 36% alpha helical, 16% beta strand, 48% random coil. (B) Shows averaged thermal-unfolding transitions obtained from CD experiments monitored at 222 nm (alpha-helical response). Unfolding transition data were fitted to a six-parameter unfolding equation,28 using Prism software to generate comparative melting temperatures (Tm) and van't Hoff enthalpies of unfolding (ΔUH(Tm)).
Table II.
Enzymatic and Thermodynamic Parameters for EphB3 Enzymes
| EphB3 WT | EphB4 A899P | ||
|---|---|---|---|
| CD thermal unfolding | Tm (°C) | 45.0 ± 0.1 | 52.2 ± 0.1 |
| ΔUH(Tm) (kJ mol−1) | 106.2 ± 3.7 | 137.7 ± 4.5 | |
| Steady-state kinetics | Km ATP (µM) | 506 ± 48 | 660 ± 46 |
| Km Subs. (µg/mL) | 943 ± 91 | 1014 ± 123 | |
| kcat (s−1) | 0.43 ± 0.01 | 0.77 ± 0.02 | |
| Vmax (nmol/min per mg) | 764.1 ± 22.9 | 1382.1 ± 41.5 | |
| CMPD1 ITC | N | 1.00 ± 0.08 | 1.170± 0.05 |
| Kd (µM) | 11.47 ± 2.83 | 8.07 ± 1.45 | |
| ΔH (kJ M−1) | −51.71 ± 13.30 | −46.40 ± 6.68 | |
| CMPD1 Inhibition | IC50 (µM) | 5.065 | 4.849 |
| 95% CIR | 1.166 | 1.206 | |
| R2 | 0.987 | 0.985 | |
| CMPD2 Inhibition | IC50 (µM) | 0.149 | 0.117 |
| 95% CIR | 1.172 | 1.211 | |
| R2 | 0.991 | 0.987 | |
| CMPD3 Inhibition | IC50 (µM) | 0.036 | 0.032 |
| 95% CIR | 1.116 | 1.190 | |
| R2 | 0.997 | 0.992 |
Chemical structures of CMPD1, 2, and 3 can be found in Ref.25, errors shown are standard errors, n ≥ 3 for all experiments.
Abbreviations: A280, absorbance at 280 nm; ADPNP, 5′-adenylyl β,γ-imidodiphosphate; Camr, chloramphenicol resistance marker; CHES, N-cyclohexyl-2-aminoethanesulfonic acid; DTT, dithiothreitol; GdnHCl, guanidine hydrochloride; IC50, half maximal inhibitory concentration; IMAC, Immobilized metal ion affinity chromatography; IPTG, isoproyl β-d-thiogalactopyranoside; ITC, isothermal titration calorimetry; Kanr, kanamycin resistance marker, LB, Luria-Bertani; Ni-NTA, nickel-nitrilotriacetic acid; OD600, optical density at 600 nM; PEG, polyethylene glycol; PTP1B, phosphotyrosine phosphatase 1 beta; RTK, receptor tyrosine kinase; TCEP, tris(2-carboxyethyl)phosphine; Tetr, tetracycline resistance marker; TEV, tobacco etch virus.
We have previously shown a very distinct chaotrope-induced unfolding-profile for the wild-type EphB3 enzyme compared with other EphB family members, monitoring tryptophan fluorescence at 345 nm in the presence of increasing GdnHCl concentration.25 In this study we have compared the EphB3 A899P unfolding-profile to that of the wild-type unfolding-profile (Fig. 4). The wild-type protein shows a distinct minimum in tryptophan fluorescence at ∼1M GdnHCl, indicating some degree of structural rearrangement, presumably resulting in the exposure of one or more of the tryptophan's to an aqueous environment. As the GdnHCl concentration is further increased, the protein appears to undergo additional restructuring events, resulting in a partial re-gain of tryptophan fluorescence and stable “third” state at ∼2M GdnHCl. As the GdnHCl increases further, up to a concentration of ∼3M, the tryptophan signal finally reaches a stable minimum, indicating complete unfolding. The unfolding-profile for the A899P mutant is markedly different; the mutant protein lacks the distinct minimum at 1M GdnHCl, but like the wild-type protein it does have the stable tryptophan signal at ∼2M and the minimal signal from ∼3M GdnHCl. This altered unfolding profile for the mutant protein now closely resembles those of the other EphB family members EphB1 and EphB2,25 indicating that the A899P mutation may be giving rise to a structural change within the C-terminal lobe of the kinase.
Figure 4.
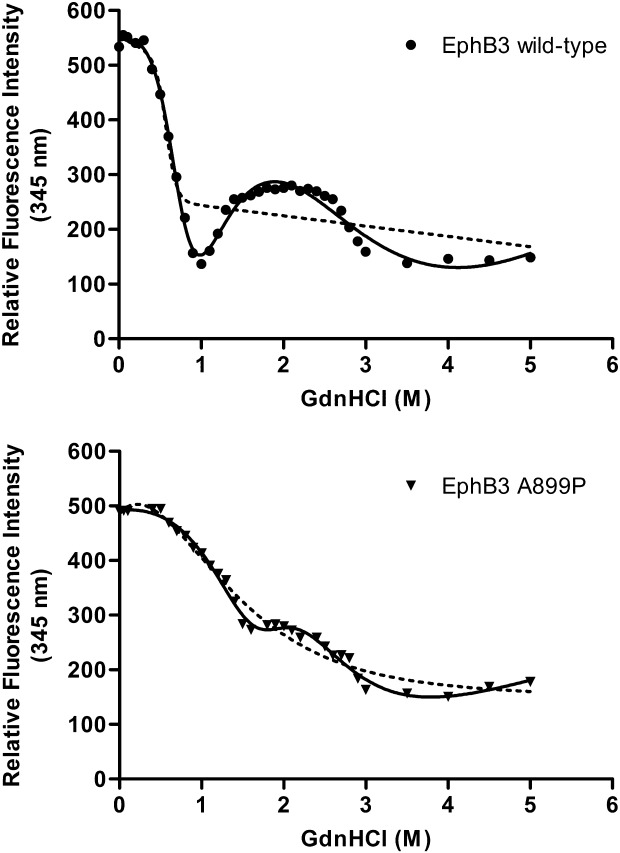
Chaotropic unfolding of EphB3 kinases. Unfolding of EphB3 wild-type and A899P kinase domains with increasing GdnHCl was monitored by following the progressive quenching of internal tryptophan fluorescence (Ex 295 nm, Em 345 nm). Units shown on the y-axis are relative fluorescence units. Curves were fitted to both two-state (dotted line) and three-state (unbroken line) models. n = 3 for all.
Kinetic analysis of EphB3 A899P
An in vitro peptide phosphorylation assay was employed to investigate the intrinsic activity of the EphB3 A899P mutant to determine whether this residue has an effect on the catalytic activity of the protein. Unphosphorylated kinase domains were incubated with the generic tyrosine-kinase substrate poly(Glu,Tyr), and the level of substrate phosphorylation was monitored over time using an ADP-production luminescence assay. This assay was used to determine the comparative Km of both kinases for ATP and substrate, as well as kcat and Vmax values (Table II). We have found that the A899P mutant showed an increase in turnover number compared with the wild-type enzyme (Table II), indicating that the increased stability afforded by the A899P mutation also affords a modest increase in activity. We also examined the inhibitor-binding profile of EphB3 A899P compared with the wild-type enzyme, using both ITC and a biochemical assay using three previously described EphB kinase inhibitors; CMPD1, 2 and 3.25 EphB3 gave affinities and IC50 (half maximal inhibitory concentration) values that were in very close agreement with those of the wild-type enzyme (Table II). This confirmed that not only is the A899P mutation sufficiently remote from the binding site so as not to compromise compound binding, but also that this enzyme would be a suitable surrogate for enzymatic and biophysical studies of EphB3 protein–ligand interactions.
EphB3 A899P crystallization and structure determination
Having demonstrated the A899P mutant to have substantially improved thermal and chaotropic stability, we attempted to obtain crystals of this protein to facilitate structure determination. A homogeneous sample of the kinase was subjected to crystallization screening by sitting-drop vapour diffusion, and screened against 192 separate conditions, using four different ligand and buffer formulations at nanoliter scale. A few proteinaceous crystal hits were observed, the best crystals appearing in 35% dioxane. These initial hits were further optimised by reduction of the dioxane concentration and the addition of 10 mM MgCl2 to obtain crystals suitable for in-house X-ray diffraction testing [Fig. 1(B)].
The structure of EphB3 A899P was solved by using the published EphB4 structure, 2VWX24 as a search model; refinement statistics for the structure can be found in Table I. A fully refined model of the EphB3 catalytic domain to 2.2Å has now been deposited in the PDB: 3ZFY [Fig. 1(A)]. Overall, the catalytic domains in the EphB family exhibit only minor structural differences. The C-terminal domains are essentially identical (average r.m.s.d. after pairwise superposition: 0.34 Å), while somewhat larger differences can be found when comparing the N-terminal lobes (average r.m.s.d. after pairwise superposition: 0.60Å), mainly due to the inherent flexibility of the N-terminal domain and differences induced by compound binding in one of the structures. A small hinge-movement between the N-and C-terminal domains can also be observed (overall average r.m.s.d. after pairwise superposition: 0.72 Å) which is also illustrated in Figure 5(A). The EphB3 structure does however highlight the position of Cys717 in the hinge loop which confirms the likelihood of steric clash with the EphB4 kinase inhibitor CMPD1 [Fig. 5(B)], which has been shown to have reduced potency against EphB3 compared with other EphB isozymes.25 Interestingly, the EphB3 structure also contains a partial ordering of the activation loop containing Tyr792 [Fig. 5(C)]. A portion of the observed activation loop including the tyrosine is lying in the substrate binding site as previously determined crystallographically in EphA3.22 The non-phosphorylated Tyr792 is pointing into the active site and appears to be coordinated by Asp758 of the HRD motif and Arg762 as would be required for catalysis.
Figure 5.

EphB3 structure observations. (A) Backbone representation of EphB catalytic domain structures [color scheme follows Fig. 1(A)], after superposition on the C-terminal lobes. (B) A ribbon overlay of the active sites of EphB4 (black) with CMPD1 (grey) (PDB: 2VWU), and EphB3 (green) highlighting the key difference at Gly699 (Cys717 in EphB3) and Ala700 (Ser706 in EphB2), the surface shown is that of EphB3; (C) surface representation of the EphB3 substrate-binding groove with the partially ordered EphB3 activation loop and HRD motif (green sticks), overlaid with part of the EPHOPT peptide (orange sticks) from the EphA3 complex structure (3FXX).
Discussion
EphB kinase crystallizability versus stability
Given the high level of sequence identity between these four enzymes (ranging from 83% between EphB2 and EphB4, to 89% between EphB1 and EphB225), one might have expected all four enzymes to be equally accessible to structure determination by X-ray crystallography, but this was not the case. EphB4 appears to be the most crystallizable of the four isozymes, despite also being the most difficult to produce recombinantly, and exhibiting the lowest stability profile.25 This does, however, agree with previously published data from large structural-biology laboratories, suggesting that stability is not the overriding factor in protein crystallizability.29
Although we have demonstrated that the EphB3 A899P mutant is substantially more stable than the EphB3 wild-type enzyme, we cannot say for certain that this increase in stability alone is the dominant factor leading to successful crystallization. For example, one could imagine that an alternative reason could be the reduced entropy of the mutant protein caused by the conformational inflexibility of the proline residue versus the alanine residue found in the wild-type enzyme. Similar types of proline mutation had been exemplified previously in other systems to aid protein stabilisation,30–32 but this one has a clear additional benefit in driving crystallization. Interestingly, EphB3 is the only one of the four wild-type kinase domains to exhibit a “salting-out” effect when placed on ice at crystallization concentrations (i.e., ∼10 mg/mL); the EphB3 kinase domain solution becomes opaque, but does not precipitate in the temperature range between 1°C and 5°C, and can be dissolved back into solution by increasing the temperature. This “salting-out” effect is not seen with the A899P mutant.
EphB active site differences and clinical exploitation
We have previously demonstrated clear differences in both the kinetic activity and inhibitor-binding profiles of the four EphB kinases.25 Access to the three-dimensional structures of each of the isozymes allows us to probe these differences in more detail; similar to the backbone of the molecules, the active sites appear to be highly conserved. As shown in Figure 5(B), nonconserved side chains mostly point toward the solvent, and hence have little influence on the shape and charge properties of the ligand-binding pocket. However, two of the nonconserved positions, 699 and 700 (EphB4 numbering), form the edge of the binding site towards the solvent. Position 699 is occupied by Gly in all except EphB3, where it is Cys, whereas 700 is Ala in all except EphB2, where it becomes Ser. As predicted, the Gly to Cys substitution renders the solvent channel much narrower, which is likely to sterically hinder the binding of molecules such as CMPD1 (an anilinoquinazoline) and CMPD2 (a 2,4-bisanilinopyrimidine), which lie across the conserved Gly in the other EphB enzymes. This Cys alters the ligand-binding profile of EphB3 relative to other kinases by inducing a sharp drop in potency for certain compounds that are otherwise good inhibitors across the rest of the family.25 Interestingly, Gly is conserved at this position within the hinge loop in all human tyrosine kinases except EphB3. This Gly is important in a number of kinases for dictating resistance/sensitivity to small molecules and is regarded as a hotspot for clinical resistance mutations to type I and type II kinase inhibitors.33–35
From the structural and ligand-binding data that we have presented, it is clear that, should it be required for clinical purposes, there may be some, albeit limited, opportunity to design EphB-selective small molecules to modulate the kinase activity of these receptors. However, this is likely to depend on which of the four enzymes one would need to select for or against. Obtaining small molecules that selectively target or avoid EphB3 should be feasible, given the presence of Cys717 in the active-site region. Similarly, targeting Ser706 of EphB2 may afford some degree of isozyme selectivity. However, as the active sites of EphB1 and EphB4 are so similar, it may be very difficult to obtain molecules that target one of these enzymes and not the other, without exploiting differences remote from the active site. This is not to say that this would be impossible as there is precedent for identifying allosteric or non-ATP competitive kinase inhibitors.36 Given that there is such complexity and disparity in the disease profiles of the various EphB isozymes, not least in the oncological setting,4 it is unclear at present whether there would be clinical benefit in: (a) producing a small-molecule EphB kinase inhibitor or activator, and (b) ensuring that this molecule is selective for one or more of the EphB isozymes over the other Eph receptors.
EphB3 activation loop conformation hints at a possible cis-activation mechanism
The constructs used for this study do not contain the juxtamembrane regions required for auto-inhibition of catalysis,19 hence the kinases are free to adopt active-like conformations. The structure of EphB3 demonstrates the ability for this kinase to bind its own activation loop within its substrate binding groove. Although this phenomena has been observed previously for the insulin receptor tyrosine kinase (PDB: 1IRK37), this is the first crystallographic evidence for such a binding event within the EphB family. The unphosphorylated activation loop tyrosine appears to be coordinated in an orientation that would allow for catalysis with its hydroxyl group within hydrogen bonding distance to the side chains of Arg762 and Asp758, and is similar to that observed for synthetic substrate peptides in EphA3 [Fig. 5(C)].22 However, this structure does not represent a fully active conformation as the catalytic lysine (Lys665) is partially disordered, and the glycine (Gly776) of the DFG motif at the start of the activation loop is in a position where it would almost certainly clash with both the α and β phosphates of ATP if it were present. The phenylalanine (Phe777) is however partially flipped out as would be required for an active conformation, but not as far as described in the active structures of EphB2 (PDB: 2HEN20) and EphA3 (PDB: 2QOC21). It is unclear whether in the presence of ATP, there would be enough flexibility in the activation loop, and in particular the five amino acids that are not visible in the electron density, to allow it to bind in the substrate grove and maintain the orientation of Tyr792 in a catalytically competent pose. There is at present, insufficient evidence in the currently available EphB structures to unequivocally support the notion of cis-phosphorylation within the Eph kinase activation pathway as described for a number of other kinases.38 Additional biochemical activity studies with very careful analysis of enzyme kinetics would be required to probe the cis versus trans autophosphorylation mechanism. Such studies might involve examining the effects of addition of a 'kinase dead' EphB mutant to an autophosphorylating EphB wild-type assay which would not be expected to substantially change the kinetics of autophosphorylation if a cis-autophosphorylation mechanism is favored.
Conclusions
The novel EphB1, EphB2, EphB3, and EphB4 kinase domain structures that we have solved now give us a more comprehensive picture of the EphB family of catalytic domains. These structures, together with those of the EphA kinases that have previously been determined, will provide researchers with more comprehensive information with which to design potent and selective Eph-kinase inhibitors. These compounds in turn will be valuable tools to enable a greater understanding of the respective Eph biological pathways and disease-linked mechanisms and potentially even facilitate clinical candidates for disease intervention.
This study also emphasises both the value of testing several homologues when attempting structural studies of specific members of protein families, and also the power of structure-based protein design, where even single point-mutations can trigger the difference between precipitation and crystallization.
Materials and Methods
All chemicals were obtained from Sigma, unless otherwise stated.
Molecular biology
E. coli expression constructs for the four wild-type EphB-kinase catalytic-domain proteins used for initial crystallization screening were synthesised in vitro by GENEART AG (Life Technologies); EphB1 residues 602–896 (UniProtKB/Swiss-Prot: EPHB1_HUMAN, P54762), EphB2 residues 604–898 (UniProtKB/Swiss-Prot: EPHB2_HUMAN, P29323), EphB3 residues 616–910 (UniProtKB/Swiss-Prot EPHB3_HUMAN, P54753) and EphB4 residues 598–892 (UniProtKB/Swiss-Prot: EPHB4_HUMAN, P54760). All sequences were codon-optimised for E. coli expression. The synthetic genes were sub-cloned into the Gateway®-adapted pT7#3.3 (Tetr) E. coli expression vector,39 using the Gateway® Cloning System (Life Technologies). The expression constructs contained a 6His purification tag, the residual Gateway® sequence, and a TEV (Tobacco Etch Virus) protease recognition sequence N-terminal to each EphB catalytic domain: MHHHHHHGSTSLYKKAGSENLYFQGSS. An additional expression vector containing a single copy of the human PhosphoTyrosine Phosphatase 1 Beta (PTP1B) protein (UniProtKB/Swiss-Prot: PTN1_HUMAN, P18031, residues 1-288) inserted into the pRSF-1b plasmid (Kanr, EMD Chemical, Merck KGaA) was also constructed: pRSF1-PTP1B.
Additional synthetic EphB3 wild-type and EphB3 A899P mutant E. coli expression constructs were created using the same protein sequence and domain boundaries as the above-described EphB3 wild-type construct. These synthetic gene fragments were restriction-ligation sub-cloned into a modified version of pET28b (Kanr, Merck) using flanking BamHI and XhoI sites. The resultant expression vectors contained the following sequence N-terminal to the EphB3 kinase domains: MHHHHHHGGGENLYFQGSS. An additional PTP1B vector for co-expression with these EphB3 constructs was also created: pT7#3.3-GST-PTP1B contained a single copy of the human PTP1B inserted into pT7#3.3 downstream of the glutathione-S-transferase (GST) gene.
Protein expression and purification
Kinase expression vectors were transformed into E. coli BL21 Star™(DE3) cells (Life Technologies) in the presence of pRSF1-PTP1B for the four original EphB kinase vectors, or pT7#3.3-GST-PTP1B for the two EphB3 constructs in pET28b. The EphB4-containing cells were also co-transformed with the GroES-GroEL containing vector pGro7 (Camr, Takara Bio).
E. coli cells were cultured in 2 l conical flasks containing LB (Luria-Bertani) broth plus antibiotics, at 37°C, 220 rpm. The temperature was reduced to 18oC prior to induction of kinase (and phosphatase) expression with 0.1 mM IPTG (ISOPROYL β-d-thiogalactopyranoside) at an average OD600 of ∼0.80 for 20 hrs. Cells were harvested by centrifugation at 12,000g. For the EphB4, pGro7-containing cultures, the addition of l-(+)-arabinose to a final concentration 1 g/L was required to induce chaperone expression.
Frozen cell pellets equivalent to 6 L of E. coli culture were re-suspended in a base buffer containing 40 mM Hepes, 500 mM NaCl, 1 mM TCEP, pH 8.0, supplemented with 10 mM imidazole, 1 mg/mL hen egg-white lysozyme, 0.1 µL/mL Benzonase® Nuclease HC (Novagen) and EDTA-free Complete Protease Inhibitor Cocktail Tablets (Roche). Cells were lysed with a single pass through a 2.2 kW TS series cell-lysis system (Constant Systems) at 25 kpsi. Lysates were clarified by centrifugation at 35,000g for 60 min and fractionated by using Ni-NTA (Nickel-NitriloTriacetic Acid) Superflow resin (Qiagen). The resin containing bound proteins was washed to A280 baseline with base buffer containing 20 mM imidazole, and eluted with, 500 mM imidazole in the same buffer. Elution fractions were pooled, and N-terminal 6His tags removed with rTEV protease (Life Technologies) during overnight dialysis at 4oC versus 40 mM Hepes, 500 mM NaCl, 5 mM imidazole, 1 mM TCEP, pH 8.0. Cleaved EphB kinase proteins were further purified by a subtractive Ni-NTA step to remove the TEV protease, 6His tags and any uncleaved material, followed by a final gel-filtration step, using a 120 mL Superdex S75 column (GE Healthcare) into crystallization buffer (50 mM Mops, 50 mM NaCl, 1 mM DTT pH 7.5). Monomeric fractions containing >95% pure EphB kinase, as judged by SDS-PAGE, were pooled and concentrated to between 9 and 11 mg/mL. Concentrated proteins were flash frozen in liquid nitrogen and stored at −80°C. All chromatographic manipulations were performed at +4°C.
Western blotting was used to detect tyrosine phosphorylation of 0.5 µg of affinity-purified kinase using 1:2000 anti-phosphotyrosine mouse monoclonal primary antibody (pY100; NEB Cell Signalling) with 1:1000 HorseRadish Peroxidase (HRP)-rabbit anti-mouse secondary antibody (Sigma), and using Supersignal West Femto ECL detection reagent (Thermo Scientific Pierce).
Crystallization and structure determination
EphB kinase samples were supplied for crystallization at between 9.5 and 11.5 mg/mL in crystallization buffer. The kinases were screened as apo-protein, and in the presence of either 1 mM Staurosporine or 1 mM ADPNP, plus 5 mM MgCl2. Protein samples were incubated ± ligand for 1 hr at 4°C and then centrifuged at 12,000g for 10 min at 4°C. Screens were performed by sitting-drop vapour-diffusion using standard in-house broad-matrix screens in MRC Innovaplate SD-2 plates (Hampton Research), using a Mosquito® drop maker (TTP Lab Tech), with an 80 µL reservoir volume. One hundred ninety two conditions were set up for each protein formulation; 1:1 protein to reservoir ratios (250 nL each), and plates were incubated at 20°C in a CrysTEL plate hotel (Emerald Biosystems), and imaged using the Crymon software (Emerald Biosystems). Initial hit conditions were further optimised to diffraction quality; 1 µL plus 1 µL sitting drops in CombiClover, Jr. crystallization plates (Emerald Biosystems Inc) using a 200 µL reservoir volume; plates were incubated at 20°C. Crystals typically appeared after 12 to 36 h, and reached maximum size with 3 days.
Final reservoir conditions identified for each of the three proteins were as follows: EphB1, 100 mM CHES pH9.5, 18% PEG-3350; EphB2, 8.8% PEG-1500, 10% PEG-8000; EphB4, 100 mM MES NaOH, 25% PEG-8000; and EphB3 A899P, 16–20% 1,4-dioxane, 10 mM MgCl2. Crystals typically appeared after 12 to 24 h, and reached maximum size within 3 days. For crystal diffraction testing at low temperature, the crystals were cryo-protected with reservoir solution containing 15 to 20% glycerol or ethylene glycol for EphB1, EphB2 and EphB4, and 20% 2,3-butanediol for EphB3 A899P. Crystals were frozen at 100 K using an Oxford Cryostream 700 series, before transferring to liquid nitrogen.
Diffraction data were collected and processed as follows: EphB1—data were collected at the ID14 beamline at ESRF to 2.5 Å resolution, integrated using XDS40 and scaled with Scala of the CCP4 software suite;41 EphB2—data were collected on a Rigaku FRE X-ray generator equipped with VariMax HR optics and a Mar345 detector, and processed using XDS and scaled with Scala; EphB4—data were collected on a Rigaku FRE X-ray generator equipped with VariMax HF optics and a Saturn944 detector, and processed using d*trek.42 EphB3—A899P data were collected at the Diamond Light Source I04-1 beamline. Data were integrated with XDS and scaled with Scala. All structures were solved with molecular replacement using Phaser.43 Models were built using Coot44 and refined with Buster.45 Data-processing and refinement statistics are listed in Table I.
Biophysical and kinetic analyses
Thermal and chaotrope-induced protein unfolding measurements, enzyme kinetics, and compound profiling experiments were conducted as described.25
Atomic coordinates
The atomic coordinates and structural factors have been deposited in the Protein Data Bank under the accession codes: 3ZFX (EphB1), 3ZFM (EphB2), 3ZEW (EphB4 + staurosporine), 3ZFY (EphB3 A899P).
Acknowledgments
The authors thank Geoff Holdgate and Dr. Gareth Davies assistance with CD and ITC analysis and Dr. Julie Tucker for help with crystallization screening.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 2.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 3.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 4.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi Y, Syeda F, Walker JR, Finerty PJ, Jr, Cuerrier D, Wojciechowski A, Liu Q, Dhe-Paganon S, Gray NS. Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2009;19:4467–4470. doi: 10.1016/j.bmcl.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiao L, Choi S, Case A, Gainer TG, Seyb K, Glicksman MA, Lo DC, Stein RL, Cuny GD. Structure–activity relationship study of EphB3 receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2009;19:6122–6126. doi: 10.1016/j.bmcl.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barlaam B, Ducray R, Brempt CLd, Plé P, Bardelle C, Brooks N, Coleman T, Cross D, Kettle JG, Read J. Inhibitors of the tyrosine kinase EphB4. Part 4: Discovery and optimization of a benzylic alcohol series. Bioorg Med Chem Lett. 2011;21:2207–2211. doi: 10.1016/j.bmcl.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, Beigi R, Xia G, Harig RA, Asatryan H, Yan SF, Zhou Y, Gu XJ, Saadat A, Zhou V, King FJ, Shaw CM, Su AI, Downs R, Gray NS, Schultz PG, Warmuth M, Caldwell JS. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc Natl Acad Sci USA. 2006;103:3153–3158. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 10.Scapin G. Structural biology in drug design: selective protein kinase inhibitors. Drug Discov Today. 2002;7:601–611. doi: 10.1016/s1359-6446(02)02290-0. [DOI] [PubMed] [Google Scholar]
- 11.Breitenlechner CB, Bossemeyer D, Engh RA. Crystallography for protein kinase drug design: PKA and SRC case studies. Biochim Biophys Acta. 2005;1754:38–49. doi: 10.1016/j.bbapap.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Himanen J, Rajashankar KR, Lackmann M, Cowan CA, Henkemeyer M, Nikolov DB. Crystal structure of an Eph receptor-ephrin complex. Nature. 2001;414:933–938. doi: 10.1038/414933a. [DOI] [PubMed] [Google Scholar]
- 13.Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S. Architecture of Eph receptor clusters. Proc Natl Acad Sci U S A. 2010;107:10860–10865. doi: 10.1073/pnas.1004148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398–402. doi: 10.1038/nsmb.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Himanen JP. Ectodomain structures of Eph receptors. Semin Cell Dev Biol. 2012;23:35–42. doi: 10.1016/j.semcdb.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 16.Bocharov EV, Mayzel ML, Volynsky PE, Goncharuk MV, Ermolyuk YS, Schulga AA, Artemenko EO, Efremov RG, Arseniev AS. Spatial structure and pH-dependent conformational diversity of dimeric transmembrane domain of the receptor tyrosine kinase EphA1. J Biol Chem. 2008;283:29385–29395. doi: 10.1074/jbc.M803089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stapleton D, Balan I, Pawson T, Sicheri F. The crystal structure of an Eph receptor SAM domain reveals a mechanism for modular dimerization. Nat Struct Biol. 1999;6:44–49. doi: 10.1038/4917. [DOI] [PubMed] [Google Scholar]
- 18.Thanos CD, Goodwill KE, Bowie JU. Oligomeric structure of the human EphB2 receptor SAM domain. Science. 1999;283:833–836. doi: 10.1126/science.283.5403.833. [DOI] [PubMed] [Google Scholar]
- 19.Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F. Structural basis for autoinhibition of the EphB2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell. 2001;106:745–757. doi: 10.1016/s0092-8674(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 20.Wiesner S, Wybenga-Groot LE, Warner N, Lin H, Pawson T, Forman-Kay JD, Sicheri F. A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 2006;25:4686–4696. doi: 10.1038/sj.emboj.7601315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis TL, Walker JR, Loppnau P, Butler-Cole C, Allali-Hassani A, Dhe-Paganon S. Autoregulation by the juxtamembrane region of the human ephrin receptor tyrosine kinase A3 (EphA3) Structure. 2008;16:873–884. doi: 10.1016/j.str.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Davis TL, Walker JR, Allali-Hassani A, Parker SA, Turk BE, Dhe-Paganon S. Structural recognition of an optimized substrate for the ephrin family of receptor tyrosine kinases. FEBS J. 2009;276:4395–4404. doi: 10.1111/j.1742-4658.2009.07147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bardelle C, Cross D, Davenport S, Kettle JG, Ko EJ, Leach AG, Mortlock A, Read J, Roberts NJ, Robins P, Williams EJ. Inhibitors of the tyrosine kinase EphB4. Part 1: Structure-based design and optimization of a series of 2,4-bis-anilinopyrimidines. Bioorg Med Chem Lett. 2008;18:2776–2780. doi: 10.1016/j.bmcl.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Bardelle C, Coleman T, Cross D, Davenport S, Kettle JG, Ko EJ, Leach AG, Mortlock A, Read J, Roberts NJ, Robins P, Williams EJ. Inhibitors of the tyrosine kinase EphB4. Part 2: Structure-based discovery and optimisation of 3,5-bis substituted anilinopyrimidines. Bioorg Med Chem Lett. 2008;18:5717–5721. doi: 10.1016/j.bmcl.2008.09.087. [DOI] [PubMed] [Google Scholar]
- 25.Overman RC, Debreczeni JE, Truman CM, McAlister MS, Attwood TK. Biochemical and biophysical characterization of four EphB kinase domains reveals contrasting thermodynamic, kinetic and inhibition profiles. Biosci Rep. 2013;33:e00040. doi: 10.1042/BSR20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 27.Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 28.Consalvi V, Chiaraluce R, Giangiacomo L, Scandurra R, Christova P, Karshikoff A, Knapp S, Ladenstein R. Thermal unfolding and conformational stability of the recombinant domain II of glutamate dehydrogenase from the hyperthermophile Thermotoga maritima. Protein Eng. 2000;13:501–507. doi: 10.1093/protein/13.7.501. [DOI] [PubMed] [Google Scholar]
- 29.Price II WN, Chen Y, Handelman SK, Neely H, Manor P, Karlin R, Nair R, Liu J, Baran M, Everett J, Tong SN, Forouhar F, Swaminathan SS, Acton T, Xiao R, Luft JR, Lauricella A, DeTitta GT, Rost B, Montelione GT, Hunt JF. Understanding the physical properties that control protein crystallization by analysis of large-scale experimental data. Nat Biotechnol. 2009;27:51–57. doi: 10.1038/nbt.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goihberg E, Dym O, Tel-Or S, Shimon L, Frolow F, Peretz M, Burstein Y. Thermal stabilization of the protozoan Entamoeba histolytica alcohol dehydrogenase by a single proline substitution. Proteins. 2008;72:711–719. doi: 10.1002/prot.21946. [DOI] [PubMed] [Google Scholar]
- 31.Zhou C, Xue Y, Ma Y. Enhancing the thermostability of α-glucosidase from Thermoanaerobacter tengcongensis MB4 by single proline substitution. J Biosci Bioeng. 2010;110:12–17. doi: 10.1016/j.jbiosc.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Tian J, Wang P, Gao S, Chu X, Wu N, Fan Y. Enhanced thermostability of methyl parathion hydrolase from Ochrobactrum sp. M231 by rational engineering of a glycine to proline mutation. FEBS J. 2010;277:4901–4908. doi: 10.1111/j.1742-4658.2010.07895.x. [DOI] [PubMed] [Google Scholar]
- 33.Scutt PJ, Chu MLH, Sloane DA, Cherry M, Bignell CR, Williams DH, Eyers PA. Discovery and exploitation of inhibitor-resistant aurora and polo kinase mutants for the analysis of mitotic networks. J Biol Chem. 2009;284:15880–15893. doi: 10.1074/jbc.M109.005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sloane DA, Trikic MZ, Chu MLH, Lamers MBAC, Mason CS, Mueller I, Savory WJ, Williams DH, Eyers PA. Drug-resistant aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem Biol. 2010;5:563–576. doi: 10.1021/cb100053q. [DOI] [PubMed] [Google Scholar]
- 35.Balzano D, Santaguida S, Musacchio A, Villa F. A general framework for inhibitor resistance in protein kinases. Chem Biol. 2011;18:966–975. doi: 10.1016/j.chembiol.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hubbard SR, Wei L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 38.Lochhead PA. Protein kinase activation loop autophosphorylation in Cis: overcoming a catch-22 situation. Sci Signal. 2009;2:pe4. doi: 10.1126/scisignal.254pe4. [DOI] [PubMed] [Google Scholar]
- 39.Tobbell DA, Middleton BJ, Raines S, Needham MR, Taylor IW, Beveridge JY, Abbott WM. Identification of in vitro folding conditions for procathepsin S and cathepsin S using fractional factorial screens. Protein Expr Purif. 2002;24:242–254. doi: 10.1006/prep.2001.1573. [DOI] [PubMed] [Google Scholar]
- 40.Kabsch W. Xds. Acta Crystallogr D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans P. Scaling and assessment of data quality. Acta Crystallogr D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 42.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D. 2004;60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.