

Abstract
Rasmussen’s encephalitis is a rare chronic neurological disorder, characterised by unilateral inflammation of the cerebral cortex, drug-resistant epilepsy, and progressive neurological and cognitive deterioration. Neuropathological and immunological studies support the notion that Rasmussen’s encephalitis is probably driven by a T-cell response to one or more antigenic epitopes, with potential additional contribution by autoantibodies. Careful analysis of the association between histopathology and clinical presentation suggests that initial damage to the brain is mediated by T cells and microglia, suggesting a window for treatment if Rasmussen’s encephalitis can be diagnosed early. Advances in neuroimaging suggest that progression of the inflammatory process seen with MRI might be a good biomarker in Rasmussen’s encephalitis. For many patients, families, and doctors, choosing the right time to move from medical management to surgery is a real therapeutic dilemma. Cerebral hemispherectomy remains the only cure for seizures, but there are inevitable functional compromises. Decisions of whether or when surgery should be undertaken are challenging in the absence of a dense neurological deficit, and vary by institutional experience. Further, the optimum time for surgery, to give the best language and cognitive outcome, is not yet well understood. Immunomodulatory treatments seem to slow rather than halt disease progression in Rasmussen’s encephalitis, without changing the eventual outcome.
Introduction
Rasmussen’s encephalitis was first described by neurosurgeon Theodore Rasmussen and his colleagues in the late 1950s.1 Since then, the variable clinical features and lack of understanding of cause have created dilemmas in clinical decision making. The 2005 European consensus on pathogenesis, diagnosis, and treatment of Rasmussen’s encephalitis remains the accepted guideline for evaluative criteria (panel 1).2,3 Progress has been made over recent years in understanding the clinical evolution and pathobiology of Rasmussen’s encephalitis. However, despite increasing evidence of an underlying immune process, the primary cause remains unknown. Targeted therapeutic strategies remain elusive. We review the evidence base and highlight questions that still need to be addressed.
Clinical presentation
Rasmussen’s encephalitis is a progressive disease characterised by drug-resistant focal epilepsy, progressive hemiplegia, and cognitive decline, with unihemispheric brain atrophy. The disorder is rare and affects mostly children or young adults. Investigators in a German study4 estimated the countrywide incidence at 2·4 cases per 10 million people aged 18 years and younger per year. Similarly, researchers in a recent UK surveillance study5 estimated an incidence of 1·7 per 10 million people aged 16 years and younger per year (a prevalence of 0·18 per 100 000 people). Sex, geographical, and ethnic predominance have not been reported.
The typical clinical course has been characterised during the past century (figure 1).6 The median age of onset is 6 years, with a range from infancy to adulthood.6-8 In some patients, a prodromal period of mild hemiparesis or infrequent seizures might precede the onset of the acute stage by up to several years. The acute stage is marked by frequent seizures arising from one cerebral hemisphere. About 50% of patients with Rasmussen’s encephalitis have epilepsia partialis continua.9-11 As the disease progresses, different focal seizure semiologies emerge, suggesting newly affected areas of inflammation in the hemisphere.7 Untreated, children will develop hemiparesis, hemianopia, and cognitive decline within a year of epilepsy onset,12 and if the language-dominant hemisphere is affected, dysphasia. Finally, there is a relatively stable residual stage with a severe fixed neurological deficit, motor and cognitive problems, and with persisting difficult-to-treat relapsing epilepsy.13
Figure 1. Natural clinical course and expected effect of immunotherapy.
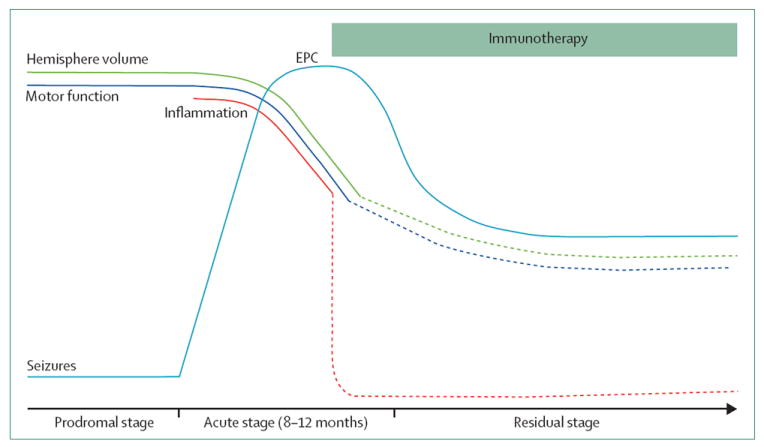
The natural clinical course of Rasmussen’s encephalitis was characterised in the past century. The disease might have a preceding prodromal stage with infrequent seizures, and presents with an acute stage of drug-resistant epilepsy. The epilepsy is characterised by very frequent seizures of different semiologies in the same patient, often epilepsia partialis continua, with the emergence of a fluctuating then permanent hemiplegia (motor function) and concurrent progressive hemispheric volume loss on neuroimaging. With the advent of immunotherapy, the natural clinical course seems to be changing. The rate of motor function and hemispheric volume loss is slowed, and seizures decrease in frequency and plateau. Cognitive deterioration is not shown because it is more variable, although usually becomes manifest during the acute phase. EPC=epilepsia partialis continua.
Some cases of Rasmussen’s encephalitis have a less common presentation. Roughly 10% of cases described in case series start in adolescent or adult life.7 The clinical course is usually slower, and final deficits are not as severe as in children;6,14-16 the semiology can be more characteristic of temporal lobe epilepsy.16,17 Disease presentations have been described with unilateral movement disorders, including hemiathetosis and hemidystonia.18,19 The existence of bilateral disease is debated but is probably very rare. Only two out of the roughly 200–300 published cases of Rasmussen’s encephalitis had evidence of bilateral disease on histopathology.20,21 Further, to the best of our knowledge, no case of contralateral involvement (even when judged by clinical criteria) after surgical treatment of unilateral Rasmussen’s encephalitis has been reported.
Rasmussen’s encephalitis without seizures might be an unrecognised cause of progressive unilateral neurological deficits in childhood.22 Otherwise typical progressive Rasmussen’s encephalitis with characteristic histopathological features has been reported in patients with delayed seizure onset, or even the absence of seizures for periods of up to 2 years.22,23 Such findings suggest that seizures are not an inevitable consequence of Rasmussen’s encephalitis.24
Neuroimaging and EEG features
MRI of the brain has become a mainstay for diagnostic assessment and follow-up in Rasmussen’s encephalitis.25,26 Usually, within months of onset of the acute stage, most patients show unilateral enlargement of the ventricular system. A T2/FLAIR hyperintense signal is often present in cortical or subcortical regions, or both, the distribution is heterogeneous, and temporal fluctuation might be related to seizure frequency in patients without epilepsia partialis continua.27 The perisylvian region is a predilection site for signal change and volume loss (figure 2). Ipsilateral atrophy of the head of the caudate nucleus is a typical but not an invariable accompanying feature of hemispheric atrophy, and can be an early sign.26 Serial MRIs typically show progression of signal change and atrophy. Recent radiological volumetric approaches describe the highest rate of volume loss in the first 8 months of disease, the acute clinical phase,6 and putamen predominance, rather than caudate predominance, in the basal ganglia.28 Some atrophy is always evident, even of the unaffected hemisphere, probably as a result of degeneration of commissural fibres.29 Functional studies using 18F-FDG PET show diffuse unilateral cerebral hypometabolism that might manifest when MRI atrophy is still at a minimum.30
Figure 2. Neuroimaging in Rasmussen’s encephalitis.

MRI brain scans of children with Rasmussen’s encephalitis, showing contrasting cases of radiological progression. (A) Progressive right hemisphere atrophy, high signal and basal ganglia loss over 1 year (from left to right) in a child with Rasmussen’s encephalitis. The disease was mostly centred near the right Sylvian fissure (arrow). (B) Slowly progressive disease with more subtle right hemisphere atrophy in a child on immunosuppressant treatment at 6 months (left), 18 months (centre), and 30 months (right) of disease course.
Widely varying abnormalities in electroencephalograms (EEG) are seen in patients with Rasmussen’s encephalitis,31 which are often related to clinical progression. No specific EEG abnormalities can distinguish Rasmussen’s encephalitis from other causes of focal epilepsy. However, from an initially normal EEG, persistent high amplitude delta activity can develop over the affected hemisphere within months of seizure onset. Epileptiform abnormalities are frequent and they often develop into electrographic seizures, but epilepsia partialis continua is not always accompanied by visually recognisable ictal surface EEG activity.31 Independent interictal abnormalities over the non-affected hemisphere emerge within 6 months in 25% of patients and in 62% within 3–5 years from seizure onset. These contralateral abnormalities are associated with, and can be a marker of, cognitive decline, but do not seem to be indicative of bilateral disease.11
Formal diagnostic criteria
A European consensus panel proposed formal diagnostic criteria for Rasmussen’s encephalitis in 2005 (panel 1).2 The most characteristic features are the progressive unilateral loss of function and contralateral hemispheric atrophy that happen over several months or a few years. Findings of previous serial and cross-sectional investigations lend confidence to the diagnosis. However, with the use of immunosuppressive treatment, the expected progressive changes, especially in the MRI hemispheric atrophy, might be slower (figure 2), making the clinician less confident in making the diagnosis of Rasmussen’s encephalitis and hence its treatment. Additional descriptive and larger cohort studies are needed to better characterise and refine the more recent clinical characteristics and course of Rasmussen’s encephalitis, particularly with increasing use of immunosuppressive treatments.
Pathobiology
The characteristic histopathological hallmarks of Rasmussen’s encephalitis are cortical inflammation, neuronal loss, and gliosis confined to one cerebral hemisphere (figure 3).1 Inflammation is multifocal within the hemisphere and progressive. Microglial and lymphocytic nodules and perivascular cuffing, neuronal death, and neuronophagia are the most common pathological features. End-stage features include cortical cavitation, marked astrogliosis, and neuronal cell loss. This progression seems to be consistent with an immune-mediated disease that is likely to be associated with both adaptive immune reactions, with T-lymphocyte responses, and innate immunity facilitated by both microglia and astroglia.32
Figure 3. Histopathology of neurodegeneration and inflammation in Rasmussen’s encephalitis.

(A–E) Cortical degeneration in Rasmussen’s encephalitis. (A) Staining for MAP2 shows intact cortical neurons on the left side while loss of MAP2 neurons is found on the right side. (B) Cortical degeneration in a later stage of the disease; most neurons are already lost. (C) The same areas stained for glial fibrillary acidic protein, showing strong activated astrocytes. (D) An almost complete loss of cortical neurons. (E) In this area, nearly complete fibrillary astrogliosis is present. (F and G) Microglia activation in the cortex is notable. (F) A microglial nodule positively stained by anti-HLA-DR. (G) Staining for CD68 shows activation of microglial cells in the cortex. (H) Staining for CD8 reveals infiltration of cytotoxic T lymphocytes in the cortex. Notably, this case is one from the collection of Dr Rasmussen (case no. 238–69D). Higher magnifications of this section in (I) and (J) show appositions of cytotoxic T cells to neurons (arrows). (K) Staining for granzyme B shows granzyme B-positive granules in close apposition to a neuron.
Any area of the brain can be affected, but recent radiological studies agree with the long-standing pathological findings of a fronto-insular predilection, with the occipital cortex less frequently affected. When the occipital cortex is involved, the patients tend to be younger with a higher burden of disease.28,32,33 Heterogeneity and variability in lesion location, progression of disease, and magnitude of pathological changes, are quite substantial within and between patients and suggest a disease process affecting different parts of the brain at different times.25,32,33
There are two patterns of cortical damage in Rasmussen’s encephalitis: a gyral pattern with involvement of large areas of the top or sulcal regions of a gyrus; and less frequently wedge-shaped or punched-out lesions. An area of pronounced cortical damage is often surrounded by normal cerebral cortex or milder stages of inflammation, explaining why a biopsy can be misleading. Four stages of neuropathological progression in Rasmussen’s encephalitis have been described,32 suggesting a continuum of cortical injury mediated by T-lymphocyte and neuroglial activation, and underpinning the role of immune and neuroglial pathogenic mechanisms.32 Additionally, morphological changes, such as focal cortical degeneration seen in late stages of cortical pathology, resemble those described in excitotoxic cortical injury,34 suggesting that other factors, such as excitotoxicity, might be involved.
Results of case series of surgical specimens have also shown dual pathology, with the finding of focal cortical dysplasia or tuberous sclerosis in association with Rasmussen’s encephalitis.35,36 More recently, inflammation has been described in association with focal cortical dysplasia type 2b lesions.37 Involvement of perilesional cortex is yet to be established, but the question arises: could Rasmussen’s encephalitis begin from inflammation around a focal cortical dysplasia or are some cases of Rasmussen’s encephalitis in fact focal cortical dysplasia type 2b with inflammation?
Neuroimmunology
Evidence for an immunopathological basis of Rasmussen’s encephalitis is growing. The immunopathological mechanisms recognised to have a role in CNS degeneration can be categorised into three types: antibody-mediated, T-cell cytotoxicity, and microglia-induced degeneration.
Antibody-mediated CNS degeneration
Although traditionally the brain is thought to be protected against the humoral immune system, data from recent studies suggest that this is not necessarily the case. Over the past 10 years it has become clear that there are a number of different CNS disorders associated with potentially pathogenic circulating antibodies to neuronal surface proteins.38,39 The first such autoantibody to be identified, against GluR3, was in Rasmussen’s encephalitis, and for some years antibody-mediated mechanisms dominated Rasmussen’s encephalitis research.40 However, GluR3 antibodies were found in only a few patients with Rasmussen’s encephalitis,41-43 and plasmapheresis helped only a few patients.44 Antibodies against other antigens, such as the alpha-7 nicotinic acetylcholine receptor or Munc-18-1, were then identified in sera from a few patients with Rasmussen’s encephalitis, but this was not subsequently reported.45-47 Munc-18 is an evolutionarily conserved neuronal protein essential for synaptic vesicle release; however, it is an intracellular protein and is therefore unlikely to be a main target.47 The MUNC18-1 gene is also termed STXBP1, and mutations in this gene are a cause of early infantile epileptic encephalopathy.
Antibodies to LGI1, AMPA receptors, and GABA-B receptors were detected in individuals with limbic encephalitis, and NMDA receptor antibodies in a more complex encephalopathy; these diseases do respond to immunotherapies, suggesting that one disease entity does not necessarily imply one pathogenic agent.38 Indeed, one case is reported of anti-NMDA receptor encephalitis mimicking the acute phase of Rasmussen’s encephalitis.48 Nevertheless, even these highly-specific cell surface-directed antibodies can occasionally be present in patients with neurodegenerative disease, in which case these antibodies are probably secondary to the pathology rather than causative. Because none of the autoantibodies have been found in more than a small number of patients with Rasmussen’s encephalitis, and responses to plasma exchange are unpredictable, the role of CNS autoantibodies in the pathogenesis of Rasmussen’s encephalitis is still unclear.
T-cell cytotoxicity
Cytotoxic T lymphocytes seem to play a major part in the pathogenesis of Rasmussen’s encephalitis. Most of the inflammatory T cells are CD8 and about 10% of these cells are granzyme B-positive cells. Such granzyme B cells were found in apposition to neurons and astrocytes, with polarisation of the cytotoxic granules to face the target cell membrane; release of granzyme B onto neurons was occasionally noted.49 Furthermore, spectra-typing of the T cells from brain lesions showed that these cells expanded from discrete, antigenic, epitope-responding, precursor T cells,50 suggesting specificity for single brain antigens. The next goal would be to find such driving antigens in the brains of patients with Rasmussen’s encephalitis. Because neurons and astrocytes are attacked by cytotoxic T cells,51 one might predict that an autoantigen was expressed by both these cell types. However, the identity of the antigen remains elusive.
Another possibility is that, rather than recognising an autoantigen, these cytotoxic T cells might recognise foreign antigens from pathogen-infected neurons and astrocytes. The presence of pathogen-infected cells in a focal area of the brain also could explain the unilateral occurrence of the disease. In the past, brain tissue from patients with Rasmussen’s encephalitis has been tested for the presence of several viruses, including Epstein-Barr virus, cytomegalovirus, herpes simplex virus, and enterovirus.52-55 None of these studies was able to show a causal association between Rasmussen’s encephalitis and a specific virus, although relatively few well-known viruses have been investigated. Finding a rare virus would be much more difficult because methods are scarce. Furthermore, infected cells under cytotoxic T-cell attack can be injured and later engulfed by microglia and macrophages, in which case the chances of finding a virus most likely occur early in the disease when surgical sampling is less frequent. Thus, the search for a Rasmussen’s encephalitis pathogen, particularly if it is rare, will be challenging.
Microglia-induced degeneration
Microglial activation is one of the neuropathological characteristics described in Rasmussen’s encephalitis.1 The degree of activation of these cells can vary in different areas of the brain,56 but follows closely the pattern of T-cell infiltration and stages of progression of cortical damage.32 Microglia, via release of factors such as interleukin 1 and other pro-inflammatory cytokines, participate in the induction of seizures in other epileptic disorders.57,58 Activated microglia can also participate in complement-mediated synaptic stripping that can increase network excitability.59,60 However, the specific pathogenic role of microglial cells in Rasmussen’s encephalitis is still unclear.
In addition to microglial activation, astrocytes are also activated in Rasmussen’s encephalitis;51 the pattern of astroglial activation follows closely the progression of cortical damage.32 Astrocytes might have a role in the pathology of several epileptic and inflammatory disorders of the brain.61 Therefore, in Rasmussen’s encephalitis, astrocytes probably have a similar role in the inflammatory response. However, astrocytes are also lost with continuing disease activity, most probably also by means of a cytotoxic T-cell reaction.51 In Rasmussen’s encephalitis, CSF albumin concentration increases with disease progression.62,63 Albumin uptake is followed by downregulation of inward-rectifying potassium (Kir 4.1) channels in astrocytes, resulting in reduced buffering of extracellular potassium and NMDA receptor-mediated neuronal excitability. As such, albumin leakage via modulation of astrocytes might enhance the initiation of seizures during the course of Rasmussen’s encephalitis.
Inflammatory gene expression
To gain further insight into the nature of the immune response in Rasmussen’s encephalitis, the relative expression levels of 86 mRNA transcripts implicated in inflammation and autoimmunity were measured by quantitative PCR in 12 Rasmussen’s encephalitis brain tissue specimens and compared with specimens from a cohort of 12 cases of cortical dysplasia. Patients with cortical dysplasia have focal seizures, display abnormalities in cortical neuron differentiation, and histopathological evidence suggests an inflammatory response is present but is less severe than in Rasmussen’s encephalitis. Findings of the analysis showed that a subset of seven functionally-related genes that code for interferon-γ, CCL5, CCL22, CCL23, CXCL9, CXCL10, and Fas ligand were expressed at higher levels in the Rasmussen’s encephalitis cohort compared with the cortical dysplasia cohort. These genes are related to the activation of helper and inducer, and memory and effector T cells, and include genes for specific chemokines that might have a role in the recruitment of these T cells to sites of inflammation.64
The problem is that none of the studies described above was done at the earliest expression of the disease, and consequently the main pathology is unknown. Many of the findings could, in theory, be secondary rather than primary to the disease process. The search for a primary immune or viral factor needs to continue because such a discovery would enable earlier diagnosis, and would enable clinicians to decide which of the phenomena described above are primary or secondary; this information is essential to establish the effectiveness of immunotherapy.
Treatment
Medical
Treatment in Rasmussen’s encephalitis aims to reduce seizure severity and frequency and improve the functional long-term outcome, as measured by both motor and cognitive performance. However, to date, treatments have only alleviated the symptoms65 and have not tackled the underlying causes.
Seizures
Antiepileptic drugs have a limited effect on seizures and disease progression in Rasmussen’s encephalitis. Epilepsia partialis continua in particular tends to be refractory to antiepileptic drugs. A realistic aim of antiepileptic drug therapy in Rasmussen’s encephalitis should be to protect the patient from the most severe seizures, namely bilateral convulsive seizures, rather than to achieve seizure freedom. Treatment should therefore be adjusted to achieve optimum seizure control with the fewest side-effects.66 Cases are reported of botulinum toxin successfully injected into the zygomaticus for facial myoclonus and into upper limb muscles for localised epilepsia partialis continua, reducing painful spasms and improving functional use of the limb.67,68 Case series of short-term intense immuno-interventions, such as steroid pulses, apheresis treatment,44 and a case report and an open-label trial of rituximab infusions13,69 have shown a temporary beneficial effect on seizure frequency. In 2011, case reports were published of seizure improvement with vagus nerve stimulation therapy and transcranial magnetic stimulation.70,71 However, most cases of Rasmussen’s encephalitis are resistant to antiepileptic drugs.
Treatment directed against the primary process
In view of the belief that Rasmussen’s encephalitis is an immune-regulated process, immunosuppressive or immunomodulatory treatments are being assessed. The effects of long-term immunotherapy for Rasmussen’s encephalitis have been described in case reports or small, uncontrolled patient series. Findings from these publications show the most positive experience with long-term corticosteroids,44,72,73 intravenous immunoglobulins,44,74-76 plasmapheresis or protein A immuno-absorption,44,77,78 and the T-cell inactivating drugs tacrolimus12 and azathioprine.79 Strategies have also been proposed that start with steroid pulse treatment and change to tacrolimus.80 A recent prospective randomised trial4 assessed the effect of long-term immunotherapy with tacrolimus or intravenous immunoglobulins. Compared with a historical control group, immunotherapy with either of these agents protected against functional or structural damage. However, the study was underpowered to assess the superiority of one of the drugs. The discrepancy between the (likely) antidegenerative effect and the apparently absent anti-seizure effect might lead to a novel artificial situation, a kind of Pyrrhic victory, in some patients. With preserved functions, these patients are difficult to refer for hemispherectomy surgery despite having intractable and severe epilepsy.
Whether such treatments improve the long-term outcome of patients, particularly cognitive endpoints, remains unclear. Certainly in the open-label case series of azathioprine treatment, cognitive decline continues; therefore, imperfectly effective immunological interventions might unacceptably postpone hemispherectomy.81 This concern seems particularly relevant in patients with very active, drug-resistant epilepsy who might otherwise be considered for a diagnosis of Rasmussen’s encephalitis. Therefore, the search continues for immunological treatments that can halt both the seizures and the functional decline (ie, loss in motor and cognitive performance). Promising candidates might come from multiple sclerosis treatments,82 including compounds reducing the likelihood of T-cell entry into the CNS (table), although the potential toxic effects and adverse effects are concerning, particularly for a disorder that affects mainly a paediatric population. Promising results have been seen in one case study of natalizumab, which blocks entry of T cells into the CNS.84
Table.
Candidate treatments for Rasmussen’s encephalitis
| Comment | Level of clinical experience (if any) | |
|---|---|---|
|
T-cell directed
| ||
| Natalizumab | Monoclonal antibody; A4-integrin cellular adhesion molecule, reduces T-cell ability to pass across blood–brain barrier | Case report, 22-year-old female, 1 year outcome of improved epilepsia partialis continua and focal seizures83 |
|
| ||
|
B-cell directed
| ||
| Rituximab | Monoclonal antibody; CD20 B-cell lysis; loss of antigen presentation, resulting in less T-cell activation | Case series of nine patients (ages 6–22 years), well-tolerated; eight patients had improvement in seizures, motor, and cognitive function at 3–22 months;69 case report of use to stabilise 20-year-old female in focal motor status epilepticus13 |
|
| ||
|
Disruption of T and B cells
| ||
| Azathioprine | Used initially as a steroid-sparing agent, azathioprine might allow continuation of the anti-seizure effect of oral steroids | Case series of 16 children, response in 13 of 16; reduction in seizure frequency; follow-up 1·6–15 years;79 might slow MRI tissue and motor function loss, but does not slow cognitive decline (personal observation, SV and JHC) |
| Fingolimod | Prevents migration of lymphocytes from nodes | No experience in Rasmussen’s encephalitis |
| Cladribine | Disrupts lymphocyte cellular processes; can penetrate CNS | No experience in Rasmussen’s encephalitis |
| Mycophenylate | Inhibits purine synthesis in lymphocytes; induces apoptosis in activated T lymphocytes; depletes tetrahydrobiopterin, a cofactor for inducible nitric oxide synthase, which is activated in inflammation | Anecdotal; patients in case reports for other treatments stated as treated13 |
| Alemtuzumab | T-cell and B-cell depletion, might also turn on neurotrophic factors | No experience in Rasmussen’s encephalitis |
|
| ||
|
Inhibition of microglia
| ||
| Minocycline and related tetracycline derivatives | Highly potent poly (ADP-ribose) polymerase (PARP) inhibitors; PARP-1 is activated by DNA damage and by cytokines such as tumour necrosis factor α (TNFα); PARP-1 interaction with NF-κB regulates expression of several pro-inflammatory mediators, including proteases, inducible nitric oxide synthase, ICAM-1, and TNFα | No experience in Rasmussen’s encephalitis |
| Perindopril | A brain-penetrating angiotensin-converting enzyme inhibitor, reported to reduce cognitive decline in Alzheimer’s disease; suppresses microglia and astrocyte activation and decreases inducible nitric oxide synthase expression83 | No experience in Rasmussen’s encephalitis |
|
| ||
|
Target excitotoxicity
| ||
| COX-2 inhibitors | Experimentally, non-selective COX inhibitor naproxen and specific COX-2 inhibitor rofecoxib ameliorate excitotoxic neuronal damage34 | No experience in Rasmussen’s encephalitis |
Surgery
Surgery still remains the only cure for the seizures caused by Rasmussen’s encephalitis. This has functional consequences because the only effective surgery remains complete disconnection of the affected hemisphere (hemidisconnection), either as (functional) hemispherectomy or hemispherotomy. Homonymous hemianopia and hemiplegia are inevitable, although both might be present before surgery. Rehabilitation to independent walking is expected although fine motor movement in the hand on the affected side is not. Small resections can sometimes be preferable to preserve function, but no investigators have reported sustained seizure freedom after limited resection in patients with Rasmussen’s encephalitis. Hemispherectomy offers one of the best chances of making patients with Rasmussen’s encephalitis seizure free (>70–80% long-term seizure-free outcome).65 The decision in older children and adults can be more difficult if the disease affects the dominant hemisphere.
Timing of surgery can be guided by the severity of epilepsy. Some advocate for early surgery to protect the contralateral normal hemisphere from repeated seizures and progressive neuropsychological loss. Few longitudinal studies of children with Rasmussen’s encephalitis have been done. Available data suggest that not all children with Rasmussen’s encephalitis show a decline in intellectual ability over time, but in those who do, the decline is associated with the appearance of contralateral, independent interictal epileptiform discharges.11 Therefore, although cognitive decline is not inevitable, the appearance of contralateral EEG abnormalities might highlight those at risk, and consequently those for whom surgery should be discussed. Group data suggest little change in psychological outcome before and after the operation.85 Moreover, findings of studies of children undergoing hemispherectomy for pathologies including Rasmussen’s encephalitis show short seizure duration is associated with better cognitive outcome,86,87 suggesting that earlier surgery should be considered to improve these outcomes. Most results show cognitive stabilisation after hemispherectomy,88-90 with better cognitive outcome in non-dominant-hemisphere surgery and poorer outcome in dominant-hemisphere surgery and in individuals with refractory seizures after surgery.80 Extrapolation of these findings to the individual patient should be done with caution because global measures, such as full-scale intelligence quotient measurement, do not adequately describe or assess functional outcome, particularly in the setting of language or memory deficit.
How much language transfer is possible from the dominant hemisphere is probably dependent on age at the start of the disease process. Disease in the hemisphere that is designated to become the hemisphere responsible for language in individuals younger than 5–6 years usually results in recruitment of the contralateral hemisphere for language functions.2 Anecdotal reports have shown transfer of language clinically or through functional MRI (fMRI).91 The question remains of whether such a process is driven more effectively through early surgery, or whether waiting will enhance the process. However, it is rare for an individual to be rendered completely aphasic by such a procedure,92 and useful aspects of language might be retained even after surgery in patients with a late onset of disease (most receptive functions are retained, whereas expressive language is restricted to telegraphic speech [two or three word] output).
Advances in fMRI for language enable an indication of the degree of transfer at any time, and this helps to predict whether useful aspects of language can be retained and help with the surgical decision-making process. The gold standard for prediction of postoperative language performance in epilepsy surgery remains the Wada test, which is done by injection of a barbiturate into the affected hemisphere. The balance has to be made between what is to be lost by waiting, and likely gained by surgery; the danger of the seizures themselves and the presumed neuropsychological compromise need to be considered. A decision aid based on the relative severity of motor deficit and seizure disorder has been suggested (figure 4).65 Although this aid might be useful as a guide, the disease process will differ between individuals and therefore such decisions need to be made on an individual basis.
Figure 4. Suggested therapeutic management of the patient with Rasmussen’s encephalitis.

*A judgment or decision to be made by the patient, carers or parents, and the treating physician. †This is a matter of consideration, not an objective measure. ‡This will mainly apply to patients with short disease duration and preserved function of the affected hemisphere. §There is no evidence to support any special agent. ¶No formal recommendation regarding the intervals can be given, and will be affected by the needs of the patient. If the course is unsatisfactory, the patient will most probably return to the treating institution needing reassessment. Modified from Bien and Schramm,65 by permission of Elsevier.
Conclusions and future directions
Rasmussen’s encephalitis is a progressive disease of one cerebral hemisphere, with management complicated by difficulties in making an early diagnosis. Although much progress has been made in establishing the likely underlying process, the triggers and markers of those at risk for Rasmussen’s encephalitis remain elusive (panel 2). The disease is complicated by variable presentations, based on age and severity, making it difficult to stratify patients for focused studies.
The hunt for the Rasmussen virus or antigen needs continuing efforts. So far, data from histopathology studies suggest that the earliest components of change include microglia and early death of glia and neurons. Findings of studies using tissue samples from cases of Rasmussen’s encephalitis support the notion that only one or a few antigens probably produce the disease in the brain. Previous attempts to find viral antigens used techniques that were not sensitive enough, and state-of-the-art analyses should be used to confirm or refute the viral hypothesis. Equally, quicker identification of patients might make it possible to show more convincingly the presence of serum or CSF autoantibodies to one or more neuronal antigens, if these do indeed exist.
Aspects that deserve further consideration are genetic and environmental interactions. To date, no clear pattern of familial inheritance in Rasmussen’s encephalitis has emerged, but in one early study93 decreased IgA concentrations and increased frequency of HLA type DR6 were more likely in patients with Rasmussen’s encephalitis and in first degree relatives than in controls, suggesting a possible genetic predisposition. More research will be needed to confirm these preliminary observations. In 2008, a mutation in SCN1A was described in association with a case of Rasmussen’s encephalitis.94
The advantage of exploring genetic avenues is that they are unidirectional and not complicated by the time from diagnosis or disease stage. One aspect that genetics might inform is the tantalising issue of why Rasmussen’s encephalitis almost always remains unilateral. Genetically determined malformations of cortical development can be unilateral and one could, for example, hypothesise a three-hit hypothesis starting with a genetic defect that affected cell migration in the fetal brain, combined with a genetically predisposed immune system that over-reacted to a rare virus infection. Further genetic studies in larger numbers of patients could be very helpful, although in such a rare disorder, international collaboration and very careful attention to the appropriate controls will be needed.
Neuroimaging has emerged as a useful biomarker of disease,28,47 but biochemical biomarkers are scarce or have only preliminarily been explored.63 Techniques such as label-free quantitative proteomic analysis of CSF and nano-high-performance liquid chromatography with electrospray-ionisation quadrupole time-of-flight mass spectrometry of brain tissue might establish something that is unique to Rasmussen’s encephalitis, and inform the development of more accessible biomarkers. The knowledge of pathobiology and treatment in other autoinflammatory grey matter diseases, which now include early-stage multiple sclerosis, might inform treatment of Rasmussen’s encephalitis. Rasmussen’s encephalitis is a rare disorder; collaborative international studies using both archival tissues and prospective collection with recruitment into clinical trials are greatly needed.
Until the causes of Rasmussen’s encephalitis are known, it is difficult to anticipate how treatments will improve. Various attempts using immunotherapy have been tried in the past decade. Some slow the progress of the disease, but none has successfully cured or even halted the progression of disease. The disease course seems to be changed, favouring more protracted and less severely hemiatrophic and hemiplegic courses, presumably as the result of immunotherapy. This change might have an effect on surgical decision making, but it is unclear whether this has substantially changed the overall outcome of the disease. In fact, there seems to be an increasingly common scenario in which immunotherapy has slowed the patient’s functional decline but not the frequent, handicapping seizures, which the patient might have endured for years. Such a situation creates a therapeutic dilemma; hemispherectomy is not favoured because of the inevitable postoperative functional deficits, but a real risk exists that treatments used to delay progression of the disease will delay definitive surgical treatment beyond the time when an optimum post-hemispherectomy outcome could be expected.
Use of immunotherapies in patients with acute encephalitis and the relevant viral or antibody biomarkers has changed clinical practice and patient outcomes. One would hope that combined descriptive clinical studies, genetic analyses, and early histopathological examination of Rasmussen’s encephalitis tissue specimens, looking for both viral and autoimmune pathology, will accelerate research efforts to identify the main causes of this disease. Development of appropriate in vitro or animal models will then make it possible to study both cause and effects, and might help to direct clinical trials.
Further systematic randomised clinical trials are needed to clarify the efficacy of new non-surgical treatment options that might be used to control seizures and preserve neurological function. In a rare disease, such studies will need to be multicentre to realistically achieve appropriate outcomes with standardised data collection. Furthermore, long-term outcome data are needed in patients to provide a comparative dataset. The RE Children’s Project has brought centres together to address these issues and facilitate a repository of tissues from patients with Rasmussen’s encephalitis so that patient material is available for research studies.
Panel 1: Disease stages, diagnostic criteria, and differential diagnoses of Rasmussen’s encephalitis2.
Three disease stages of Rasmussen’s encephalitis
Prodromal stage: Non-specific, low seizure frequency, and mild hemiplegia
Acute stage: Frequent seizures, often epilepsia partialis continua; progressive hemiparesis, hemianopia, cognitive deterioration, and aphasia (if dominant hemisphere affected)
Residual stage: Permanent and stable neurological deficits and continuing seizures
Diagnostic criteria
Part A (all three)
Clinical: Focal seizures (with or without epilepsia partialis continua) and unilateral cortical deficits
Electroencephalogram: Unihemispheric slowing with or without epileptiform activity and unilateral seizure onset
-
MRI: Unihemispheric focal cortical atrophy and at least one of the following:
Grey or white matter T2/FLAIR hyperintense signal
Hyperintense signal or atrophy of the ipsilateral caudate head
Or
Part B (two of three)
Clinical: Epilepsia partialis continua or progressive* unilateral cortical deficits
MRI: Progressive* unihemispheric focal cortical atrophy
Histopathology: T-cell-dominated encephalitis with activated microglial cells typically, but not necessarily, forming nodules and reactive astrogliosis; numerous parenchymal macrophages, B cells, or plasma cells or viral inclusion bodies exclude the diagnosis of Rasmussen’s encephalitis
Differential diagnoses
Unihemispheric epileptic syndromes
Cortical dysplasia
Hemimegalencephaly
Tuberous sclerosis
Sturge-Weber syndrome
Stroke
Hemiconvulsion–hemiplegia–epilepsy syndrome
Tumour
Epilepsia partialis continua due to metabolic disorders
-
Diabetes mellitus
Ketotic or non-ketotic hyperglycaemia
Type 1 diabetes and anti-GAD-65 antibodies
Renal or hepatic encephalopathy
Metabolic or degenerative progressive neurological diseases
MELAS and other mitochondriopathies
Alpers’ syndrome
Kufs’ disease
Inflammatory and infectious diseases
Cerebral vasculitis in systemic connective tissue disease (eg, lupus erythematosus)
Unihemispheric cerebral vasculitis mimicking Rasmussen’s encephalitis
Subacute sclerosing panencephalitis and other delayed subacute measles encephalitis with or without immunodeficiency
Paraneoplastic syndrome
Onconeural antibodies (anti-Hu)
Russian Spring Summer meningoencephalitis
Multiple sclerosis
Creutzfeldt-Jakob disease
HIV
Cat scratch disease
Other
Proconvulsive drugs (eg, metrizimide, penicillin, and azlocillin-cefotaxim)
Bone marrow transplant
Gliomatosis cerebri
*At least two sequential clinical examinations or MRI studies are needed to meet the respective criteria.
Panel 2: Key questions for future research in Rasmussen’s encephalitis.
What causes Rasmussen’s encephalitis?
The cause of Rasmussen’s encephalitis is unknown. On the basis of the earliest immunological response, the inflammation in the brain seems to be driven by an antigen, which could be foreign (an infectious agent) or autoimmune. Another possibility could be that Rasmussen’s encephalitis is the consequence of dysfunction of the immune response to the presentation of an otherwise minor antigen. That dysfunction could be genetic in origin. These possibilities can be explored in hypothesis-driven studies.
Why does Rasmussen’s encephalitis only affect one cerebral hemisphere?
The answer to this question would depend on the source of the presumed antigen that initiates Rasmussen’s encephalitis. If the antigen is a foreign infectious agent, this could explain why Rasmussen’s encephalitis is unihemispheric but without a side preference. If Rasmussen’s encephalitis is an autoimmune disease, attention might need to be focused on recently discovered genes and proteins related to individual cerebral hemisphere brain development expressed only on one side.
What approaches might be used to advance research and treatment?
Efforts directed by the professionals and families of the RE Children’s Project are addressing the key questions regarding identification of the cause of Rasmussen’s encephalitis. With up-to-date molecular techniques, studies are underway to elucidate whether antigens are associated with Rasmussen’s encephalitis and if these antigens are foreign or autoimmune. Studies will examine, with exome sequencing, whether signs exist of germline or somatic mutations in the brains of patients with Rasmussen’s encephalitis. If infectious antigens can be effectively ruled out, empirical treatments with many already FDA-approved anti-inflammatory drugs could be tried in patients with Rasmussen’s encephalitis. The response to particular drugs might provide clues to the cause of Rasmussen’s encephalitis. For any of these approaches to be successful, large numbers of patients with Rasmussen’s encephalitis need to be identified for studies, a challenge for a very rare brain disease. Here again, the RE Children’s Project could be key—a non-profit international organisation bringing together patients, their families, and key centres interested in Rasmussen’s encephalitis research and treatment development with social media.
Search strategy and selection criteria.
We searched PubMed for articles published in English between Jan 1, 2005, and Sept 30, 2013, with the search terms “Rasmussen encephalitis”, “Rasmussen syndrome”, and “Rasmussen disease”, and reviewed the references. We also referred to our personal databases of papers and information; therefore, data for this review also came from references contained within older relevant articles and from clinical data of individual cases or case series described in published abstracts. From the available resources, we selected sources of particular relevance to basic science understanding and to treatment in Rasmussen’s encephalitis.
Acknowledgments
The collaboration leading to this work was initiated at an international conference on Rasmussen’s encephalitis in Oct, 2010, sponsored by the RE Children’s Project (www.rechildrens.org). GWM was supported in part by NIH RO1NS38992 and the RE Children’s Project (Seth Wohlberg). CAK was supported in part by NIH R01CA125244-01A2 and R01CA154256, the Joan S Holmes Memorial Research Fund, and the RE Children’s Project. All authors have received travel and conference support from the RE Children’s Project. We would like to thank Deirdre Pinto, Associate Researcher, RE Children’s Project, for her very helpful comments on the manuscript.
SV has sat on Advisory Panels for Eisai and received honoraria for speaking engagements from Cyberonics (Zaventem, Belgium) and Eisai (Hatfield, UK) with remuneration paid to her department. CGB gave scientific advice to Eisai (Frankfurt, Germany) and UCB (Monheim, Germany), undertook industry-funded travel with support of Eisai (Frankfurt, Germany), UCB (Monheim, Germany), Desitin (Hamburg, Germany), and Grifols (Frankfurt, Germany), and obtained honoraria for speaking engagements from Eisai (Frankfurt, Germany), UCB (Monheim, Germany), Desitin (Hamburg, Germany), Diamed (Köln, Germany), Fresenius Medical Care (Bad Homburg, Germany). His employer (Krankenhaus Mara, Bielefeld, Germany) runs a lab for the detection of autoantibodies; external senders are charged for antibody diagnostics. He received research support from Diamed (Köln, Germany), Fresenius Medical Care (Bad Homburg, Germany), Astellas Pharma (München, Germany) and Octapharma (Langenfeld, Germany). CAK was partly supported by NIH RO1CA125244-01A2 and RO1CA154256, the Joan S Holmes Memorial Research Fund, and the RE Children’s Project. FEJ has investigator-initiated research grants from Eisai Inc (USA) and Lundbeck (Deerfield, USA).
AV and Oxford University hold patents and receive royalties and payments for antibody assays. AV is a consultant for Athena Diagnostics (Worcester, USA) and received a grant from Euroimmun AG (Wimbledon, UK). GWM was partly supported by NIH RO1NS38992 and the RE Children’s Project (Seth Wohlberg). JHC holds an endowed Chair through University College London (London, UK). She has sat on Advisory Panels for Eisai (Hatfield, UK) and Viropharma (Maidenhead, UK) for which remuneration has been paid to her department. She has received money to the Department as an educational grant from UCB (Slough, UK) and Eisai (Hatfield, UK) for a Clinical Training Fellowship in Epilepsy. She holds grants for research from Action Medical Research, Epilepsy Research UK, and the Great Ormond Street Hospital Children’s Charity, and she co-supervises a PhD student funded by Vitaflo (Liverpool, UK). She worked as Clinical Advisor to the update of the NICE guidelines on the diagnosis and management of epilepsy (2009–12) for which remuneration was made to her department.
Footnotes
Contributors
All authors contributed to the scientific literature search and the writing and revision of the paper. SV prepared the first draft. SV, CGB, JB, and GWM prepared the figures. All authors approved the final version for submission.
Conflicts of interest
CAP and JB have no conflicts of interest to declare.
Contributor Information
Sophia Varadkar, Epilepsy Unit, Great Ormond Street Hospital for Children NHS Foundation Trust and UCL Institute of Child Health, London, UK.
Prof Christian G Bien, Epilepsy Centre Bethel, Krankenhaus Mara, Bielefeld, Germany.
Prof Carol A Kruse, Department of Neurosurgery, Brain Research Institute, David Geffen School of Medicine, University of California, Los Angeles, CA, USA.
Prof Frances E Jensen, Department of Neurology, Perelman School of Medicine, University of Pennsylvania, PA, USA.
Prof Jan Bauer, Department of Neuroimmunology, Center for Brain Research, Medical University of Vienna, Vienna, Austria.
Carlos A Pardo, Department of Neurology and Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Prof Angela Vincent, Nuffield Department of Clinical Neurosciences, University of Oxford, UK.
Prof Gary W Mathern, Departments of Neurosurgery and Psychiatry and Biobehavioral Medicine, David Geffen School of Medicine, Mattel Children’s Hospital, University of California, Los Angeles, CA, USA.
Prof J Helen Cross, Neurosciences Unit, UCL Institute of Child Health, Great Ormond Street Hospital for Children NHS Foundation Trust, London, and Young Epilepsy, Lingfield, UK.
References
- 1.Rasmussen T, Olszewski J, Lloyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology. 1958;8:435–45. doi: 10.1212/wnl.8.6.435. [DOI] [PubMed] [Google Scholar]
- 2.Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain. 2005;128:454–71. doi: 10.1093/brain/awh415. [DOI] [PubMed] [Google Scholar]
- 3.Olson HE, Lechpammer M, Prabhu SP, et al. Clinical application and evaluation of the Bien diagnostic criteria for Rasmussen encephalitis. Epilepsia. 2013;54:1753–60. doi: 10.1111/epi.12334. [DOI] [PubMed] [Google Scholar]
- 4.Bien CG, Tiemeier H, Sassen R, et al. Rasmussen encephalitis: Incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia. 2013;54:543–50. doi: 10.1111/epi.12042. [DOI] [PubMed] [Google Scholar]
- 5.Lamb K, Scott WJ, Mensah A, et al. Prevalence and clinical outcome of Rasmussen encephalitis in children from the United Kingdom. Dev Med Child Neurol. 2013;55(suppl 1):14. [Google Scholar]
- 6.Bien CG, Widman G, Urbach H, et al. The natural history of Rasmussen’s encephalitis. Brain. 2002;125:1751–59. doi: 10.1093/brain/awf176. [DOI] [PubMed] [Google Scholar]
- 7.Oguni H, Andermann F, Rasmussen TB. The natural history of the syndrome of chronic encephalitis and epilepsy: a study of the MNI series of forty-eight cases. In: Andermann F, editor. Chronic encephalitis and epilepsy–Rasmussen’s syndrome. Boston: Butterworth-Heinemann; 1991. pp. 7–35. [Google Scholar]
- 8.Granata T, Gobbi G, Spreafico R, et al. Rasmussen’s encephalitis: early characteristics allow diagnosis. Neurology. 2003;60:422–25. doi: 10.1212/wnl.60.3.422. [DOI] [PubMed] [Google Scholar]
- 9.Thomas JE, Reagan TJ, Klass DW. Epilepsia partialis continua. A review of 32 cases. Arch Neurol. 1977;34:266–75. doi: 10.1001/archneur.1977.00500170020003. [DOI] [PubMed] [Google Scholar]
- 10.Obeso JA, Rothwell JC, Marsden CD. The spectrum of cortical myoclonus. From focal reflex jerks to spontaneous motor epilepsy. Brain. 1985;108:193–24. doi: 10.1093/brain/108.1.193. [DOI] [PubMed] [Google Scholar]
- 11.Longaretti F, Dunkley C, Varadkar S, et al. Evolution of the EEG in children with Rasmussen’s syndrome. Epilepsia. 2012;53:1539–45. doi: 10.1111/j.1528-1167.2012.03565.x. [DOI] [PubMed] [Google Scholar]
- 12.Bien CG, Gleissner U, Sassen R, et al. An open study of tacrolimus therapy in Rasmussen encephalitis. Neurology. 2004;62:2106–09. doi: 10.1212/01.wnl.0000128044.94294.87. [DOI] [PubMed] [Google Scholar]
- 13.Thilo B, Stingele R, Knudsen K, et al. A case of Rasmussen encephalitis treated with rituximab. Nat Rev Neurol. 2009;5:458–62. doi: 10.1038/nrneurol.2009.98. [DOI] [PubMed] [Google Scholar]
- 14.Gray F, Serdaru M, Baron H, et al. Chronic localised encephalitis (Rasmussen’s) in an adult with epilepsia partialis continua. J Neurol Neurosurg Psychiatry. 1987;50:747–51. doi: 10.1136/jnnp.50.6.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villani F, Pincherle A, Antozzi C, et al. Adult-onset Rasmussen’s encephalitis: anatomical-electrographic-clinical features of 7 Italian cases. Epilepsia. 2006;47(suppl 5):41–46. doi: 10.1111/j.1528-1167.2006.00876.x. [DOI] [PubMed] [Google Scholar]
- 16.Hart YM, Andermann F, Fish DR, et al. Chronic encephalitis and epilepsy in adults and adolescents: a variant of Rasmussen’s syndrome? Neurology. 1997;48:418–24. doi: 10.1212/wnl.48.2.418. [DOI] [PubMed] [Google Scholar]
- 17.Hennessy MJ, Koutroumanidis M, Dean AF, et al. Chronic encephalitis and temporal lobe epilepsy: a variant of Rasmussen’s syndrome? Neurology. 2001;56:678–81. doi: 10.1212/wnl.56.5.678. [DOI] [PubMed] [Google Scholar]
- 18.Bhatjiwale MG, Polkey C, Cox TC, Dean A, Deasy N. Rasmussen’s encephalitis: neuroimaging findings in 21 patients with a closer look at the basal ganglia. Pediatr Neurosurg. 1998;29:142–48. doi: 10.1159/000028709. [DOI] [PubMed] [Google Scholar]
- 19.Frucht S. Dystonia, athetosis, and epilepsia partialis continua in a patient with late-onset Rasmussen’s encephalitis. Mov Disord. 2002;17:609–12. doi: 10.1002/mds.10131. [DOI] [PubMed] [Google Scholar]
- 20.Chinchilla D, Dulac O, Robain O, et al. Reappraisal of Rasmussen’s syndrome with special emphasis on treatment with high doses of steroids. J Neurol Neurosurg Psychiatry. 1994;57:1325–33. doi: 10.1136/jnnp.57.11.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tobias SM, Robitaille Y, Hickey WF, et al. Bilateral Rasmussen encephalitis: postmortem documentation in a five-year-old. Epilepsia. 2003;44:127–30. doi: 10.1046/j.1528-1157.2003.36602.x. [DOI] [PubMed] [Google Scholar]
- 22.Bien CG, Elger CE, Leitner Y, et al. Slowly progressive hemiparesis in childhood as a consequence of Rasmussen encephalitis without or with delayed-onset seizures. Eur J Neurol. 2007;14:387–90. doi: 10.1111/j.1468-1331.2007.01684.x. [DOI] [PubMed] [Google Scholar]
- 23.Korn-Lubetzki I, Bien CG, Bauer J, et al. Rasmussen encephalitis with active inflammation and delayed seizures onset. Neurology. 2004;62:984–86. doi: 10.1212/01.wnl.0000115393.67403.53. [DOI] [PubMed] [Google Scholar]
- 24.Antel JPM, Rasmussen TM. Rasmussen’s encephalitis and the new hat. Neurology. 1996;46:9–11. doi: 10.1212/wnl.46.1.9. [DOI] [PubMed] [Google Scholar]
- 25.Bien CG, Urbach H, Deckert M, et al. Diagnosis and staging of Rasmussen’s encephalitis by serial MRI and histopathology. Neurology. 2002;58:250–57. doi: 10.1212/wnl.58.2.250. [DOI] [PubMed] [Google Scholar]
- 26.Chiapparini L, Granata T, Farina L, et al. Diagnostic imaging in 13 cases of Rasmussen’s encephalitis: can early MRI suggest the diagnosis? Neuroradiology. 2003;45:171–83. doi: 10.1007/s00234-002-0923-7. [DOI] [PubMed] [Google Scholar]
- 27.Yamazaki E, Takahashi Y, Akasaka N, Fujiwara T, Inoue Y. Temporal changes in brain MRI findings in Rasmussen syndrome. Epileptic Disord. 2011;13:229–39. doi: 10.1684/epd.2011.0464. [DOI] [PubMed] [Google Scholar]
- 28.Wagner J, Schoene-Bake JC, Bien CG, et al. Automated 3D MRI volumetry reveals regional atrophy differences in Rasmussen encephalitis. Epilepsia. 2012;53:613–21. doi: 10.1111/j.1528-1167.2011.03396.x. [DOI] [PubMed] [Google Scholar]
- 29.Larionov S, Konig R, Urbach H, et al. MRI brain volumetry in Rasmussen encephalitis: the fate of affected and “unaffected” hemispheres. Neurology. 2005;64:885–87. doi: 10.1212/01.WNL.0000152895.23010.52. [DOI] [PubMed] [Google Scholar]
- 30.Fiorella DJ, Provenzale JM, Edward CR, Crain BJ, Al Sugair A. 18F-fluorodeoxyglucose positron emission tomography and MR imaging findings in Rasmussen encephalitis. Am J Neuroradiol. 2001;22:1291–99. [PMC free article] [PubMed] [Google Scholar]
- 31.So N, Gloor P. Electroencephalographic and electrocorticographic findings in chronic encephalitis of the Rasmussen type. In: Andermann F, editor. Chronic encephalitis and epilepsy Rasmussen’s syndrome. Boston: Butterworth-Heinemann; 1991. pp. 37–45. [Google Scholar]
- 32.Pardo CA, Vining EP, Guo L, et al. The pathology of Rasmussen syndrome: stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia. 2004;45:516–26. doi: 10.1111/j.0013-9580.2004.33103.x. [DOI] [PubMed] [Google Scholar]
- 33.Robitaille Y. Neuropathologic aspects of chronic encephalitis. In: Andermann F, editor. Chronic encephalitis and epilepsy Rasmussen’s syndrome. Boston: Butterworth-Heinemann; 1991. pp. 79–110. [Google Scholar]
- 34.Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- 35.Hart YM, Andermann F, Robitaille Y, et al. Double pathology in Rasmussen’s syndrome: a window on the etiology? Neurology. 1998;50:731–35. doi: 10.1212/wnl.50.3.731. [DOI] [PubMed] [Google Scholar]
- 36.Takei H, Wilfong A, Malphrus A, et al. Dual pathology in Rasmussen’s encephalitis: a study of seven cases and review of the literature. Neuropathology. 2010;30:381–91. doi: 10.1111/j.1440-1789.2009.01079.x. [DOI] [PubMed] [Google Scholar]
- 37.Iyer A, Zurolo E, Spliet WGM, et al. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia. 2010;51:1763–73. doi: 10.1111/j.1528-1167.2010.02547.x. [DOI] [PubMed] [Google Scholar]
- 38.Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10:759–72. doi: 10.1016/S1474-4422(11)70096-5. [DOI] [PubMed] [Google Scholar]
- 39.Lancaster E, Dalmau J. Neuronal autoantigens - pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8:380–90. doi: 10.1038/nrneurol.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science. 1994;265:648–51. doi: 10.1126/science.8036512. [DOI] [PubMed] [Google Scholar]
- 41.Wiendl HM, Bien CGM, Bernasconi PP, et al. GluR3 antibodies: prevalence in focal epilepsy but no specificity for Rasmussen’s encephalitis. Neurology. 2001;57:1511–14. doi: 10.1212/wnl.57.8.1511. [DOI] [PubMed] [Google Scholar]
- 42.Watson R, Jiang Y, Bermudez I, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology. 2004;63:43–50. doi: 10.1212/01.wnl.0000132651.66689.0f. [DOI] [PubMed] [Google Scholar]
- 43.Mantegazza R, Bernasconi P, Baggi F, et al. Antibodies against GluR3 peptides are not specific for Rasmussen’s encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J Neuroimmunol. 2002;131:179–85. doi: 10.1016/s0165-5728(02)00261-8. [DOI] [PubMed] [Google Scholar]
- 44.Granata T, Fusco L, Gobbi G, et al. Experience with immunomodulatory treatments in Rasmussen’s encephalitis. Neurology. 2003;61:1807–10. doi: 10.1212/01.wnl.0000099074.04539.e0. [DOI] [PubMed] [Google Scholar]
- 45.Watson R, Jepson JE, Bermudez I, et al. Alpha7-acetylcholine receptor antibodies in two patients with Rasmussen encephalitis. Neurology. 2005;65:1802–04. doi: 10.1212/01.wnl.0000191566.86977.04. [DOI] [PubMed] [Google Scholar]
- 46.Alvarez-Baron E, Bien CG, Schramm J, et al. Autoantibodies to Munc18, cerebral plasma cells and B-lymphocytes in Rasmussen encephalitis. Epilepsy Res. 2008;80:93–97. doi: 10.1016/j.eplepsyres.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 47.Yang R, Puranam RS, Butler LS, et al. Autoimmunity to munc-18 in Rasmussen’s encephalitis. Neuron. 2000;28:375–83. doi: 10.1016/s0896-6273(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 48.Greiner H, Leach JL, Lee KH, Krueger DA. Anti-NMDA receptor encephalitis presenting with imaging findings and clinical features mimicking Rasmussen syndrome. Seizure. 2011;20:266–70. doi: 10.1016/j.seizure.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 49.Bien CG, Bauer J, Deckwerth TL, et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol. 2002;51:311–18. doi: 10.1002/ana.10100. [DOI] [PubMed] [Google Scholar]
- 50.Schwab N, Bien CG, Waschbisch A, et al. CD8+ T cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain. 2009;132:1236–46. doi: 10.1093/brain/awp003. [DOI] [PubMed] [Google Scholar]
- 51.Bauer J, Elger CE, Hans VH, et al. Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann Neurol. 2007;62:67–80. doi: 10.1002/ana.21148. [DOI] [PubMed] [Google Scholar]
- 52.Friedman H, Ch’ien L, Parham D. Virus in brain of child with hemiplegia, hemiconvulsions, and epilepsy. Lancet. 1977;310:666. doi: 10.1016/s0140-6736(77)92540-5. [DOI] [PubMed] [Google Scholar]
- 53.Walter GF, Renella RR. Epstein-Barr virus in brain and Rasmussen’s encephalitis. Lancet. 1989;1:279–80. doi: 10.1016/s0140-6736(89)91292-0. [DOI] [PubMed] [Google Scholar]
- 54.Power C, Poland SD, Blume WT, Girvin JP, Rice GP. Cytomegalovirus and Rasmussen’s encephalitis. Lancet. 1990;336:1282–84. doi: 10.1016/0140-6736(90)92965-k. [DOI] [PubMed] [Google Scholar]
- 55.Jay V, Becker LE, Otsubo H, et al. Chronic encephalitis and epilepsy (Rasmussen’s encephalitis): detection of cytomegalovirus and herpes simplex virus 1 by the polymerase chain reaction and in situ hybridization. Neurology. 1995;45:108–17. doi: 10.1212/wnl.45.1.108. [DOI] [PubMed] [Google Scholar]
- 56.Wirenfeldt M, Clare R, Tung S, et al. Increased activation of Iba1+ microglia in pediatric epilepsy patients with Rasmussen’s encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiol Dis. 2009;34:432–40. doi: 10.1016/j.nbd.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–43. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 58.Balosso S, Maroso M, Sanchez-Alavez M, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain. 2008;131:3256–65. doi: 10.1093/brain/awn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 61.Aronica E, Ravizza T, Zurolo E, Vezzani A. Astrocyte immune responses in epilepsy. Glia. 2012;60:1258–68. doi: 10.1002/glia.22312. [DOI] [PubMed] [Google Scholar]
- 62.Ivens S, Kaufer D, Flores L, et al. TGF-b receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–47. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- 63.Takahashi Y, Mine J, Kubota Y, Yamazaki E, Fujiwara T. A substantial number of Rasmussen syndrome patients have increased IgG, CD4+ T cells, TNFa, and Granzyme B in CSF. Epilepsia. 2009;50:1419–31. doi: 10.1111/j.1528-1167.2008.01977.x. [DOI] [PubMed] [Google Scholar]
- 64.Owens GC, Huynh M, Chang JW, et al. Differential expression of Interferon-ã and chemokines genes distinguishes Rasmussen encephalitis from cortical dysplasia and provides evidence for an early Th1 immune response. J Neuroinflammation. 2013;10:56. doi: 10.1186/1742-2094-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bien CG, Schramm J. Treatment of Rasmussen encephalitis half a century after its initial description: promising prospects and a dilemma. Epilepsy Res. 2009;86:101–12. doi: 10.1016/j.eplepsyres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 66.Dubeau F, Sherwin AL. Pharmacologic principles in the management of chronic focal encephalitis. In: Andermann F, editor. Chronic encephalitis and epilepsy Rasmussen’s syndrome. Boston: Butterworth-Heinemann; 1991. pp. 179–92. [Google Scholar]
- 67.Browner N, Azher SN, Jankovic J. Botulinum toxin treatment of facial myoclonus in suspected Rasmussen encephalitis. Mov Disord. 2006;21:1500–02. doi: 10.1002/mds.20991. [DOI] [PubMed] [Google Scholar]
- 68.Lozsadi DA, Hart IK, Moore AP. Botulinum toxin A improves involuntary limb movements in Rasmussen syndrome. Neurology. 2004;62:1233–34. doi: 10.1212/01.wnl.0000118283.51400.7a. [DOI] [PubMed] [Google Scholar]
- 69.Laxer KD, Wilfong A, Morris GLI, Andermann F. Pilot study of Rituximab to treat chronic focal encephalitis. Epilepsia. 2008;49:121. [Google Scholar]
- 70.Grujic J, Bien CG, Pollo C, Rossetti AO. Vagus nerve stimulator treatment in adult-onset Rasmussen’s encephalitis. Epilepsy Behav. 2011;20:123–25. doi: 10.1016/j.yebeh.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 71.San-Juan D, Calcaneo JD, Gonzalez-Aragon MF, et al. Transcranial direct current stimulation in adolescent and adult Rasmussen’s encephalitis. Epilepsy Behav. 2011;20:126–31. doi: 10.1016/j.yebeh.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 72.Bahi-Buisson N, Nabbout R, Plouin P, et al. Recent advances in pathogenic concepts and therapeutic strategies in Rasmussen’s encephalitis. Rev Neurol (Paris) 2005;161:395–405. doi: 10.1016/s0035-3787(05)85069-6. [DOI] [PubMed] [Google Scholar]
- 73.Hart Y, Andermann F, Fish D, et al. The medical treatment of chronic encephalitis and epilepsy. In: Wolf P, editor. Epileptic seizures and syndromes. London: John Libbey & Company; 1994. pp. 399–404. [Google Scholar]
- 74.Hart YM, Cortez M, Andermann F, et al. Medical treatment of Rasmussen’s syndrome (chronic encephalitis and epilepsy): effect of high-dose steroids or immunoglobulins in 19 patients. Neurology. 1994;44:1030–36. doi: 10.1212/wnl.44.6.1030. [DOI] [PubMed] [Google Scholar]
- 75.Villani F, Spreafico R, Farina L, et al. Positive response to immunomodulatory therapy in an adult patient with Rasmussen’s encephalitis. Neurology. 2001;56:248–50. doi: 10.1212/wnl.56.2.248. [DOI] [PubMed] [Google Scholar]
- 76.Leach JP, Chadwick DW, Miles JB, Hart IK. Improvement in adult-onset Rasmussen’s encephalitis with long-term immunomodulatory therapy. Neurology. 1999;52:738–42. doi: 10.1212/wnl.52.4.738. [DOI] [PubMed] [Google Scholar]
- 77.Antozzi C, Granata T, Aurisano N, et al. Long-term selective IgG immuno-adsorption improves Rasmussen’s encephalitis. Neurology. 1998;51:302–05. doi: 10.1212/wnl.51.1.302. [DOI] [PubMed] [Google Scholar]
- 78.Andrews PIF, Dichter MAM, Berkovic SFF, Newton MRF, McNamara JOM. Plasmapheresis in Rasmussen’s encephalitis. Neurology. 1996;46:242–46. doi: 10.1212/wnl.46.1.242. [DOI] [PubMed] [Google Scholar]
- 79.Varadkar S, Chong WK, Robinson R, et al. Azathioprine therapy in Rasmussen Syndrome. Epilepsy Currents. 2012;12(suppl 1):417. [Google Scholar]
- 80.Takahashi Y, Yamazaki E, Mine J, et al. Immunomodulatory therapy versus surgery for Rasmussen syndrome in early childhood. Brain Devel. 2013;35:778–85. doi: 10.1016/j.braindev.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 81.Daniel RT, Villemure JG. Experience with immunomodulatory treatments in Rasmussen’s encephalitis. Neurology. 2004;63:1761–62. doi: 10.1212/wnl.63.9.1761-a. [DOI] [PubMed] [Google Scholar]
- 82.Barten LJ, Allington DR, Procacci KA, Rivey MP. New approaches in the management of multiple sclerosis. Drug Des Devel Ther. 2010;4:343–66. doi: 10.2147/DDDT.S9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dong YF, Kataoka K, Tokutomi Y, et al. Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J. 2011;25:2911–20. doi: 10.1096/fj.11-182873. [DOI] [PubMed] [Google Scholar]
- 84.Bittner S, Simon OJ, Göbel K, et al. A case of Rasmussen’s encephalitis treated with natalizumab. Neurology. 2013;81:395–97. doi: 10.1212/WNL.0b013e31829c5ceb. [DOI] [PubMed] [Google Scholar]
- 85.Pulsifer MB, Brandt J, Salorio CF, et al. The cognitive outcome of hemispherectomy in 71 children. Epilepsia. 2004;45:243–54. doi: 10.1111/j.0013-9580.2004.15303.x. [DOI] [PubMed] [Google Scholar]
- 86.Jonas R, Nguyen S, Hu B, et al. Cerebral hemispherectomy: hospital course, seizure, developmental, language, and motor outcomes. Neurology. 2004;62:1712–21. doi: 10.1212/01.wnl.0000127109.14569.c3. [DOI] [PubMed] [Google Scholar]
- 87.Thomas SG, Chacko AG, Thomas MM, et al. Outcomes of disconnective surgery in intractable pediatric hemispheric and subhemispheric epilepsy. Int J Pediatr. 2012;2012:527891. doi: 10.1155/2012/527891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Althausen A, Gleissner U, Hoppe C, et al. Long-term outcome of hemispheric surgery at different ages in 61 epilepsy patients. J Neurol Neurosurg Psychiatry. 2013;84:529–36. doi: 10.1136/jnnp-2012-303811. [DOI] [PubMed] [Google Scholar]
- 89.Vora N, Cross JH, Robinson R, et al. Epilepsy Surgery in Rasmussen Syndrome. Epilepsy Currents. 2012;12:296. [Google Scholar]
- 90.Vining E, Freeman J, Pillas D, et al. Why Would You Remove Half a Brain? The Outcome of 58 Children After Hemispherectomy—The Johns Hopkins Experience: 1968 to 1996. Pediatrics. 1997;100:163–71. doi: 10.1542/peds.100.2.163. [DOI] [PubMed] [Google Scholar]
- 91.Hertz-Pannier L, Chiron C, Jambaque I, et al. Late plasticity for language in a child’s non-dominant hemisphere: a pre- and post-surgery fMRI study. Brain. 2002;125:361–72. doi: 10.1093/brain/awf020. [DOI] [PubMed] [Google Scholar]
- 92.Boatman D, Freeman J, Vining E, et al. Language recovery after left hemispherectomy in children with late-onset seizures. Ann Neurol. 1999;46:579–86. doi: 10.1002/1531-8249(199910)46:4<579::aid-ana5>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 93.Andermann E, Oguni H, Guttmann RD, et al. Genetic aspects of chronic encephalitis. In: Andermann F, editor. Chronic encephalitis and epilepsy: Rasmussen’s syndrome. Boston: Butterworth-Heinemann; 1991. pp. 167–75. [Google Scholar]
- 94.Ohmori I, Ouchida M, Kobayashi K, et al. Rasmussen encephalitis associated with SCN 1 A mutation. Epilepsia. 2008;49:521–26. doi: 10.1111/j.1528-1167.2007.01411.x. [DOI] [PubMed] [Google Scholar]