

Abstract
RACK1 is a member of the WD repeat family of proteins and is involved in multiple fundamental cellular processes. An intriguing feature of RACK1 is its ability to interact with at least 80 different protein partners. Thus, the structural features enabling such interactomic flexibility are of great interest. Several previous studies of the crystal structures of RACK1 orthologs described its detailed architecture and confirmed predictions that RACK1 adopts a seven-bladed β-propeller fold. However, this did not explain its ability to bind to multiple partners. We performed hydrogen-deuterium (H-D) exchange mass spectrometry on three orthologs of RACK1 (human, yeast, and plant) to obtain insights into the dynamic properties of RACK1 in solution. All three variants retained similar patterns of deuterium uptake, with some pronounced differences that can be attributed to RACK1's divergent biological functions. In all cases, the most rigid structural elements were confined to B-C turns and, to some extent, strands B and C, while the remaining regions retained much flexibility. We also compared the average rate constants for H-D exchange in different regions of RACK1 and found that amide protons in some regions exchanged at least 1000-fold faster than in others. We conclude that its evolutionarily retained structural architecture might have allowed RACK1 to accommodate multiple molecular partners. This was exemplified by our additional analysis of yeast RACK1 dimer, which showed stabilization, as well as destabilization, of several interface regions upon dimer formation.
Keywords: receptor for activated C kinase, WD repeats, scaffolding protein, cell signaling, hydrogen deuterium exchange, mass spectrometry, protein dynamics
Introduction
Receptor for activated C kinase (RACK1) is a member of the WD-repeat (tryptophan-aspartate) family of proteins. RACK1 was initially identified as an intracellular receptor for protein kinase CβII (PKC)1 but is now known to interact with at least 80 partners. RACK1 is highly conserved among all eukaryotic species. It is found in a wide range of organisms, including the alga Chlamydomonas reinhardtii, nematodes, insects, fungi, plants, yeasts, and vertebrates.2–7 RACK1 is a component of ribosomes. Mass spectrometry analysis, later confirmed by cryoelectron microscopy mapping and the crystal structure of the eukaryotic ribosome, showed that RACK1 is in close proximity to the mRNA exit channel of the 40S ribosomal subunit.8–10 RACK1 plays a role in translation regulation and brings ribosomes to translation sites, such as focal adhesions.11 It also exists as a cytosolic pool of free form RACK1 that is not bound to ribosomes.12,13 In humans, RACK1 (referred to here as “hRACK1”) is present in almost every tissue but is most abundantly expressed in liver, spleen, and brain. By interacting with a broad spectrum of kinases, phosphodiesterases, ion channels, and membrane-bound receptors, hRACK1 is thought to be a key player in many important signal transduction pathways and cellular processes, such as cell proliferation, adhesion and migration, circadian rhythm, and apoptosis. Changes in hRACK1 homeostasis lead to brain development disorders, tumors, heart failure, muscle atrophy, and addiction; thus, it is often considered as a disease marker.14–17
Yeast contains an ortholog of RACK1, called Asc1 in Saccharomyces cerevisiae (referred to here as “yRACK1”). The yRACK1 amino acid sequence is 52% identical to that of hRACK1 [Fig. 1(A)]. yRACK1 is involved in translation regulation, iron homeostasis, pheromone response, cell-wall integrity, and oxidative stress.18 It functions as Gβ in trimeric G protein, which is involved in the glucose response pathway, and it is important for yeast cell growth and dimorphism.19,20 RACK1A from Arabidopsis thaliana (referred to here as “atRACK1”) shares 66% identity with hRACK1 [Fig. 1(A)] and is known to be engaged in multiple hormonal responses and developmental signaling pathways, as well as in environmental stress response.21,22
Figure 1.
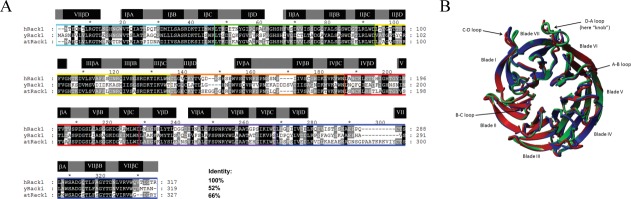
RACK1 orthologs. (A) Sequence alignment of RACK1 orthologs from H. sapiens (hRACK1), S. cerevisiae (yRACK1), and A. thaliana, isoform A (atRACK1). Percentage sequence identity is shown at end of the alignment. Characteristic WD repeats are highlighted with colored rectangles. β-propeller blades are highlighted by gray rectangles that contain A, B, C, D β-strands (black rectangles), as assigned based on hRACK1crystal structures available in PDB (4AOW). (B) Superposition of the crystal structures of hRACK1 (4AOW, blue), yRACK1 (3FRX, green,) and atRACK1 (3DM0, red). Characteristic structural elements such as blades and loops are highlighted.
The crystal structures of human, yeast, and plant RACK1 have been solved (Protein Data Bank [PDB] ID: 4AOW, 3FRX, 3RFH, 3RFG, and 3DM0).23–26 The structures of hRACK1 and atRACK1 show the highest similarity, with a root-mean-square deviation (RMSD) of 0.811 Å for 296 aligned residues. Despite only 52% sequence identity between human and yeast proteins, their RMSD values also do not differ substantially (0.869 Å over 299 aligned residues) [Fig. 1(B)]. hRACK1 and its orthologs adopt a seven-bladed β-propeller structure with significant homology to the β subunit of heterotrimeric G protein.7 Each blade comprises a four-stranded antiparallel β-sheet (strand A–D), with strand A positioned near the central channel of the protein. The full WD repeat is not equivalent to a single blade, but spans the A-B-C strands of one blade and the fourth of the previous one. WD repeats of RACK1 proteins have sequences variable in length, with a characteristic motif usually flanked by GH and WD dipeptides on both sides. The blades are connected with loops linking strand D to strand A of the next blade, which, together with the B-C loop, are exposed on the top face of the propeller. The other loops (loops A-B and C-D) are located on the opposite side. Loop D-A between blades 6 and 7 adopts a flexible knob-like structure (Fig. 1). In contrast, loop B-C folds into a conserved tight turn with a characteristic aspartate residue in the middle position, which forms a salt bridge with the histidine residue in the GH dipeptide (in 1–5 propeller blades).
RACK1 has been reported to interact with as many as 80 molecular partners, but its mode of interaction is largely unknown. It is thus of great interest to elucidate structural determinants at the molecular level that allow for such versatility. To some extent, the general building plan of the β-propeller structure and the WD-repeats themselves may explain this phenomenon. The central hole is thought to accommodate small molecules, while the outer strands can interact with bigger partners.27 Moreover, structural studies of yRACK1 have shown that this protein can dimerize, creating a greater binding surface and allowing it to accept more molecules.24 There have been two examples of in vivo dimerization of hRACK1. Dimeric protein is required for regulation of the N-methyl-d-aspartate (NMDA) receptor by Fyn kinase in the brain and for oxygen-independent degradation of hypoxia-inducible factor (HIF-1). In the latter case, Ser146 phosphorylation is essential for dimerization.28,29 hRACK1-Gβ heterodimerization and its role in the integration of signals in GPCR-mediated transduction pathways has also been reported.30–32
We carried out a structural analysis of RACK1 from three different organisms with an alternative analytical technique, namely, hydrogen-deuterium exchange coupled to mass spectrometry (HDXMS), to expand what is known about the structural features enabling RACK1 to bind multiple partners. HDXMS provides information regarding the exchange kinetics of amide protons at different regions of a protein and indicates both highly structured and flexible regions with high reproducibility and precision.33 This method maps the dynamics of protein structure in solution at low micromolar concentration, without meticulous and time-consuming data analysis procedures. In the three protein variants, we mapped regions with substantial protection, as well as regions with considerable levels of deuteration, which correspond to regions of relaxed structure. We compared the average rates of hydrogen-deuterium exchange kinetics in RACK1 peptides and found large differences in these rates at different protein regions; these differences were maintained in general across the species under study. Our results indicate the intertwined structure of a relatively small and stable protein core and the remaining regions of different levels of flexibility.
Results
We expressed and purified proteins from human, yeast (S. cerevisiae), and plant (A. thaliana). The oligomeric state after purification was assessed by size exclusion chromatography and analytical ultracentrifugation. Under our experimental conditions, hRACK1 and atRACK1 eluted from the SEC column as a monomer, whereas we observed two SEC fractions for yRACK1, which correspond to monomeric and dimeric species [Supporting Information Fig. 1(A)]. Analytical ultracentrifugation experiments corroborated these findings [Supporting Information Fig. 1(B)]. The molecular masses of the monomers were in agreement with masses calculated from amino acid sequences. However, the molecular mass of yRACK1 dimer was slightly lower than expected. The elongated shape of the dimer may explain this observation. Our results are consistent with previous data, which showed that yRACK1 can form a dimer.24
Hydrogen-deuterium exchange monitored by MS
Pilot studies were conducted to establish good sequence coverage of peptic peptides; in the case of hRACK1, peptic fragments covered about 97% of the sequence. Exchange patterns were analyzed at different incubation periods (10 s, 1 min, 20 min, and 1 h). We chose to present the results of HDXMS experiments obtained after 10 s of incubation (if not stated otherwise in the text) for clarity, as these data best illustrate differences in the dynamics of RACK1 regions [Fig. 2(A–C)]. We also overlaid these results on the crystal structures of each protein (PDB ID: human, 4AOW; S. cerevisiae, 3FRX; A. thaliana, 3DM0) and color-coded them according to the percentage of deuteration of the shortest available peptide corresponding to a given region [Fig. 3(A–C)].
Figure 2.
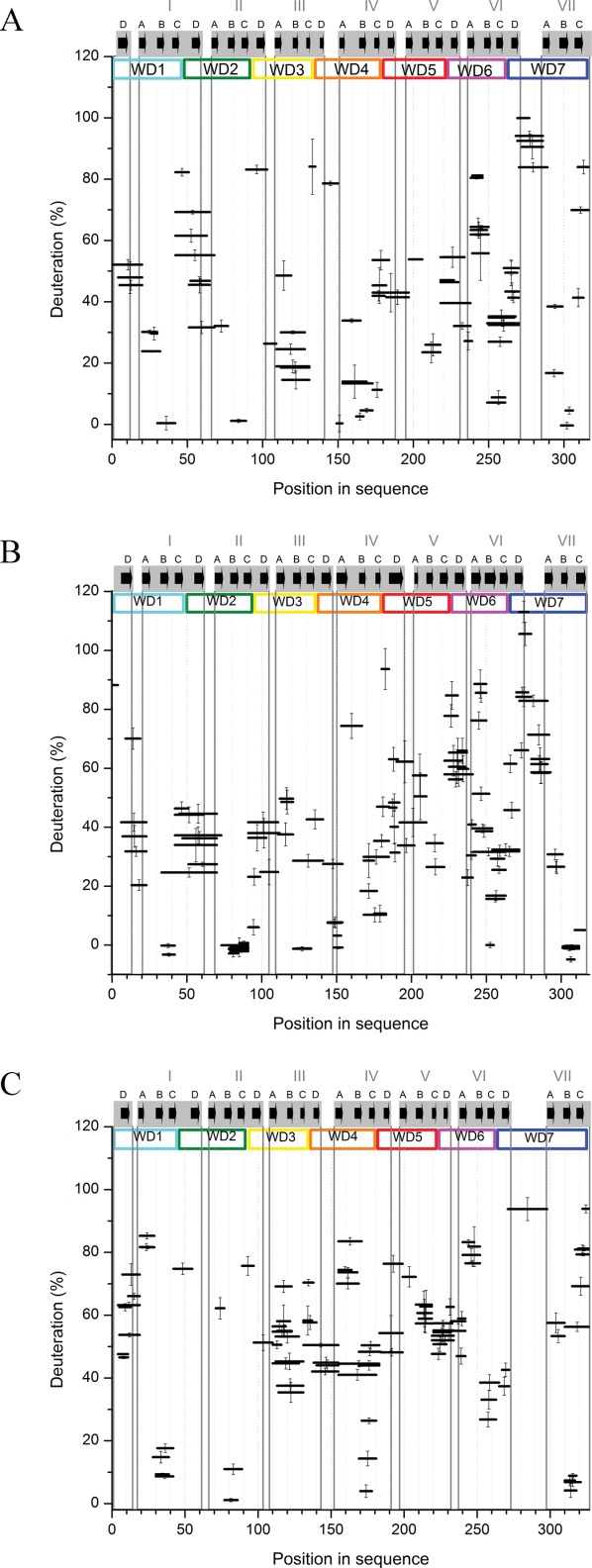
Percentage of deuteration of peptic fragments from human (A), yeast (B), and plant (C) RACK1 at 10 s exchange time. Position of a peptide in the sequence is shown on the horizontal axis, represented by a horizontal bar with length equal to the length of the peptide. Position of the bar at the vertical axis marks the fraction exchanged after 10 s. y-Axis error bars are standard deviations calculated from three independent experiments. WD repeats are marked and colored as in Figure 1(A). β-Propeller blades are highlighted by gray rectangles that contain A, B, C, and D β-strands (black arrows), as assigned based on RACK1 crystal structures available in PDB (4AOW, 3RFH, and 3DM0).
Figure 3.
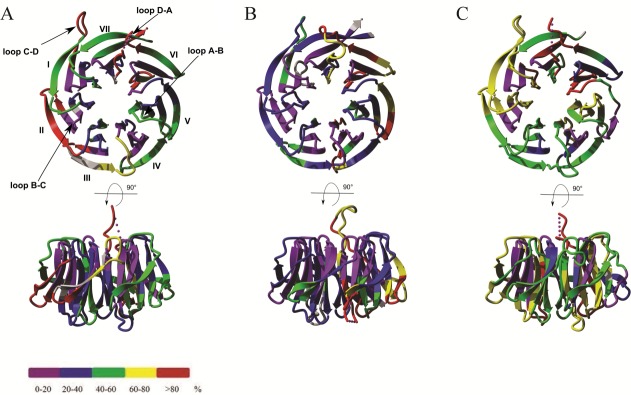
Overlay of 10 s hydrogen-deuterium exchange results on crystal structures from (A) hRACK1 (4AOW), (B) yRACK1 (3FRX), and (C) atRACK1 (3DM0) at 10 s exchange time. Structures are color coded according to HDXMS results of the shortest available peptide: violet, strongly protected (0–20%); blue, protected (20–40%); green, moderately protected (40–60%); yellow, moderately flexible (60–80%); and red, flexible (>80%). Gray color represents regions not covered by peptic peptides in the sequence.
In all three proteins, the C-terminal blades were more relaxed than the N-terminal portion of the proteins after 10 s of exchange. There were only a few relatively short regions characterized by low deuteration levels, which correspond to those elements of structure for which the exchange is very slow. These were mainly confined to B-C turns and some parts of adjacent strands B and C (later referred to as “B-C hairpins”) of the majority of blades, whereas the remaining parts were not fully protected [Figs. 2(A–C) and 3(A–C)].
HDXMS of hRACK1
The regions of slowest exchange included the B-C hairpins of blades I, II, IV, VI, and VII in hRACK1 [Figs. 2(A) and 3(A)]. The fastest exchange was observed for peptides covering the C-terminal peptide located at strand C of blade VII and loop D-A between blades VI and VII, which adopts a knob-like structure. The fastest exchange within the blade structures was observed for peptic fragments, which cover the characteristic WD dipeptide (or equivalent) on strand C and loops C-D, as exemplified in blades I, II, III, IV, and VII in hRACK1. Loop C-D in blade V was as flexible as loop A-B in the same blade. There was a general pattern in which the direct vicinity of the most protected B-C hairpins neighbored the least protected C-D turns in five out of seven blades. For blade V, we observed no region-specific protection. In addition, strong protection was not observed for the peptides that cover loop B-C of blade III. These peptic peptides were quite long and spanned not only loop B-C, but also loop A-B and strand B; thus, they do not contain information about the B-C turn alone. Loops A-B from the whole structure represented moderately protected regions, except in blades V and VI. Loop A-B of blade VI was the least protected within the blade, and it was almost fully deuterated after 10 s of exchange.
A majority of the strongly protected regions remain unexchanged even after longer incubations in D2O [Fig. 4(A)]. To integrate the data obtained after 10 s, 1 min, 20 min, and 1 h of incubation, we calculated the average rate constants of exchange (kex) for all peptides of hRACK1 [Fig. 4(B)], assuming the simplest approximation of a single rate constant for exchange. Exemplary results of the fitting procedures are illustrated in Supporting Information Figure 2. This analysis revealed marked differences in the exchange rate constants, spanning four orders of magnitude. While the majority of rate constants were at the level of 10/min or higher, there were a few in the range below 0.01/min or even 0.001/min. All of the slowest rate constants were assigned to a few peptides corresponding to B-C hairpins of blades I, II, IV, VI, and VII and complemented by two other peptides from positions 150 to 153 and 163 to 167 of blade IV. For comparison, we have calculated the intrinsic rate constants kint for all identified Rack1 peptides [Fig. 4(B)] expected for their fully exposed state at pH 8.0, room temperature. These values do not differ by more than sevenfold with values ranging from 630/min to 4000/min. This leads to more than 104-fold difference in protection factors, defined as a ratio of kint/kex, within hRACK1 sequence [Fig. 4(C)]. Expectedly, the largest protection factors mark B-C hairpins, spanning also A-B hairpin in blade IV and including blade V B-C hairpin. The observed lack of full protection in B-C hairpin of blade V [Fig. 2(A)] can thus be explained by a relatively fast intrinsic exchange in this region, higher than anywhere along the sequence. In contrast, the protection factors in blade III B-C hairpin stay at the relatively low level.
Figure 4.
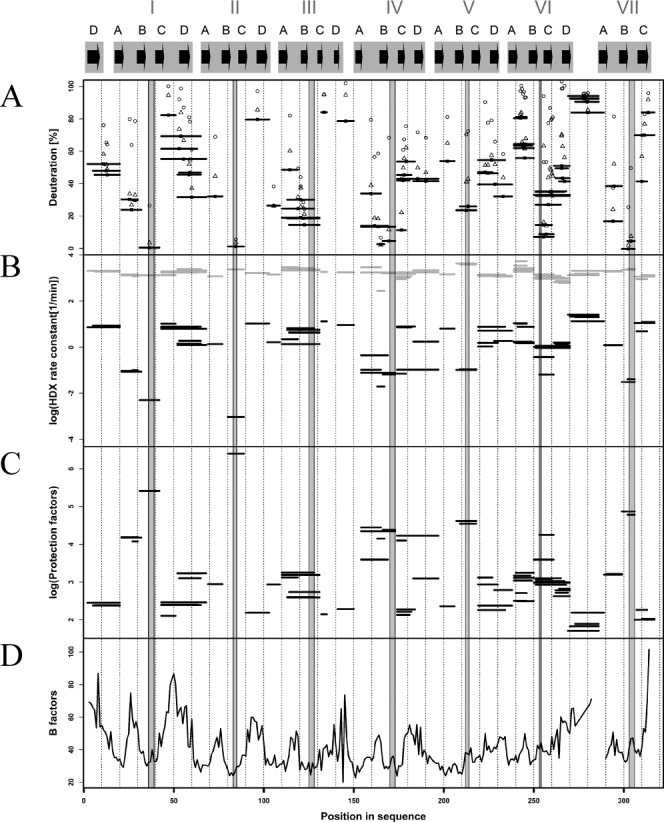
Regions of fast and slow hydrogen-deuterium exchange in hRACK1, correlated with B-factor of its crystal structure. The gray shaded regions represent B-C loops alone. (A) Percentage of deuteration of peptic fragments from hRACK1 at 10 s [horizontal bars as in Fig. 2(A)], 1 min (open triangles), 20 min (filled circles), and 1 h (open circles) of exchange. (B) average rate of exchange kex (log scale) obtained by a monoexponential fit for each peptide, marked by the position of a black horizontal bar with length equal to the length of the peptide on the vertical axis. Position of each peptide in the sequence is shown on the horizontal axis. Intrinsic rates of exchange kint for each peptide are also shown marked by gray bars. Note a few peptides, characterized by low kex values, leading to very high protection factor (P) values, as shown in log scale in (C). (D) B-factor value profile for hRACK1 crystal structure, with several minima correlating with regions of lowest kex and highest P values.
Islands of strong stability in hRACK1 appeared to be small and intertwined with much larger regions of variable flexibility, especially those exposed to interaction with molecular partners. The placement of stability anchors correlates well with the minima in the B-factor profile [Fig. 4(D)]; however, only studies carried out in solution, such as HDXMS, properly underscore the dynamic aspects dominating the RACK1 structure. The overall scenario of a stiff protein core strictly limited to a relatively small set of B-C hairpins that specifically position the remaining protein regions of significant flexibility is a highlight of the RACK1 structure.
Major differences in hydrogen-deuterium exchange patterns of hRack1 and its orthologs
We monitored the exchange kinetics of peptic peptides covering 97% and 99% of yRACK1 and atRACK1, respectively. In general, atRACK1 appeared to be more flexible compared to its human and yeast orthologs. Nevertheless, the characteristic pattern of H-D exchange observed in hRACK1 was largely retained in the other two proteins (Fig. 2). Peptides that form the knob-like structure were in all cases among the most rapidly exchanging segments, and those from B-C hairpins were among the most highly protected. The overall flexibility was high, similar to hRACK1, and the protein structure was maintained by an internal ring of short B-C turns. On the other hand, there were also significant differences in exchange patterns, which in many cases correlated with the known functional differences of RACK1 variants. The differences in H-D exchange of peptides were verified by additional analysis of their intrinsic rate constants (Supporting Information Fig. 3).
Strand A and loop A-B within blade I showed greater susceptibility to exchange in atRACK1 than in its human counterpart. Small differences in intrinsic rates (kint = 1312/min for hRACK1 and 1163/min for atRACK1) in this region cannot account for differences in deuteration levels larger than a few %. Coverage for the peptic fragments was poor for the analogous region in yRACK1 [Fig. 2(B)].
In yRACK1, loop A-B and strands B and C, as well as loop C-D, of blade II were relatively strongly protected, whereas in hRACK1 and atRACK1, only B-C hairpins showed low levels of deuteration. Relatively high exchange in A-B loop in atRACK1 can be rationalized by the highest intrinsic rates (4000/min) in this region, two times higher as compared to yRACK1 (1860/min). In contrast, intrinsic rate is the lowest (1200/min) in hRACK1. In spite of this, the deuteration level in hRACK1 is higher than in yRACK1, implying a much stronger structural protection in loop A-B of blade II in yRACK1. Blade III of atRACK1 showed in general a higher susceptibility for exchange and a more homogeneous exchange level, without distinct protection of B-C region, as compared to hRACK1 and yRACK1. Strong protection was observed for the peptide covering the B-C hairpin of blade III in yRACK1 only; however, peptic peptides from this region in both hRACK1and atRACK1 were quite long and spanned other structural elements of blade III. Intrinsic rates in this region are similar for each protein leading to average values of 2400/min, 1730/min, 1360/min for hRACK1, atRACK1, yRACK1, respectively. atRACK1 kint values are intermediate and cannot explain stronger exchange, at least when compared with hRACK1. This leads to the conclusion that the structural protection of blade III in hRACK1 is stronger than in atRACK1.
Blade IV had a distinguishable, highly dynamic loop A-B in yRACK1 and atRACK1, but this loop showed only approximately 30% deuteration in hRACK1. Intrinsic rate in hRACK1 is the highest (two-fold higher than in yRACK1) indicating a stronger structural protection for hRACK1. Another prominent difference was a very high exchange in loop C-D of yRACK1. Its high flexibility and plasticity are important in the formation of yRACK1 dimer.24 Intrinsic rate in this region is slightly higher in yRACK1 than in the other two variants (1995/min, 1628/min, 1513/min for yRACK1, hRACK1, atRACK1, respectively), but this difference could not account for the differences of deuteration level larger than 10%.
Blade V displayed an overall moderate protection for all three orthologs. Interestingly, for atRACK1, the B-C region of blade V was much more strongly exchanged as compared to all B-C hairpins of the three orthologs. The differences in intrinsic rates in this region (1445/min, 4078/min, 2021/min for yRACK1, hRACK1, atRACK1, respectively) cannot account for the enhanced exchange in this region of atRACK1, implying its larger flexibility.
The deuteration profile of blade VI did not differ substantially among the three RACK1 orthologs. The most pronounced difference in deuteration levels within blade VII was observed in the C-terminal region. N and C termini of RACK1 proteins were arranged in a “Velcro” closure in blade VII that helps circularize the propeller. Interestingly, substantial protection in the region of the C terminus was observed only in the case of yRACK1, while this region was strongly exchanged in hRACK1 and atRACK1. In yRACK the intrinsic rate for the very C-terminal peptide is the smallest (890/min, 1318/min, 2040/min for yRACK1, hRACK1, atRACK1, respectively). These differences however are not sufficient to explain the large difference in exchange (>70%). The protein structure more strongly protects this region from exchange in yRACK1, which is in agreement with the more twisted, extended interactions between both termini observed in the yRACK1 crystal structure.26
Dimerization mode of yRACK1 in solution
During our experiment, we were able to isolate monomeric and dimeric species of yRACK1. We performed hydrogen-deuterium exchange studies on both species and prepared a difference plot by subtracting the percentage of deuteration of the monomer (% DMONOMER) from that of the dimer (% DDIMER) [Fig. 5(A)].
Figure 5.
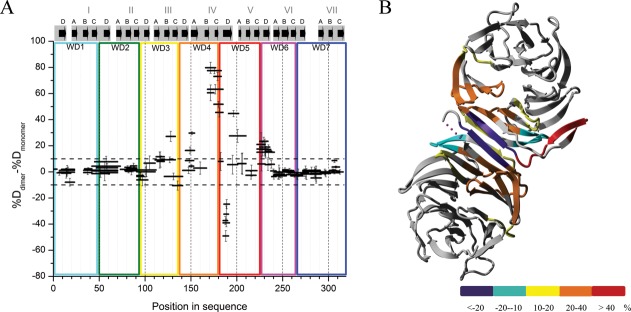
Differences in fraction of exchanged amide protons for peptic peptides between yRACK1 monomers and dimers. (A) Subtraction plot representing differences in exchange between peptides of dimeric and monomeric yRACK1. WD repeats, blades, and strands of β-propeller are marked as in Figure 2(B). (B) Differences in amide proton exchange between monomeric and dimeric yRACK1 overlaid onto crystal structure of its dimer (3RFH). Differences upon dimer formation are color coded as follows: violet, strong stabilization (less than −20%); cyan, stabilization (−20 to −10%); yellow, weak destabilization (10 to 20%); orange, moderate destabilization (20 to 40%); and red, high destabilization (more than 40%).
There was a strong stabilizing effect at strand D in blade IV. There were also several regions that became destabilized in the dimer, either more strongly, as in the B-C loop region in blade IV, or to a lesser extent, as in some regions in blades III and V. A previous study solved the crystal structure of the yRACK1 dimer and revealed an unusual means of dimer formation, in which strands B and C of blade IV were ejected outside the propeller core and the two remaining strands (A and D) from both protomers created a new blade IV, composed of A1-D1-D2-A2 strands.24 We could thus overlay our results onto the crystal structure of dimeric yRACK1 (PDB ID: 3RFH) and color code it according to the observed exchange differences [Fig. 5(B)]. The region most protected upon dimer formation was represented by five peptides that cover the 186-FQIEADF-192 sequence of strand D in blade IV. This corresponds well with the formation of eight new hydrogen bonds in the dimer main chain. The most dynamic fragment (position 169–183) was contained within strands B and C and the connecting B-C turn of the same blade. Here, the difference between monomer and dimer was around 80% after 10 s of deuteration. High exposure (approximately 40–50%) was also observed for peptides from loop C-D of blade IV and for loop D-A connecting blades IV and V. The peptide (position 149–154) from loop D-A that is located between blades III and IV and strand A from blade IV showed approximately 30% higher deuteration in the dimer than in the monomer. We also mapped other regions that underwent slight conformational changes upon dimerization. For example, peptides from blade III covering the B-C turn and the peptide from strand D of blade V were more relaxed, whereas peptide 131–141 from blade III showed some structural stabilization in the dimer. Small differences in H-D exchange (approximately 15% increase in dimer) were observed for the peptide containing His147, which has an important role in stabilizing the dimer structure. Peptides containing this residue were strongly protected in the monomeric form [Fig. 2(B)]. In a monomer, His147 forms hydrogen bonds with the conserved Asp175, as well as with Ser171 of blade IV, whereas His147 flips out in the dimer but interacts with Gln139 of the other molecule.24 Interestingly, a fragment covering the end of strand A, the whole of loop A-B, and the start of strand B of blade IV (residues 154–167), which is flipped out from the regular structure in the crystal, showed very small differences in exchange (only 3%). This peptide was already very flexible in monomeric yRACK1, and its exposure in the D2O environment did not change appreciably upon formation of the dimer [Fig. 2(B)].
This experiment illustrates the high level of plasticity in the RACK1 structure, which can undergo substantial changes upon interaction with its partners. In the dimer, not only does part of the structure become hidden in the interface, but a large region becomes exposed, presenting a new scaffolding platform for further interactions with other proteins.
Discussion
The three orthologs showed a largely conserved pattern of highly variable levels of deuterium exchange, indicating that the general placement of stable and flexible regions was retained among the species under study. Nevertheless, we also mapped some variations in protein dynamics in solution that may be associated with functions of RACK1 that vary among the species.
The regions of strong protection in all three RACK1 variants were limited to a small inner core ring of six B-C hairpins in nearly all blades, excluding blade V, which was only moderately protected [Fig. 2(A–C)]. Thus, in solution, the majority of protein structural elements (e.g., external strands A and D) found by crystal structure studies to be part of the well-defined β-propeller blades retained substantial flexibility.
Previous studies have indicated that the B-C turns from blades I and II are involved in binding to ribosomal RNA. Mutations Arg38Asp and Lys40Glu in the RDK conserved motif, positioned in the B-C turn of blade I, almost completely abolish binding of yRACK1 to ribosomes.9,26 In our study, we observed very high protection of the peptides covering these residues. As mentioned above, only the B-C turn of blade V was more flexible in all three proteins compared to the remaining blades [Fig. 2(A–C)]. Closer investigation of the crystal structures showed that the distance between Asp and His residues, which is supposed to form a stabilizing salt bridge, is larger (5.11 Å) in blade V of yRACK1, which could explain its higher mobility. However, in hRACK1 and atRACK1, the distance remained approximately 3 Å; thus, the reason for its relative flexibility is unclear in these proteins.
The organization of salt bridges in blades VI and VII is different from those in blades I–V. The aspartate residue is absent in blade VI, whereas in blade VII, it forms a salt bridge with an arginine residue on strand C. Despite this, we observed equally high levels of protection in B-C hairpins of blades VI and VII compared to the other blades. On the other hand, in all three proteins, the C-terminal part that spans blades V, VI, and VII contained more flexible fragments than the N-terminal part. WD repeats 5, 6, and 7, which span blades IV, V, VI, and VII, are thought to be the main docking station for locating other proteins.34 Our results indicate a link between the observed enhanced plasticity in the region and its ability to accommodate other molecules.
The most flexible region in RACK1 covers a knob-like structure of loop D-A between blades VI and VII. In the crystal structures of hRACK1 and atRACK1, these parts were disordered and not visible, whereas in monomeric yRACK1, this large insertion folded into a well-ordered structure.23,25,26 Our results are consistent with previous findings, as the short peptides that cover the knob in yRACK1 showed slightly lower deuterium uptake than in hRACK1. This combined with slightly higher intrinsic rate in yRACK1 [Fig. 2 and Supporting Information Fig. S3(A,B)] indicate stronger structural protection in yeast protein. In atRACK1, there was only one long peptide in the knob, which also covered strands D and A of the adjacent blades. The function of this protrusion is unknown, but it is considered to be important and to play a role in accommodating other molecules, such as protein kinase C.14 Moreover, the hRACK1 sequence 270-LKQEVIST-277, located in the knob, has the consensus ΨKxExxS (where Ψ is a large hydrophobic residue and x is any amino acid), which is often present in transcription factors. This knob motif contains a putative sumoylation site (270-LKQE-273), as well as Ser276, which can be phosphorylated; thus, the so-called “phospho-sumoyl” switch has been proposed to modulate hRACK1 function.35
The localization of protein kinase C–hRACK1 binding has not been established. Peptide competition studies have shown that fragment 99-RRFVGHTKDV-108, spanning the D-A loop between blades II and III, together with part of the outer strand of blade II, are potential targets for PKC binding.36 Others have shown that peptides from the inner strands of blades III (107-DVLSVAF-113) and VI (234-DIINALCF-241) can inhibit PKCβ.7 Recently, affinity grid-based cryoelectron microscopy studies have identified PKC not only in proximity to blade III, but also to blade IV of RACK1.37 A PKC ortholog has not been found in plants. S. cerevisiae expresses PKC, but there have been no reports of any connection between yRACK1 and the PKC signaling pathway.38 Looking for differences in deuterium uptake in the aforementioned potential PKC binding regions, we found one differential peptide, 141-TVQDESHSEW-150, that spans loop D-A between blades III and IV. This peptide shows higher exposure in hRACK1 than in yRACK1 and atRACK1, which could not be explained by small differences in the intrinsic rates; thus, it may be an alternative/additional region involved in binding/accommodation of PKC. The peptic peptides from blade VI of human protein (228-YTLDGGDIINA-238, 235-IINAL-238, and 239-LCFSPNRY-246) contained residues previously identified as part of the potential PKC-binding peptide 234-DIINALCF-241. The first two peptides are rather moderately protected, whereas the third one is highly unprotected, indicating that at least its C-terminal residues are flexible. The structural flexibility of this fragment may also be associated with other functions. Tyrosine 246, located in this region, or/and Tyr228, are potential phosphorylation sites for Src kinase. Both tyrosines are conserved, but only peptides with Tyr246 (Tyr248 in atRACK1 and Tyr250 in yRACK1) are very susceptible to exchange and therefore easily accessible to modification. It was previously shown that phosphorylation of Tyr246 is essential for hRACK1 binding to Src kinase, whereas Tyr228 is not.39,40 A recent study identified the same Tyr246 of hRACK1 as the critical site for binding of RACK1 to the β-actin mRNA/ZBP1 complex.41 Thus, high flexibility of the peptides with Tyr246 is required. Although plants lack classic tyrosine kinases, tyrosine phosphorylation and dephosphorylation events occur quite often.42 The conserved tyrosines of atRACK1 can be phosphorylated by dual-specificity STY kinases that were previously proposed to modify plant tyrosines.43 In yeast, the situation is similar. There are no bona fide tyrosine protein kinases, but phosphotyrosines are present.44 Spk1p (Rad53p) kinase phosphorylates proteins on serines, threonines, and tyrosines, as well as on poly(Tyr-Glu) patches in yeast.45 High flexibility of some peptides containing Ser/Thr/Tyr residues could be an important prerequisite for phosphorylation to occur.46 Moreover, flexible regions that flank phosphorylation sites are often involved in the substrate-kinase recognition process.47 Many other kinases can potentially phosphorylate hRACK1 in cells. For example, c-Abl kinase modifies Tyr52 located on the C-D loop of blade I, and its phosphorylation regulates association with focal adhesion kinase.48 The peptides that cover Tyr52 are also flexible in hRACK1 and show high deuterium uptake [Fig. 2(A)]. This residue is conserved in atRACK1, and the corresponding peptide shows high levels of deuteration [Fig. 2(C)]. In yeast, Tyr52 is not conserved but is replaced by Phe54; peptides that contain this residue are more strongly protected [Fig. 2(B)], and the effect cannot be justified by small differences in intrinsic rates in this region. On the other hand, phosphorylation/dephosphorylation of hRACK1 on Tyr302 is required to mediate the binding of PP2A and the cytoplasmic tail of β1-integrin; nevertheless, the peptides that cover this residue are strongly protected in hRACK1 [Fig. 2(A)].49
The dimerization interface for yRACK1 in solution, which we identified in the present study, correlates very well with the known crystal structure of yRACK1 dimer [Fig. 5(A,B)]. Blade IV is most strongly engaged in stabilizing the dimer, but as a consequence of dimer formation, its B-C hairpin, which is very stable in the monomeric form, becomes destabilized and exposed to solvent, accompanied by a few regions from adjacent blades III and V. This underscores the high plasticity of the RACK1 structure, which may undergo a significant reconstruction upon binding of its ligands, thus expanding the spectrum of possible molecular partners even further. hRACK1 did not form a dimer under our experimental conditions. It was shown previously that Ser146 phosphorylation is required to form dimeric species of RACK1.29 The peptide containing Ser146 is located in loop D-A between blades III and IV in the region affected by dimer formation in yRACK1. Under our experimental conditions, we were also unable to obtain dimeric species of atRACK1, although we tried different expression and purification strategies. Thus, the dimerization mode in solution could be assessed only for yRACK1.
In conclusion, we mapped the stiff and plastic regions of RACK1 in solution and compared patterns of hydrogen deuterium exchange among three RACK1 orthologs and between monomeric and dimeric yRACK1. Our results reveal an evolutionarily retained building plan for RACK1, in which large regions of plasticity are precisely positioned by the intertwining small stable core. This supports a more fluidic structure of external regions of the polypeptide chain than previously implicated by the crystal structures, in which A and D strands are structurally well defined. By contrast, we found that in solution, their stabilizing hydrogen bonds are broken much more frequently than in the neighboring B and C strands. This could be an important determinant that enables this relatively small molecule to accommodate numerous diverse molecular partners. The general dynamic patterns were similar in the three evolutionarily distant variants of the protein. However, our results also allowed us to localize differences in structural dynamics among the species under study, which may be linked to their function. The observed differences suggest that these proteins evolved to interact in a species-specific way with different molecules, so that they play roles in different cellular functions. One such example is PKC, which interacts only with hRACK1. Another example is heterotrimeric G-protein, which does not form a canonical interaction with atRACK1.50
Little is known about the specific interactions between hRACK1 and its binding partners in solution. For example, the exact position of the residues involved in PKC binding to RACK1 is still unknown. Our study shows that HDXMS may be helpful in identifying regions in both proteins that are affected upon complex formation. In a broader sense, our study provides a basis for further research into the multiple binding modes of RACK1 with different molecular partners and their structural consequences.
Materials and Methods
Protein expression and purification
hRACK1, yRACK1, and atRACK1 genes were synthesized and cloned into pET28a vector bearing kanamycin resistance cassette (GenScript). His-tagged proteins were expressed in C43 Escherichia coli strain. Bacterial clones containing hRACK1 construct were grown in 2 l of LB medium, induced with 1.7 mM IPTG (final concentration), and expressed for 3 h at 30°C. atRACK1 protein was produced in 100 mL of autoinduction medium (Super Broth Base including trace elements; Formedium) for 24 h at 37°C. Cells were then harvested and frozen at −20°C. hRACK1 and atRACK1 were purified in the same way. Cell pellets were resuspended in buffer A (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 20% glycerol) supplemented with 1 mg/mL lysozyme, 10 µg/mL DNaseI, and 1 mM PMSF. After disruption with sonication, cells were centrifuged at 15,000g for 1 h. Soluble fraction was loaded onto Ni-NTA column (Qiagen) equilibrated with buffer A. Beads were washed with five-column volumes of buffer B (20 mM Tris-HCl pH 7.5, 200 mM NaCl, 50 mM imidazole, 10% glycerol). Protein was eluted with buffer C containing 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 500 mM imidazole, and 10% glycerol. yRACK1 was expressed and purified as described previously, with some modifications.24 Cells were grown in 2 L of 2 × YT medium and induced with 1mM IPTG for 16h at 20°C. The pellet was resuspended in buffer D (50 mM HEPES-NaOH pH 7.6, 300 mM NaCl, 25 mM imidazole, 10% glycerol) supplemented with lysozyme, DNase I, and PMSF. After sonication and centrifugation, supernatant was loaded onto Ni-NTA column (Qiagen) equilibrated with buffer D. Beads were washed with five column volumes of buffer E (50 mM HEPES-NaOH pH 7.6, 1M NaCl, 50 mM imidazole, 10% glycerol). Protein was eluted with buffer F containing 50 mM HEPES-NaOH pH 7.6, 200 mM NaCl, 500 mM imidazole, 10% glycerol. After concentration, protein samples were applied to a Superdex 200 10/300 GL column (GE Healthcare) to perform size exclusion chromatography (SEC).
Assessing oligomeric state of RACK1
Size exclusion chromatography
SEC was used as a purification step as well as to estimate the oligomeric state of RACK1 proteins. SEC experiments were done on Superdex 200 10/300 GL columns connected to Äkta FPLC system (GE Healthcare). Samples were run in a buffer containing 20 mM Tris-HCl pH 7.5, 150 mM NaCl, and 5% glycerol, with flow rate 0.5 mL/min. Column was calibrated with standard proteins conalbumin (75 kDa) and carbonic anhydrase (29 kDa) purchased from GE Healthcare.
Analytical ultracentrifugation
Analytical ultracentrifugation sedimentation velocity experiments were performed in a Beckman-Coulter ProteomeLab XL-I analytical ultracentrifuge equipped with AN-60Ti rotor and 12 mm double-sector Epon cells, loaded with 400 µL of protein (A280 = 0.5) and 405 µL of buffer in reference cell. atRACK1 and yRACK1 were run in a buffer containing 20 mM Tris-HCl pH7.5 and 150 mM NaCl. For hRACK1, the buffer was supplemented with 5% glycerol due its high precipitation in a buffer that lacked glycerol. Experiments were carried out at 20°C and 50,000 rpm, using continuous scan mode and 0.003 cm radial spacing. Scans were collected at 4 min intervals at 280 nm, without averaging. Data were analyzed by Sedfit software51 using 200-point resolution. Density and viscosity of the buffer and the partial specific volume of the proteins was calculated with SEDNTERP (version 1.09, http://www.jphilo.mailway.com/download.htm).
Hydrogen-deuterium exchange of three orthologs
Hydrogen-deuterium exchange studies were performed as described previously, with some modifications listed in the text.52 Briefly, to determine the sequence coverage by peptic peptides of RACK1 proteins, 5 µL of each protein stock (50–80 µM) was diluted 10-fold by adding 45 µL of H2O reaction buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1 mM EDTA). The sample was then acidified by mixing with 10 µL of H2O stop buffer (2M glycine, 4M guanidine hydrochloride [Gdn-HCL], 1M tris-(2-carboxyethyl)phosphine [TCEP] pH 2.5) and digested on an immobilized pepsin column (Porozyme; ABI) with 0.07% formic acid in water as a mobile phase (flow rate 200 µL/min). Digested peptides were passed to the C18 trapping column (ACQUITY BEH C18 VanGuard Pre-column, Waters) and then directed onto a reverse-phase column (Acquity UPLC BEH C18 column; Waters) with a 6 to 40% gradient of acetonitrile in 0.1% formic acid at 40 µL/min using nanoACQUITY Binary Solvent Manager. All fluidics, valves, and columns were maintained at 0.5°C with HDX Manager, but the pepsin column was kept at 13°C inside the temperature-controlled digestion compartment of the HDX Manager. C18 column outlet was coupled directly to the ion source of SYNAPT G2 HDMS (Waters) working in Ion Mobility mode. For protein identification, mass spectra were acquired in MSE mode over the m/z range of 50 to 2000. Spectrometer parameters were as follows: ESI positive mode, capillary voltage 3 kV, sampling cone voltage 35 V, extraction cone voltage 3 V, source temperature 80°C, desolvation temperature 175°C, and desolvation gas flow 800 L/h. Peptides were identified using ProteinLynx Global Server software (PLGS, Waters). The list of identified peptides was passed to the DynamX 2.0 program (Waters).
Hydrogen-deuterium exchange experiments were carried out as described above for nondeuterated samples, but H2O was replaced with D2O. After mixing 5 µL of protein stock with 45 µL of D2O reaction buffer, the exchange reactions were carried out for specific time points (10 s, 1 min, 20 min, and 1 h) at room temperature. The exchange was quenched by reducing pH by adding the reaction mixture to stop buffer cooled on ice. The sample was then incubated for 2 min on ice and immediately injected. Two control experiments were carried out to take into account in-and out-exchange artifacts. Briefly, to calculate minimum exchange (IN control), D2O reaction buffer was added to stop buffer cooled on ice before the addition of protein stock, kept for 2 min on ice, and subjected to pepsin digestion and LC-MS analysis. The deuteration level in an in-exchange experiment was denoted as 0% exchange (Mex0). For the OUT control, 5 µL of protein stock was mixed with 45 µL of D2O reaction buffer, incubated overnight, then mixed with stop buffer and analyzed as described above. The deuteration level in an out-exchange control experiment was calculated and denoted as 100% exchange (Mex100).
HDXMS data analysis
Deuteration levels for each peptide resulting from the exchange were calculated with DynamX 2.0 software, based on the peptic peptide list obtained from the PLGS program, which was further filtered in the DynamX 2.0 program with the following acceptance criteria: minimum intensity threshold of 1000 and minimum products per amino acids of 0.3. Analyses of the isotopic envelopes after exchange were carried out with the following parameters: retention time deviation ± 15 s, m/z deviation ± 12.5 ppm, drift time deviation ± 2 time bins. Final data were exported to Excel (Microsoft Office) for calculations. Percentage of deuterium uptake was calculated with a formula [Eq. (1)] that takes into consideration the minimum (Mex0) and maximum exchange (Mex100) of a given peptide:
| (1) |
Error bars for percentage of deuteration (%D) are represented by standard deviations of three independent experiments. The difference in exchange between dimeric and monomeric species of yRACK1 was calculated by subtracting percentage of deuteration of the monomer (%DMONOMER) from that of the dimer (%DDIMER) [Fig. 5(A)]. Here, the error was estimated as the square root of the sum of their variances.
Calculations of rate constant for hydrogen-deuterium exchange
Rate constants of hydrogen-deuterium exchange for a given peptide were derived from nonlinear least-squares curve fitting of an exponential function [Eq. (2)] to the data points using the Levenberg-Marquardt algorithm:
| (2) |
where D%(t) is deuteration in percentage after a given time (t), k is the hydrogen-deuterium exchange rate constant for a particular peptide, and Dmax is the free parameter that describes maximal deuteration for a given peptide. The nls.lm function from the “MINPACK” library implemented in R was used.53 The intrinsic exchange rate constants kint were calculated with use of the SPHERE program.54
Sequence alignment and structural information
ClustalX55,56 was used for sequence alignment, and further editing was performed with GeneDoc.57 Graphical representations of the results were done with OriginPro 8.0 software (OriginLab) and the R program (http://www.R-project.org; September 2013). All figures containing structural information were prepared in YASARA software (downloaded from http://yasara.org; May 2012) Superposition of the three RACK1 orthologs was done in YASARA, which runs on the MUSTANG algorithm.58
Glossary
- Asc1
absence of growth suppressor of Cyp1
- HDXMS
hydrogen-deuterium exchange monitored by mass spectrometry
- RACK1
receptor for activated C kinase 1
- SEC
size exclusion chromatography
- WD repeat
tryptophan aspartate repeat.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Mochly-Rosen D, Khaner H, Lopez J. Identification of intracellular receptor proteins for activated protein kinase C. Proc Natl Acad Sci USA. 1991;88:3997–4000. doi: 10.1073/pnas.88.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schloss JA. A Chlamydomonas gene encodes a G protein beta subunit-like polypeptide. Mol Gen Genet. 1990;221:443–452. doi: 10.1007/BF00259410. [DOI] [PubMed] [Google Scholar]
- 3.Kuo WN, Jones DL, Ku TW, Weeks KD, Jordon PM, Dopson NC. Immunoreactivity of PKC gammalambda and RACK1 in baker's yeast, lobster and wheat germ. Biochem Mol Biol Int. 1995;36:957–963. [PubMed] [Google Scholar]
- 4.Kwak JM, Kim SA, Lee SK, Oh SA, Byoun CH, Han JK, Nam HG. Insulin-induced maturation of Xenopus oocytes is inhibited by microinjection of a Brassica napus cDNA clone with high similarity to a mammalian receptor for activated protein kinase C. Planta. 1997;201:245–251. doi: 10.1007/s004250050063. [DOI] [PubMed] [Google Scholar]
- 5.Vani K, Yang G, Mohler J. Isolation and cloning of a Drosophila homolog to the mammalian RACK1 gene, implicated in PKC-mediated signalling. Biochim Biophys Acta. 1997;1358:67–71. doi: 10.1016/s0167-4889(97)00079-7. [DOI] [PubMed] [Google Scholar]
- 6.Bini L, Heid H, Liberatori S, Geier G, Pallini V, Zwilling R. (YEAR) Two-dimensional gel electrophoresis of Caenorhabditis elegans homogenates and identification of protein spots by microsequencing. Electrophoresis. 18:557–562. doi: 10.1002/elps.1150180337. [DOI] [PubMed] [Google Scholar]
- 7.Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci USA. 1994;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Link a J, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 9.Sengupta J, Nilsson J, Gursky R, Spahn CMT, Nissen P, Frank J. Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nat Struct Mol Biol. 2004;11:957–962. doi: 10.1038/nsmb822. [DOI] [PubMed] [Google Scholar]
- 10.Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 2011;331:730–736. doi: 10.1126/science.1198308. [DOI] [PubMed] [Google Scholar]
- 11.Nilsson J, Sengupta J, Frank J, Nissen P. Regulation of eukaryotic translation by the RACK1 protein: a platform for signalling molecules on the ribosome. EMBO Rep. 2004;5:1137–1141. doi: 10.1038/sj.embor.7400291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baum S, Bittins M, Frey S, Seedorf M. Asc1p, a WD40-domain containing adaptor protein, is required for the interaction of the RNA-binding protein Scp160p with polysomes. Biochem J. 2004;380:823–830. doi: 10.1042/BJ20031962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ceci M, Gaviraghi C, Gorrini C, Marchisio PC, Sala LA, Offenha N, Biffo S. Release of eIF6 (p27 BBP) from the 60S subunit allows 80S ribosome assembly. Nature. 2003;8:579–584. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 14.Adams DR, Ron D, Kiely P. RACK1, A multifaceted scaffolding protein: structure and function. Cell Commun Signal. 2011;9:22. doi: 10.1186/1478-811X-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi S, Deng Y-Z, Zhao J-S, Ji X-D, Shi J, Feng Y-X, Li G, Li J-J, Zhu D, Koeffler HP, Zhao Y, Xie D. RACK1 promotes non-small-cell lung cancer tumorigenicity through activating sonic hedgehog signaling pathway. J Biol Chem. 2012;287:7845–7858. doi: 10.1074/jbc.M111.315416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, Meng J, Du Y, Huang Y, Jin Y, Zhang J, Wang B, Zhang Y, Sun M, Tang J. RACK1 promotes the proliferation, migration and invasion capacity of mouse hepatocellular carcinoma cell line in vitro probably by PI3K/Rac1 signaling pathway. Biomed Pharmacother. 2013;67:313–319. doi: 10.1016/j.biopha.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Cao X-X, Xu J-D, Liu X-L, Xu J-W, Wang W-J, Li Q-Q, Chen Q, Xu Z-D, Liu X-P. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int J Cancer. 2010;127:1172–1179. doi: 10.1002/ijc.25120. [DOI] [PubMed] [Google Scholar]
- 18.Rachfall N, Schmitt K, Bandau S, Smolinski N, Ehrenreich A, Valerius O, Braus GH. RACK1/Asc1p, a ribosomal node in cellular signaling. Mol Cell Proteomics. 2013;12:87–105. doi: 10.1074/mcp.M112.017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeller CE, Parnell SC, Dohlman HG. The RACK1 ortholog Asc1 functions as a G-protein beta subunit coupled to glucose responsiveness in yeast. J Biol Chem. 2007;282:25168–25176. doi: 10.1074/jbc.M702569200. [DOI] [PubMed] [Google Scholar]
- 20.Valerius O, Kleinschmidt M, Rachfall N, Schulze F, López Marín S, Hoppert M, Streckfuss-Bömeke K, Fischer C, Braus GH. The Saccharomyces homolog of mammalian RACK1, Cpc2/Asc1p, is required for FLO11-dependent adhesive growth and dimorphism. Mol Cell Proteomics. 2007;6:1968–1979. doi: 10.1074/mcp.M700184-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Chen J-G, Ullah H, Temple B, Liang J, Guo J, Alonso JM, Ecker JR, Jones AM. RACK1 mediates multiple hormone responsiveness and developmental processes in Arabidopsis. J Exp Bot. 2006;57:2697–2708. doi: 10.1093/jxb/erl035. [DOI] [PubMed] [Google Scholar]
- 22.Kundu N, Dozier U, Deslandes L, Somssich IE, Ullah H. Arabidopsis scaffold protein RACK1A interacts with diverse environmental stress and photosynthesis related proteins. Plant Signal Behav. 2013;8:1–7. doi: 10.4161/psb.24012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz Carrillo D, Chandrasekaran R, Nilsson M, Cornvik T, Liew CW, Tan SM, Lescar J. Structure of human Rack1 protein at a resolution of 2.45 Å. Acta Crystallogr F. 2012;68:867–872. doi: 10.1107/S1744309112027480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yatime L, Hein KL, Nilsson J, Nissen P. Structure of the RACK1 dimer from Saccharomyces cerevisiae. J Mol Biol. 2011;411:486–498. doi: 10.1016/j.jmb.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 25.Ullah H, Scappini EL, Moon AF, Williams LV, Armstrong DLEE. Structure of a signal transduction regulator, RACK1, from Arabidopsis thaliana. Protein Sci. 2008;17:1771–1780. doi: 10.1110/ps.035121.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coyle SM, Gilbert W, V, Doudna J. Direct link between RACK1 function and localization at the ribosome in vivo. Mol Cell Biol. 2009;29:1626–1634. doi: 10.1128/MCB.01718-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fülöp V, Jones DT. Beta propellers: structural rigidity and functional diversity. Curr Opin Struct Biol. 1999;9:715–721. doi: 10.1016/s0959-440x(99)00035-4. [DOI] [PubMed] [Google Scholar]
- 28.Thornton C, Tang K-C, Phamluong K, Luong K, Vagts A, Nikanjam D, Yaka R, Ron D. Spatial and temporal regulation of RACK1 function and N-methyl-d-aspartate receptor activity through WD40 motif-mediated dimerization. J Biol Chem. 2004;279:31357–31364. doi: 10.1074/jbc.M402316200. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, V, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, Liu JO, Semenza GL. Calcineurin promotes hypoxia-inducible factor 1alpha expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem. 2007;282:37064–37073. doi: 10.1074/jbc.M705015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen S, Spiegelberg BD, Lin F, Dell EJ, Hamm HE. Interaction of Gbetagamma with RACK1 and other WD40 repeat proteins. J Mol Cell Cardiol. 2004;37:399–406. doi: 10.1016/j.yjmcc.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 31.Chen S, Dell EJ, Lin F, Sai J, Hamm HE. RACK1 regulates specific functions of Gbetagamma. J Biol Chem. 2004;279:17861–17868. doi: 10.1074/jbc.M313727200. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Lin F, Hamm HE. RACK1 binds to a signal transfer region of G betagamma and inhibits phospholipase C beta2 activation. J Biol Chem. 2005;280:33445–33452. doi: 10.1074/jbc.M505422200. [DOI] [PubMed] [Google Scholar]
- 33.Jaswal SS. Biological insights from hydrogen exchange mass spectrometry. Biochim Biophys Acta. 2013;1834:1188–1201. doi: 10.1016/j.bbapap.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 34.McCahill A, Warwicker J, Bolger GB, Houslay MD, Yarwood SJ. The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol Pharmacol. 2002;62:1261–1273. doi: 10.1124/mol.62.6.1261. [DOI] [PubMed] [Google Scholar]
- 35.Yang X-J, Grégoire S. A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol Cell. 2006;23:779–786. doi: 10.1016/j.molcel.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 36.Grosso S, Volta V, Sala L, Vietri M, Marchisio PC, Ron D, Biffo S. PKCbetaII modulates translation independently from mTOR and through RACK1. Biochem J. 2008;415:77–85. doi: 10.1042/BJ20080463. [DOI] [PubMed] [Google Scholar]
- 37.Sharma G, Pallesen J, Das S, Grassucci R, Langlois R, Hampton CM, Kelly DF, des Georges A, Frank J. Affinity grid-based cryo-EM of PKC binding to RACK1 on the ribosome. J Struct Biol. 2013;181:190–194. doi: 10.1016/j.jsb.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melamed D, Bar-Ziv L, Truzman Y, Arava Y. Asc1 supports cell-wall integrity near bud sites by a Pkc1 independent mechanism. PLoS One. 2010;5:e11389. doi: 10.1371/journal.pone.0011389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang BY, Chiang M, Cartwright CA. The interaction of Src and RACK1 is enhanced by activation of protein kinase C and tyrosine phosphorylation of RACK1. J Biol Chem. 2001;276:20346–20356. doi: 10.1074/jbc.M101375200. [DOI] [PubMed] [Google Scholar]
- 40.Chang BY, Harte R, Cartwright C. RACK1: a novel substrate for the Src protein-tyrosine kinase. Oncogene. 2002;21:7619–7629. doi: 10.1038/sj.onc.1206002. [DOI] [PubMed] [Google Scholar]
- 41.Ceci M, Welshhans K, Ciotti MT, Brandi R, Parisi C, Paoletti F, Pistillo L, Bassell GJ, Cattaneo A. RACK1 is a ribosome scaffold protein for β-actin mRNA/ZBP1 complex. PLoS One. 2012;7:e35034. doi: 10.1371/journal.pone.0035034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De la Fuente van Bentem S, Hirt H. Protein tyrosine phosphorylation in plants: more abundant than expected? Trends Plant Sci. 2009;14:71–76. doi: 10.1016/j.tplants.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Rudrabhatla P, Reddy MM, Rajasekharan R. Genome-wide analysis and experimentation of plant serine/threonine/tyrosine-specific protein kinases. Plant Mol Biol. 2006;60:293–319. doi: 10.1007/s11103-005-4109-7. [DOI] [PubMed] [Google Scholar]
- 44.Brinkworth RI, Munn AL, Kobe B. Protein kinases associated with the yeast phosphoproteome. BMC Bioinformatics. 2006;7:47. doi: 10.1186/1471-2105-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stern DF, Zheng P, Beidler DR, Zerillo C. Spk1, a new kinase from Saccharomyces cerevisiae, phosphorylates proteins on serine, threonine, and tyrosine. Mol Cell Biol. 1991;11:987–1001. doi: 10.1128/mcb.11.2.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue B, Uversky VN. Structural characterizations of phosphorylatable residues in transmembrane proteins from Arabidopsis thaliana. Landes Biosci Intrinsically Disord Proteins. 2013;1:1–10. doi: 10.4161/idp.25713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ubersax Ja, Ferrell JE. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 48.Kiely Pa, Baillie GS, Barrett R, Buckley D, Adams DR, Houslay MD, O'Connor R. Phosphorylation of RACK1 on tyrosine 52 by c-Abl is required for insulin-like growth factor I-mediated regulation of focal adhesion kinase. J Biol Chem. 2009;284:20263–20274. doi: 10.1074/jbc.M109.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiely P, Baillie GS, Lynch MJ, Houslay MD, O'Connor R. Tyrosine 302 in RACK1 is essential for insulin-like growth factor-I-mediated competitive binding of PP2A and beta1 integrin and for tumor cell proliferation and migration. J Biol Chem. 2008;283:22952–22961. doi: 10.1074/jbc.M800802200. [DOI] [PubMed] [Google Scholar]
- 50.Chen J-G, Ullah H, Temple B, Liang J, Guo J, Alonso JM, Ecker JR, Jones AM. RACK1 mediates multiple hormone responsiveness and developmental processes in Arabidopsis. J Exp Bot. 2006;57:2697–2708. doi: 10.1093/jxb/erl035. [DOI] [PubMed] [Google Scholar]
- 51.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sitkiewicz E, Tarnowski K, Poznański J, Kulma M, Dadlez M. Oligomerization interface of RAGE receptor revealed by MS-monitored hydrogen deuterium exchange. PLoS One. 2013;8:e76353. doi: 10.1371/journal.pone.0076353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Anon CRAN—Package minpack.lm. Available from: http://cran.r-project.org/web/packages/minpack.lm/index.html. Accessed on March 2, 2013.
- 54.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 56.Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;2:2–3. doi: 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- 57.Nicholas KB, Nicholas HB, Jr, Deerfield DWI. GeneDoc: analysis and visualization of genetic variation. EMBNEW News 4–14. 1997 [Google Scholar]
- 58.Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. MUSTANG: a multiple structural alignment algorithm. Proteins. 2006;574:559–574. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.