

Abstract
Innate lymphoid cells (ILCs) regulate stromal, epithelial and immune cells, but their impact on B cells remains unclear. We identified RORγt+ ILCs nearby the marginal zone (MZ), a splenic compartment containing innate-like B cells that respond to circulating T cell-independent (TI) antigens. Spenic ILCs established a bidirectional crosstalk with MAdCAM-1+ marginal reticular cells by providing tumor necrosis factor (TNF) and lymphotoxin, and activated MZ B cells via BAFF, CD40 ligand and the Notch ligand, Delta-like 1. Splenic ILCs further helped MZ B cells and their plasma cell progeny by co-opting neutrophils through the release of GM-CSF. Consequently, ILC depletion impaired both pre- and post-immune TI antibody responses. Thus, ILCs integrate stromal and myeloid signals to orchestrate innate-like antibody production at the interface between the immune and circulatory systems.
INTRODUCTION
The spleen is a highly perfused organ specialized in host defense against blood-borne pathogens. Interposed between the follicles of the splenic white pulp and the circulation, the marginal zone (MZ) contains B cells enmeshed with macrophages and dendritic cells (DCs) in a stromal reticular cell network1-3. All of these cells provide an efficient immunosurveillance of the circulatory system by readily interacting with circulating antigens from commensal or pathogenic microbes owing to the slow flow rate of the blood passing through the MZ4. Following antigen capture, macrophages, DCs and possibly neutrophils of the innate immune system expose antigen to MZ B cells, a unique subset of antibody-producing lymphocytes that develop from transitional B cells in response to NOTCH2 signals5.
Lymphoid sites positioned between the host and the environment contain innate-like B and T cells that belong to the adaptive immune system, but share several properties with effector cells of the innate immune system. Mucosal and serosal membranes include innate-like B-1 cells that generate a first line of protection through early production of low-affinity immunoglobulin M (IgM) to bacteria6. When microbes breach the mucosal barrier and enter the general circulation, innate-like MZ B cells provide a second line of protection via low-affinity IgM and IgG that bridge the temporal gap required for the slower production of high-affinity IgG by follicular (FO) B cells4.
Similar to B-1 cells, MZ B cells express clonally distributed and somatically recombined but rather unspecific B cell receptor (BCR) molecules encoded by poorly diversified immunoglobulin (Ig) genes4, 6. MZ B cells also express non-clonally distributed and germline-encoded Toll-like receptors (TLRs)7, a subfamily of nonspecific microbial sensors generally known as pattern recognition receptors. Typically expressed by effector cells of the innate immune system, TLRs activate MZ B cells after recognizing conserved microbial molecular signatures in cooperation with BCRs8. The activation of MZ B cells is further enhanced by B cell-stimulating cytokines released by DCs, macrophages and neutrophils9, 10.
Besides innate-like lymphocytes, mucosal surfaces include innate lymphoid cells (ILCs) that express neither somatically recombined antigen receptors nor conventional surface lineage molecules11. These ILCs require the transcriptional repressor inhibitor of DNA 2 (Id2) and the cytokine interleukin-7 (IL-7) for their development and generate cytokine secretion patterns that mirror those of T helper (TH) cells of the adaptive immune system12, 13. Similar to pro-inflammatory TH1 cells, group 1 ILCs (ILC1) release interferon-γ (IFN-γ) and require the transcription factor T-bet for their development as do natural killer (NK) cells of the innate immune system14. ILC2, which include natural helper cells and nuocytes, secrete IL-5 and IL-13 and require the transcription factor GATA-3, thus resembling pro-inflammatory TH2 cells15-17. Finally, ILC3 require the transcription factors retinoic acid receptor-related orphan receptor-γt (RORγt) and aryl hydrocarbon receptor (AhR) and include mucosal NK-22 cells, which secrete IL-22 and thus mimic non-inflammatory TH22 cells18-21, as well as fetal and mucosal lymphoid tissue inducer (LTi) cells, which produce IL-22 and IL-17 and thus resemble pro-inflammatory TH17 cells22-24. While NK-22 cells express natural cytotoxicity receptors (NCRs) usually associated with NK cells and mediate mucosal homeostasis by targeting epithelial cells via IL-22 (refs. 25-27), LTi cells lack NCRs and promote fetal lymphoid organogenesis and post-natal mucosal immunity by targeting stromal cells via lymphotoxin (LT) and tumor necrosis factor (TNF)28-30.
Mucosal NK-22 cells, also defined as NCR+ ILC3 to distinguish them from inflammatory NCR– ILC3 characterized by constitutive IL-17, IL-22 and activation-induced IFN-γ production31, 32, express B cell-activating factor of the TNF family (BAFF)20, a cytokine used by DCs, macrophages and neutrophils to help MZ B cells and plasma cells in a T cell-independent (TI) manner1, 9, 10. BAFF and its homologue a proliferation-inducing ligand (APRIL) are related to CD40 ligand (CD40L), a TNF family member used by T follicular helper (TFH) cells to activate FO B cells33. Given their involvement in mucosal TI antibody production29, 34, ILCs could regulate humoral immunity also in the MZ, a lymphoid area that is continually exposed to antigen as are mucosal membranes.
Here we identified ILCs with mucosa-like properties in the MZ and perifollicular zone of the spleen. These ILCs required survival signals from marginal reticular cells (MRCs), a MZ subset of stromal cells that responded to TNF and LT from ILCs. In addition to stimulating MZ B cells and plasma cells via BAFF, APRIL, CD40L and the NOTCH2 ligand Delta-like 1 (DLL1), splenic ILCs co-opted MZ B cell-helper neutrophils via granulocyte monocyte-colony stimulating factor (GM-CSF). Consequently, ILC depletion hampered MZ B cell production of antibodies to TI antigens.
RESULTS
Splenic ILCs have type-3 mucosal features
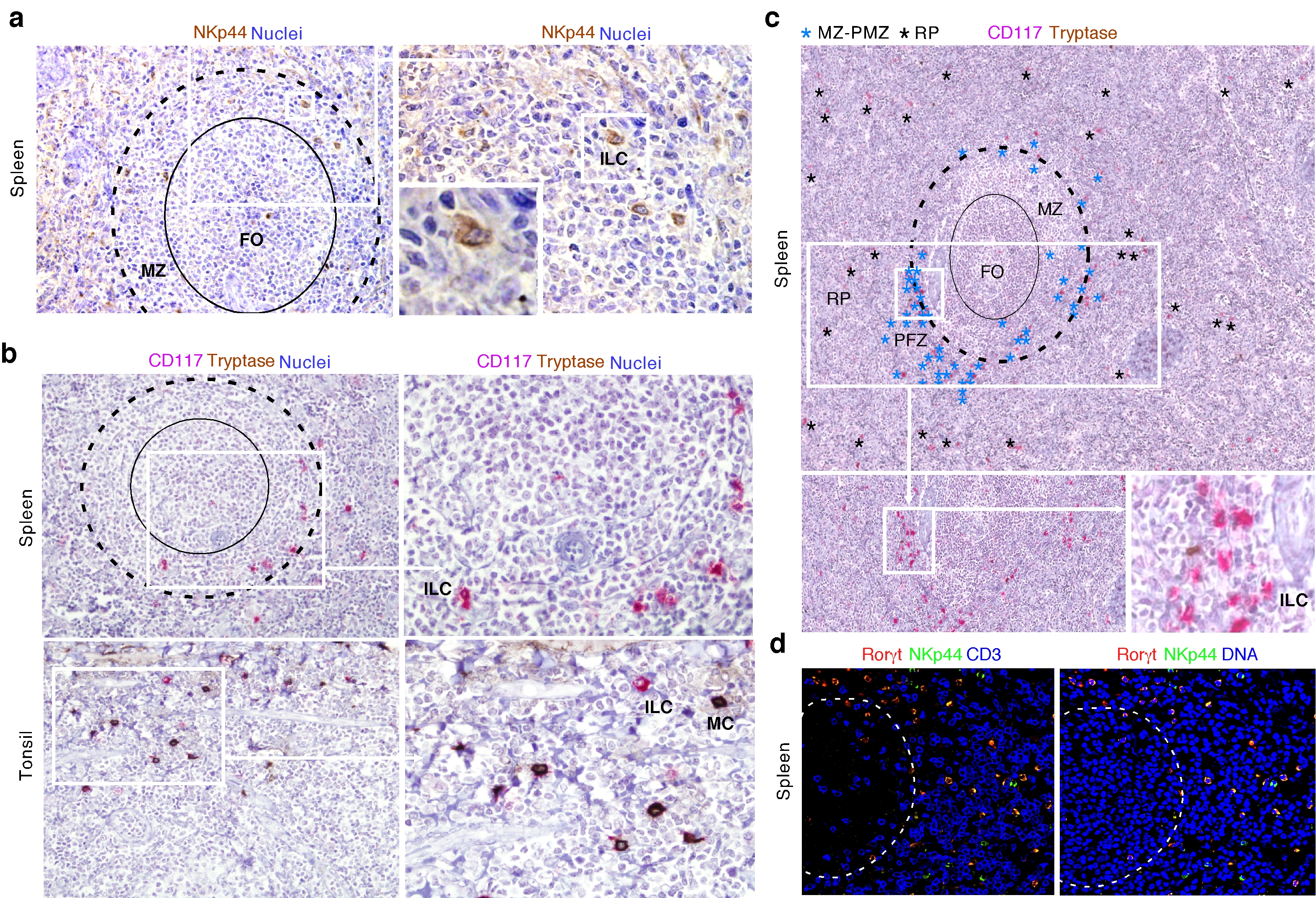
The MZ is continually exposed to blood-borne antigens and may thus require homeostatic signals from ILCs as mucosal membranes do4. Flow cytometry showed that histologically normal adult human spleens contained cells that lacked common lymphoid and myeloid lineage (Lin)-associated molecules, but expressed the IL-7 receptor CD127 and the stem cell growth factor receptor CD117 (Fig. 1a and Supplementary Fig. 1a). These ILCs resembled mucosal NCR+ ILC3s19, 20, because they expressed the NK cell molecules CD56, CD96, CD161, NKp44 and NKp46, the CCL20 chemokine receptor CCR6, the activation molecules CD25 and CD69, and NOTCH2 (Fig. 1a and Supplementary Figs. 1b). Splenic ILCs expressed neither the CD8 co-receptor, which is associated with cytotoxic T cells, nor the integrin CD103, which is expressed by some mucosal lymphocytes, nor the CD4 co-receptor and the prostaglandin D2 receptor CRTH2 (Fig. 1a and Supplementary Fig. 1b), which are expressed by mouse LTi cells and human ILC2, respectively16, 22. Albeit expressing as much CD161 as NK cells, splenic ILCs showed more NKp44, CCR6 and CD96 but less NKp46 than NK cells did (Supplementary Fig. 1c).
Figure 1. Human splenic ILCs express a mucosa-like group-3 phenotype and occupy MZ and perifollicular areas.

(a) Flow cytometry of CD4, CD25, CD56, CD69, CD96, CD161, NKp44, NKp46 and CCR6 on splenic Lin–CD117+CD127+ ILCs (red gate). Gray shading, negative control. (b) qRT-PCR of RORC (RORγt), AHR (AhR), IL22 (IL-22), TNF (TNF), LTA (LT-α), LTB (LT-β), PRF1 (Perforin-1) and IFNG (IFN-γ) mRNAs from splenic or tonsillar ILCs and NK cells and from splenic macrophages (Mϕ), B cells and T cells. Results are normalized to ACTB (β-actin) mRNA and presented as relative expression (RE) compared with that of fresh splenic NK cells. Error bars, s.e.m.; *P <0.05 (two-tailed unpaired Student's t test). (c) Flow cytometry of intracellular RORγt and T-bet in splenic ILCs (red lines) and NK cells (blue lines). (d) Flow cytometry of Lin–CD117+CD127+ ILCs in splenic and tonsillar lymphocytes. *P < 0.05 (Mann-Whitney U-test). (e) Flow cytometric analysis of frequency of viable Annexin-V–propidium iodide (PI)– ILCs after culture of splenic and tonsillar ILCs with medium alone (Ctrl), IL-1β, IL-7 or IL-23 for 72 h. (f) ELISA of IL-22 from splenic and tonsillar ILCs cultured as in (e). (g) IFA of spleen stained for MR (purple), RORγt (green) and DNA-binding 4’-6-diamidine-2’-phenylindole (DAPI; blue). RP, red pulp. Original magnification, ×10. FO: center of the follicle (h) Immunohistochemical quantification of CD117+Tryptase– ILCs from MZ-PFZ and red pulp (RP) areas in nine microscopic ×20 fields from two spleens. *P< 0.05 (Wilcoxon Matched-pairs signed Rank test). Data summarize three measurements from three pooled experiments with 1 donor in each (spleen and tonsil) (b), display values from thirty seven spleens and twelve tonsils (d), or show one of four experiments with similar results (a,c,e,f,g).
Gene expression studies performed through quantitative reverse transcription-polymerase chain reaction (qRT-PCR) showed that splenic ILCs expressed the transcription factors RORγt, AhR and Id2, the cytokines IL-22, IL-26, LT-α, LT-β and TNF and the IL-23 receptor, whereas splenic NK cells, T cells and macrophages did not, except B cells that expressed abundant LT-β (Fig. 1b and Supplementary Fig. 1d). Compared to mucosal ILCs from tonsils, splenic ILCs expressed comparable RORγt and AhR, but less IL-22 and more LT-α, LT-β and TNF (Fig. 1b). These latter cytokines are also hallmarks of LTi cells together with IL-17 (refs. 23, 30). However, splenic ILCs lacked IL-17, which was instead detected in T cells (Supplementary Fig. 1d). Of note, splenic ILCs included CD56+ and CD56– subsets with an overlapping ILC3 gene expression profile that did not include IFN-γ and Perforin-1, which were instead abundant in NK cells (Fig. 1b and Supplementary Fig. 1e,f). Flow cytometry showed that splenic RORγt+ ILCs lacked T-bet, a transcription factor that was instead expressed by RORγt– NK cells (Fig. 1c).
Flow cytometry and enzyme-linked immunosorbent assay (ELISA) demonstrated that splenic ILCs were more abundant than mucosal ILCs (Fig. 1d) and displayed NCR+ ILC3 properties19, 20. Indeed, splenic ILCs received anti-apoptotic signals from the stromal cytokine IL-7 and the innate cytokines IL-1β and IL-23 and secreted IL-22 in response to IL-1β and IL-23 as mucosal ILCs did (Fig. 1e,f). Finally, tissue immunofluorescence analysis (IFA) and immunohistochemistry (IHC) showed that RORγt+NKp44+CD117+tryptase– ILCs occupied MZ and perifollicular zone (PFZ) areas and were distinct from CD117+tryptase+ mast cells and CD3+RORγt–NKp44– T cells (Fig. 1g,h and Supplementary Fig. 2a-d). Some ILCs were also found in the periarteriolar lymphoid sheath (not shown). Thus, the human spleen contains mucosa-like ILCs different from NK cells and positioned nearby MZ B cells.
Splenic ILCs receive survival signals from MRCs
Given that LTi cells interact with stromal cells via LT and TNF22, 29, 30, we sought to identify stromal cells in the human MZ by tissue IFA, IHC and flow cytometry. Fibroblast-like cells expressing mucosal addressing cell adhesion molecule-1 (MAdCAM-1) were detected in the MZ nearby IgD+ B cells and some RORγt+ ILCs (Fig. 2a and Supplementary Fig. 3a). These MAdCAM-1+ cells did not express the coagulation protein von Willebrand factor (vWF), which characterizes endothelial cells, whereas MAdCAM-1– sinusoid-lining cells (SLCs) from the red pulp did (Fig. 2a). MAdCAM-1+ cells also lacked the endothelial molecules platelet endothelial cell adhesion molecule-1 (PECAM-1) and CD34 as well as the leukocyte molecule CD45, but expressed the stromal molecules Thy-1 (or CD90), CD141 (thrombomodulin), intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and α-smooth muscle actin (Fig. 2b and Supplementary Fig. 3b,c), and were thus considered equivalent to mouse MRCs3.
Figure 2. Human splenic ILCs establish a bidirectional crosstalk with MRCs.

(a) IFA of spleens stained for MAdCAM-1 (green or red), IgD (blue), vWF (red) and/or RORγt (green). Original magnification, ×40 (upper panels), ×10 (upper inset) and ×63 (bottom-left panel) and zoom ×2 (bottom-right panel). (b) Flow cytometry of PECAM-1, CD34, CD45, Thy-1, CD141, ICAM-1, VCAM-1, TLR3, TLR4 and TLR9 expression by ex vivo expanded MRCs. Gray shading, negative control. (c) qRT-PCR of IL1B (IL-1β), IL7 (IL-7), IL23 (IL-23), CCL20 (CCL20), ICAM1 (ICAM-1) and MADCAM1 (MadCAM-1) mRNAs in MadCAM-1+ MRC-enriched (red box), MR+ SLC-enriched (blue box) and control (ctrl) MR– (black box) areas microdissected from immunohistochemically stained splenic tissue (original magnification, ×20). qRT-PCR results are normalized to ACTB (β-actin) mRNA and presented as relative expression (RE) compared with that of MR– areas. (d) Flow cytometry of integrins α4 and β7 on freshly isolated splenic ILCs (left) or ILCs cultured with IL-1β and IL-7 for 72 h (right). (e,f) Flow cytometry of ICAM-1 and VCAM-1 on MRCs incubated for 72 h with or without ILCs in the presence or absence of control (Ctrl), anti-TNF, anti-LTαβ or anti-TNF plus anti-LTαβ antibodies. (g,h) Flow cytometric analysis of frequency of Annexin-V–propidium iodide– ILCs from splenic ILCs cultured for 72 h with or without MRCs in the presence of absence of a transwell, a control (Ctrl) antibody or anti-IL-7. Error bars, s.e.m.; *P <0.05 (two-tailed unpaired Student's t test). Data summarize three experiments with one donor in each (c,h) or show one of four experiments with similar results (a,b,d,e,f,g).
Human MRCs expressed TLR3, which binds double-stranded RNA, TLR4, which binds lipopolysaccharide (LPS), and TLR9, which binds unmethylated CpG-rich DNA (Fig. 2b), pointing to a role of microbial products in the regulation of MRCs. Laser capture microdissection followed by qRTPCR demonstrated that MRC-enriched MAdCAM-1+ tissue contained more ILC survival factors such as IL-1β, IL-7 and IL-23 and more ILC-recruiting factors such as CCL20 (a CCR6 ligand) than SLC-enriched mannose receptor (MR, an endocytic receptor)+ tissue or control MR– tissue (Fig. 2c).
As shown by flow cytometry, splenic ILCs up-regulated the MAdCAM-1-binding integrin α4β7 in response to IL-7 and IL-1β, pointing to a role of MAdCAM-1-α4β7 interaction in the perifollicular positioning of ILCs. Conversely, MRCs up-regulated ICAM-1 and VCAM-1 in response to splenic ILCs or a combination of LT and TNF or LPS (Fig. 2d and Supplementary Fig. 3d,e). Blocking experiments and qRT-PCR indicated that ILCs activated MRCs via LT and TNF, whereas MRCs up-regulated IL-7, CCL20 and MAdCAM-1 in response to LT and TNF (Fig. 2e and Supplementary Fig. 3f,g). Of note, transwell assays showed that MRCs supported ILC survival via contact-dependent and contact-independent signals that included IL-7 (Fig. 2g,h). Finally, splenic macrophages and DCs expressed more IL-1β and IL-23 than MRCs (Supplementary Fig. 3h), suggesting that ILCs establish a bi-directional interplay with MRCs in splenic niches that may also entail innate immune cells.
Splenic ILCs and MRCs deliver B cell-helper signals
Given their MZ and perifollicular location, human splenic ILCs may regulate MZ B cells. As shown by tissue IFA and qRT-PCR, splenic RORγt+ ILCs were positioned nearby CD20+ B cells and, compared to other splenic leukocyte subsets or mucosal ILCs, expressed more BAFF, CD40L and DLL1 (Fig. 3a,b), three major B cell and plasma cell helper factors5, 9, 10, 35. Unlike splenic macrophages, splenic ILCs expressed little or no APRIL (Fig. 3b), a BAFF-related plasma cell survival factor36.
Figure 3. Human splenic ILCs express the MZ B cell-helper factors BAFF, CD40L and DLL1 and activate MZ B cells in cooperation with MRCs.

(a) IHC of spleen stained for RORγt (brown) and CD20 (purple). Arrowheads point to RORγt+ cells. Original magnification, ×40. (b) qRT-PCR of TNFSF13B (BAFF), TNFSF13 (APRIL), CD40LG (CD40L) and DLL1 (DLL1) mRNAs from splenic or tonsillar ILCs and NK cells and from splenic macrophages (Mϕ), B cells and T cells. Results are normalized to ACTB (β-actin) mRNA and presented as relative expression (RE) compared with that of fresh splenic NK cells. (c) Flow cytometry of BAFF, APRIL, CD40L and DLL1 on splenic ILCs exposed to IL-1β plus IL-7 for 72 h. Gray shading, negative control. (d) ELISA of soluble BAFF and APRIL from splenic ILCs and NK cells cultured as in (c) as well as Mϕ. (e) Flow cytometric analysis of the frequency of viable Annexin-V–propidium iodide (PI)– and divided carboxyfluorescein diacetate succinimidyl ester (CFSE)low splenic MZ (blue bars) and FO (pink bars) B cells after incubation with medium alone (ctrl) or ILCs for 5 d. (f) ELISA of IgM, IgG and IgA from splenic MZ B cells incubated for 5 d with medium alone (Ctrl), ILCs, MRCs and/or CpG. (g) Frequency of CD27hiCD38hi plasmablasts in splenic MZ B cells cultured for 5 d as in (f). Error bars, s.e.m.; *P <0.05 (two-tailed unpaired Student's t test). Data summarize 3 measurements from 3 pooled experiments with one donor in each (b,d) or show one of three experiments with similar results (a,c,e,f with s.e.m of triplicate, g).
As shown by flow cytometry and ELISA, splenic ILCs expressed surface BAFF, CD40L and DLL1 and secreted BAFF, whereas APRIL was mainly secreted by macrophages (Fig. 3c,d). Accordingly, splenic ILCs stimulated the survival, proliferation, IgM secretion and plasmablast differentiation of IgDloCD27+ MZ but not IgDhiCD27– FO B cells (Fig. 3e-g). Having shown that MRCs provide survival and activation signals to ILCs, we verified whether MRCs augmented the B cell-helper function of ILCs. Compared to MZ B cells exposed to ILCs or MRCs alone, MZ B cells exposed to both ILCs and MRCs not only underwent more IgM secretion and plasmablast differentiation, but also acquired the ability to release IgG and IgA, particularly in the presence of CpG DNA (Fig. 3f,g). Thus, human splenic ILCs may integrate stromal and innate immune signals to orchestrate antibody production.
Splenic ILCs help MZ B cells via BAFF, CD40L and DLL1
Next, we used flow cytometry and ELISA to determine the mechanism by which human splenic ILCs help MZ B cells. BAFF-R-Ig, a soluble decoy receptor that blocks the binding of BAFF to B cells, attenuated the survival of MZ B cells exposed to ILCs (Fig. 4a). Of note, MZ B cells underwent plasmablast differentiation and IgM secretion in response to ILCs but not ILC-conditioned medium (Fig. 4b,c), suggesting the involvement of contact-dependent signals from membrane-bound BAFF, CD40L and/or DLL1. Accordingly, BAFF-R-Ig, CD40-Ig or a NOTCH signaling inhibitor mitigated ILC-induced IgM secretion, particularly when these inhibitors were used in combination (Fig. 4d).
Figure 4. Human splenic ILCs help MZ B cells via BAFF, CD40L and DLL1.

(a,b) Flow cytometric analysis of the frequency of viable DAPI– MZ B cells from MZ B cells cultured for 5 d with or without splenic ILCs in the presence or absence of control Ig or BAFF-R-Ig. SSC-A, side scatter area. (b) Frequency of CD27hiCD38hi plasmablasts from splenic MZ B cells cultured for 5 d with or without splenic ILCs or ILC conditioned medium (ILC-CM). (c) ELISA of IgM from splenic MZ B cells cultured as in (b). (d) ELISA of IgM from splenic MZ B cells cultured for 5 days with medium alone (Ctrl) or ILCs in the presence or absence of control Ig and dimethyl sulfoxide vehicle (Vehi), BAFF-R-Ig, CD40-Ig, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT, a NOTCH inhibitor) or a combination of BAFF-R-Ig plus CD40-R-Ig plus DAPT (All). Summary of data from 7 donors shown as % of maximal induction (100%) after MZ B cell stimulation with ILCs and control vehicle (Vehi). (e) Frequency of CD27hiCD38hi plasmablasts from splenic MZ B cells cultured for 5 days with control OP9 cells or DLL1-OP9 cells in the presence or absence of BAFF. (f) Frequency of CD27hiCD38hi plasmablasts from splenic MZ B cells cultured for 5 d with or without control (ctrl) OP9 cells or DLL1-OP9 cells in the presence or absence of BAFF and/or DAPT. (g) ELISA of IgM from splenic MZ B cells cultured as in (e). Error bars, s.e.m.; *P <0.05 (two-tailed unpaired Student's t test). Data summarize four (a), three (b,c,e) and seven (d) experiments with one donor in each or show one of three experiments with similar results (f, g with s.e.m of triplicate).
Although required for the early development of MZ B cells, NOTCH2 remains expressed on mature MZ B cells and may thus modulate their function5. Indeed, when combined with BAFF, DLL1-expressing OP9 stromal cells stimulated plasma cell differentiation and IgM secretion through NOTCH signals in MZ B cells (Fig. 4e-g). In contrast, BAFF alone or combined with DLL1-negative OP9 cells had little or no stimulatory effect (Fig. 4e-g). Thus, besides eliciting MZ B cell survival and activation, human splenic ILCs promote plasmablast differentiation and survival via BAFF, CD40L and DLL1.
Splenic ILCs activate perifollicular neutrophils via GM-CSF
In human spleens, MZ B cells interact with neutrophils (termed NBH cells) possibly originating from circulating precursors (termed NC cells)10. Given the involvement of RORγt in GM-CSF expression by TH17 cells and the important role of GM-CSF in neutrophil activation and survival37, we wondered whether splenic ILCs expressed GM-CSF. Tissue IHC showed CD117+ ILCs nearby CD66 (CEACAM-1)+ NBH cells positioned around the MZ (Fig. 5a). qRT-PCR, ELISA and flow cytometry demonstrated the expression of abundant GM-CSF in splenic ILCs, but not other leukocyte subsets (Fig. 5b,c). Splenic ILCs further expressed the neutrophil-recruiting chemokine IL-8 (or CXCL8), which was also produced by macrophages and NK cells, albeit in lesser amounts (Fig. 5b,c).
Figure 5. Human splenic ILCs activate MZ B cell-helper neutrophils via GM-CSF.

(a) IHC of spleen stained for CD117 (red) and CD66 (brown). Original magnification, ×20 or ×40 (inset). (b) Left: qRT-PCR of CSF2 (GM-CSF) and IL8 (IL-8) mRNAs from splenic ILCs, NK cells, macrophages (Mϕ), B cells and T cells. CSF2 and IL8 data are normalized to ACTB (β-actin) mRNA and presented as relative expression (RE) compared with that of fresh splenic NK cells. Right: ELISA of GM-CSF and IL-8 from splenic ILCs and NK cells cultured for 48 h with IL-1β and IL-7. (c) Flow cytometry of intracellular GMCSF and IL-8 in splenic CD127+ (Lin–CD117+) ILCs activated with or without PMA (P) plus ionomycin (I) for 4 h. (d) Flow cytometry of CD69, CD11b, CD24 and CD62L on Nc cells cultured for 24 h with or without splenic ILCs in the presence or absence of control IgG1 or blocking anti-GM-CSF antibodies. Gray shading: isotype control. (e) Flow cytometry of viable DAPI– Nc cells cultured with medium alone (Ctrl), GM-CSF, ILCs or ILC-derived conditioned medium (ILC-CM) in the presence or absence of IgG1 or anti-GM-CSF antibodies. (f) qRT-PCR of TNFSF13 (APRIL) mRNA from Nc cells cultured for 24 h with medium alone (Ctrl), GM-CSF or ILC-CM. Results are normalized to ACTB mRNA and presented as RE compared with that of Nc cells cultured with medium alone. (g) ELISA of IgA from splenic MZ B cells cultured for 5 d with or without Nc cells, ILCs and/or ILC-CM. (h) ELISA of IgM, IgA and IgG from splenic MZ B cells cultured for 5 d with or without NBH cells, ILC-CM and/or NBH cells preconditioned with ILC-CM. (i) IFA of elastase (green) and DAPI-stained DNA (blue) in Nc cells cultured for 3 h either with medium alone (Ctrl), GM-CSF, ILC-CM or LPS (original magnification, ×63) and quantification of NET-forming Nc cells from nine ×10 fields from two independent experiments. Error bars, s.e.m.; *P <0.05 (two-tailed unpaired Student's t test). Data summarize 2-3 experiments with one donor in each (b,e,f,g,h,i) or show one of four experiments with similar results (a,c,d).
Given that GM-CSF enhances the B cell-helper function of neutrophils10, we used flow cytometry, ELISA, IFA and qRT-PCR to establish whether GM-CSF-producing splenic ILCs induce NBH cell-like properties to NC cells. In the presence of splenic ILCs, NC cells not only acquired phenotypic traits typical of NBH cells10, such as up-regulation of the activation molecules CD69, CD11b and CD24 and down-regulation of the adhesion molecule CD62 ligand (CD62L), but also received survival signals from GMCSF (Fig. 5d,e). When incubated with splenic ILCs or their conditioned medium, NC cells up-regulated APRIL and thus stimulated more IgA production in MZ B cells (Fig. 5f,g). ILC-conditioned medium also augmented the capacity of NBH cells to induce IgM, IgG and IgA in MZ B cells (Fig. 5h). Similar to GMCSF or LPS, ILC-conditioned medium stimulated NC cells to form neutrophil extracellular traps (NETs) (Fig. 5i), which are antigen-trapping projections often emanating from NBH cells10. Thus, human splenic ILCs may amplify MZ B cell responses by stimulating perifollicular neutrophils via GM-CSF.
Splenic ILCs enhance TI antibody production
To elucidate the in vivo contribution of splenic ILCs to humoral immunity, we initially took advantage of Rorc–/– mice, which lack the RORγt transcription factor required for the development of ILC3 (refs. 22, 28). Rorc–/– mice also lack TH17 cells, a subset of CD4+ T cells that regulate neutrophils via IL-17 and GM-CSF37-39. Flow cytometry showed that Rorc–/– mice had fewer splenic Lin–CD117+CD127+ ILCs than Rorc+/+ mice (Fig. 6a). In these wild type mice, flow cytometry and qRT-PCR demonstrated that splenic ILCs included both CD4+ and CD4– subsets that expressed more RORγt, IL-22, LT-α and TNF than did other splenic leukocytes (Fig. 6b and Supplementary Fig. 4a). In contrast to human splenic ILCs, mouse splenic ILCs lacked BAFF and CD40L, but expressed APRIL and DLL1 (Fig. 6b).
Figure 6. Mouse splenic ILCs express the plasma cell-helper factors APRIL and DLL1 and enhance TI IgG3 responses.

(a) Flow cytometric analysis of frequency and absolute numbers of splenic Lin– CD117+CD127+ ILCs from Rorc+/+ (n = 3) and Rorc–/– mice (n = 3). (b) qRT-PCR of Rorc (RORγt) Il22 (IL-22) Tnf (TNF), Lta (LT-α), Tnfsf13 (APRIL), Tnfsf13b (BAFF), CD40lg (CD40L) and Dll1 (DLL1) mRNAs from splenic ILCs, macrophages (Mϕ), DCs, neutrophils, NK cells, T cells or B cells. Results are normalized to Gapdh (glyceraldehyde 3-phosphate dehydrogenase) mRNA and presented as relative expression (RE) compared to the expression level in NK cells. (c) Flow cytometric analysis of frequency and absolute numbers of splenic IgG3E+IgG3Ilo B cells, IgG3EhiIgG3I+ plasmablasts (PBs) and IgG3E+IgG3Ihi plasma cells (PCs) from Rorc+/+ (n = 3) and Rorc–/– mice. E, extracellular; I, intracellular. (d) IFA of IgM (green), IgG3 (red) and MOMA-1 (blue) in spleens from Rorc+/+ and Rorc–/– mice. Original magnification, ×20. (e,f) ELISA of total (e) and PC-reactive (f) serum IgG3 from Rorc+/+ (n = 6-7) and Rorc–/– (n = 7-8) mice. (g) Frequency of IgG3-expressing B cells, PBs and PCs from spleens of Rorc+/–Cd3e–/– and Rorc–/–Cd3e–/– mice. (h) Serum IgG3 from Rorc+/–Cd3e–/– (n = 9) and Rorc–/–Cd3e–/– (n = 6) mice. Error bars, s.e.m. (b), s.d. (a,c); *P <0.05 (one-tailed unpaired Student's t test). Data show one of four experiments with similar results (a,d,g; c: cytograms) or summarize results from 3-8 animals per each group (a,c: bars; b,e,f,h).
We then verified whether Rorc–/– mice had an altered MZ. As shown by tissue IFA, Rorc–/– mice had no perturbations of either MOMA1+ metallophilic macrophages, ER-TR9+ MZ macrophages and ICAM-1+ marginal sinus cells from the MZ or IgM+ B cells from the MZ and white pulp or F4/80+ macrophages and VCAM-1+ endothelial cells from the red pulp (Supplementary Fig. 4b). Flow cytometry, tissue IFA and ELISA demonstrated that spleens from non-immunized Rorc–/– mice contained conserved B cells, but fewer plasmablasts and plasma cells expressing IgG3 (Fig. 6c,d and Supplementary Fig. 5a), an antibody produced by MZ B cells through a TI pathway under pre-immune conditions or after immunization40, 41. Besides less total serum IgG3, Rorc–/– mice had less serum IgG3 to the bacterial TI antigen phosphorylcholine (PC) (Fig. 6e,f). To ascertain the relative contribution of ILCs and T cells to pre-immune IgG3 responses, Rorc+/– and Rorc–/– mice were crossed with T cell-deficient Cd3e–/– mice. In agreement with the TI nature of pre-immune IgG3 production40, 41, non-immunized Rorc+/–Cd3e–/– mice had normal pools of splenic B cells, plasma cells and plasmablasts expressing IgG3 (Fig. 6g). Compared to these mice, Rorc–/–Cd3e–/– mice showed conserved B cells, but fewer plasmablasts and plasma cells expressing IgG3 and less pre-immune serum IgG3 (Fig. 6g,h).
Unlike pre-immune serum IgG3, pre-immune serum IgM was augmented in Rorc–/– mice together with splenic IgMhiB220int/loCD5int B-1 cells but not B220+CD21+CD23+ FO B cells and B220+CD21hiCD23– MZ B cells (Supplementary Fig. 5b,c). The expansion of IgM-producing B-1 cells in Rorc–/– mice may result from enhanced migration of B-1 cells from the peritoneal cavity to the spleen due to the lack of gut-associated lymphoid tissue and increased systemic translocation of gut bacteria22, 28, 34, 42. Nonetheless, Rorc–/– mice had less pre-immune serum IgM to PC (Supplementary Fig. 5d), suggesting a role of ILCs in at least some TI IgM responses.
To dissect the MZ B cell-helper function of ILCs in a more physiological model, an anti-Thy-1.2 antibody was used to deplete ILCs in Thy-1-disparate Rag1–/– chimeric mice generated as described in a recently published study43. Compared to mice treated with a control antibody, mice treated with anti-Thy-1.2 showed fewer splenic ILCs, fewer IgG3-expressing plasmablasts and plasma cells, and reduced pre-immune serum IgG3 (Fig. 7a-c and Supplementary Fig. 5e). Furthermore, anti-Thy-1.2-treated mice had impaired post-immune IgG3 responses to 2,4,6-trinitrophenyl (TNP)-Ficoll (Fig. 7d), a TI antigen that induces antibody production in innate-like B cells, including MZ B cells40.
Figure 7. Mouse splenic ILCs help plasmablasts and plasma cells emerging from TI IgG3 responses.

(a) Flow cytometric analysis of frequency and absolute numbers of splenic Lin–CD117+CD127+ ILCs from Thy-1-disparate chimeric Rag1–/– mice treated with control (ctrl) (n = 7) or anti-Thy.1.2 antibodies (n = 7). (b) Frequency and absolute numbers of splenic IgG3E+IgG3Ilo B cells, IgG3EhiIgG3I+ plasmablasts (PBs) and IgG3E–IgG3Ihi plasma cells (PCs) from Ctrl or ILC-depleted mice obtained as in (a). E, extracellular; I, intracellular. (c,d) ELISA of pre-immune total (c) and PC-reactive (d) serum IgG3 from Ctrl or ILC-depleted mice obtained as in (a). (e,f) Frequency and absolute numbers of splenic Lin– CD117+CD127+ ILCs (e) and IgG3-expressing splenic B cells, PBs and PCs (f) from ILC+ (n = 3) or ILC– (n = 3) bone marrow chimeric mice. (g) Pre-immune total serum IgG3 from ILC+ (n = 4) and ILC– (n = 3) mice, age-matched Rorc+/+ mice (n = 10), and Rorc–/– mice (n = 5). Error bars, s.d.; *P <0.05 (one-tailed unpaired Student's t test). Data show one of four experiments with similar results (a,b,e,f: cytograms) or summarize results from 3-10 animals per each group (a,b,e,f: bars; c,d,g).
The involvement of splenic ILCs in TI antibody production was also demonstrated by reconstituting lethally irradiated Rorc–/– mice with a mix of bone marrow cells from Rag1–/– mice – which generate ILCs but not T and B cells – and Rorc–/– mice – which generate T and B cells but not ILCs. ILC-sufficient (ILC+) chimeric mice developed splenic ILCs, whereas control ILC– mice generated by reconstituting irradiated Rorc–/– mice with bone marrow cells from Rorc–/– mice did not (Fig. 7e). Compared to ILC+ mice, ILC– mice showed fewer splenic plasmablasts and plasma cells expressing IgG3 and less pre-immune serum IgG3 (Fig. 7f,g). In all mouse models, IgM responses were only marginally affected (not shown), indicating that splenic ILCs predominantly help IgG3-expressing plasmablasts and plasma cells emerging from MZ B cell responses to TI antigens.
Splenic ILCs regulate neutrophil homeostasis
We next determined the role of mouse splenic ILCs in the homeostasis of NBH cells. Tissue IFA and flow cytometry showed that ILC– bone marrow chimeric mice had fewer splenic Ly6G+CD11b+ neutrophils and extrafollicular IgG3-producing plasmacytes than control ILC+ mice (Fig. 8a,b). Splenic neutrophils were also decreased in Rorc–/– mice with either a T cell-sufficient or T cell-deficient background (Supplementary Fig. 6a-c), indicating that splenic ILCs provide helper signals to neutrophils independently of T cells. To ascertain the contribution of splenic neutrophils to pre-immune IgG3 production, ILC-reconstituted (ILC+) bone marrow chimeric mice were depleted of splenic neutrophils with an anti-Ly6G antibody (Fig. 8c). These mice had normal splenic ILCs and IgG3-expressing B cells and plasmablasts, but fewer splenic IgG3-expressing plasma cells and decreased pre-immune serum IgG3 compared to ILC+ mice treated with a control antibody (Fig. 8d-f). Thus, unlike ILCs, neutrophils may predominantly control terminally differentiated IgG3-secreting cells.
Figure 8. Mouse splenic neutrophils help plasma cells emerging from TI IgG3 responses and receive homeostatic signals from ILCs via GM-CSF.

(a) IFA of IgG3 (green), Ly6G (red) and IgM (blue) in spleens from ILC+ or ILC– bone marrow chimeric mice. Original magnification, ×20. (b) Flow cytometric analysis of the frequency of splenic Ly6G+CD11b+ neutrophils from ILC+ (n = 3) or ILC– (n = 3) mice. (c) Frequency and absolute numbers of splenic Ly6G+CD11b+ neutrophils from ILC+ mice treated with control (Ctrl) (n = 4) or anti-Ly6G antibodies (n = 4). (d,e) Frequency and absolute numbers of splenic Lin–CD117+CD127+ (d) as well as splenic IgG3E+IgG3Ilo B cells, IgG3EhiIgG3I+ plasmablasts (PBs) and IgG3E+IgG3Ihi plasma cells (PCs) from ILC+ mice treated as in (c). E, extracellular; I, intracellular. (f) ELISA of total serum IgG3 from ILC+ mice treated as in (c). (g) qRT-PCR of Csf2 (GM-CSF) mRNA from splenic ILCs, macrophages (Mϕ), DCs, neutrophils, NK cells, T cells or B cells. Results are normalized to Gapdh (glyceraldehyde 3-phosphate dehydrogenase) mRNA and presented as relative expression (RE) compared to the expression level in NK cells. (h) Frequency and absolute numbers of splenic Ly6G+CD11b+ neutrophils from Csf2+/+ (n = 8) and Cfs2–/– (n = 12) mice before and after transfer of B16Cfs2 cells overexpressing GM-CSF (n = 3). (i) Frequency of splenic neutrophils in ILC-sufficient Rag1–/–Il2rg+/+ mice or ILC-insufficient Rag1–/–Il2rg–/– mice reconstituted or not with gut ILCs from Csf2+/+ or Cfs2–/– mice (n = 3). Error bars, s.e.m. (g-i), s.d. (b-e); *P <0.05 (one-tailed unpaired Student's t test). Data summarize results from at least two experiments with 3 mice in each group (b,c,d,e,h: bars; f,g,i) or show one of at least three experiments with similar results (a and b,c,d,e,h: cytograms).
Finally, we determined whether splenic ILCs regulate neutrophils via GM-CSF. As shown by qRTPCR, splenic ILCs from wild-type mice expressed more GM-CSF than other leukocyte subsets did, including macrophages and NK cells (Fig. 8g). Compared to controls, GM-CSF (also known as colony-stimulating factor 2)-deficient Csf2–/– mice had reduced pre-immune serum antibodies to bacterial PC and fewer splenic neutrophils, which increased after inoculation of GM-CSF-expressing B16 melanoma cells (Fig. 8h and Supplementary Fig. 6d). Also Rag1–/–Il2rg–/– mice, which lack the IL-2 receptor common γ chain mandatory for IL-7-dependent ILC development13, had fewer splenic neutrophils compared to Rag1–/–Il2rg+/+ controls (Fig. 8i). Moreover, adoptive transfer of ILCs from Csf2+/+ but not Csf2–/– mice increased splenic neutrophils in Rag1–/–Il2rg–/– mice (Fig. 8i). Thus, in addition to helping MZ B cells and plasma cells through BAFF (or APRIL), CD40L and DLL1, splenic ILCs co-opt neutrophils via GM-CSF to enhance TI antibody production (Supplementary Fig. 7a,b).
DISCUSSION
We have shown that the MZ and PFZ of the spleen contained group-3 mucosa-like ILCs that released LT and TNF to establish a bi-directional crosstalk with stromal MRCs. In addition to stimulating MZ B cells and plasma cells through BAFF, APRIL, CD40L and DLL1, splenic ILCs co-opted neutrophils with MZ B cell- and plasma cell-helper functions through GM-CSF, thereby sustaining TI antibody production.
Mucosal and serosal membranes contain evolutionarily primitive lymphocyte subsets that include B-1 cells, γδ T cells, invariant NKT cells and mucosa-associated invariant T cells44. By sensing conserved microbial signatures through somatically recombined and germline-encoded receptors, these innate-like lymphocytes rapidly activate protective programs that crossover the conventional boundaries between the innate and adaptive immune systems44. Mucosal membranes also contain ILCs that lack somatically recombined receptors and yet mount prompt T cell-like responses13. Here, we found that splenic ILCs enhanced antibody production to TI antigens by activating innate-like B cells positioned at the interface between the immune and circulatory systems.
Similar to mucosal ILC3 (refs. 19, 20), human splenic ILCs contained both RORγt and AhR transcription factors, expressed NCRs along with various activation molecules, and secreted IL-22 in response to IL-23. In addition, human splenic ILCs activated MZ-based stromal MRCs via LT and TNF and thus also resembled fetal or mucosal LTi cells28-30, 45. However, unlike LTi cells23, 30, human splenic ILCs did not express IL-17, raising questions as to the ontogenetic relationship between these ILC subsets. Mouse splenic ILCs had a phenotype and gene expression profile similar to those of human splenic ILCs, but expressed CD4, thus showing a closer affiliation with LTi cells.
Human splenic ILCs not only expressed the gut homing receptor α4β7, but also interacted with MRCs equipped with MAdCAM-1, an α4β7 ligand usually associated with gut endothelial cells34. Given their additional expression of CCL20 and the presence of the CCL20 receptor CCR6 on splenic ILCs, MRCs might be involved in the positioning of ILCs within and around the MZ. Consistent with their stroma phenotype and reticular morphology, MRCs lacked endothelial molecules and exhibited strong responsiveness to LT and TNF from ILCs, thus resembling MAdCAM-1+ stromal cells lining the mouse marginal sinus46. Conversely, MRCs delivered both contact-dependent and contact-independent survival signals to ILCs, including IL-7. Remarkably, MRCs increased IL-7 expression in response to LT and TNF, pointing to the presence a bi-directional functional crosstalk between MRCs and ILCs in the MZ and PFZ of the spleen. Additional ILC survival factors such as IL-1β and IL-23 were predominantly expressed by splenic DCs and macrophages, suggesting that ILCs inhabit a splenic niche that entails survival signals from both stromal and immune cells. Of note, MRCs received further activation signals from TLR ligands such as LPS, lending support to the possibility that commensal antigens from the intestinal mucosa help the post-natal development and function of the splenic MZ4, 10, 47, 48.
In humans, splenic ILCs enhanced the survival of MZ B cells through a contact-independent mechanism involving soluble BAFF, but triggered the differentiation of MZ B cells into plasmablasts through contact-dependent signals provided by membrane-bound BAFF, DLL1 and CD40L. Of note, DLL1 synergized with soluble BAFF to induce IgM secretion, possibly owing to the pro-survival effect of soluble BAFF on plasmablasts35. Given that DLL1 also cooperates with BAFF to stimulate the early development of MZ B cells5, there is the possibility that DLL1 on ILCs operates at both early and late stages of MZ B cell differentiation with distinct signaling programs. Of note, splenic ILCs further helped MZ B cells by co-opting NBH cells through GM-CSF. This cytokine may also enhance the MZ B cell-stimulating function of DCs and possibly macrophages1, 4, which may explain why a relative small number of ILCs is sufficient to enhance TI antibody responses.
Despite expressing CD40L, a class switch-inducing factor typically detected on TFH cells33, human splenic ILCs did not trigger IgG or IgA production in MZ B cells. Nonetheless, MZ B cells induced IgA and IgG when exposed to ILCs in the presence of MRCs and microbial TLR ligands. Accordingly, small amounts of these bacterial components can be detected in splenic perifollicular areas together with plasma cells releasing IgM, IgG or IgA10. Thus, similar to gut LTi cells29, splenic ILCs may integrate stromal and innate immune signals to rapidly generate antibodies for antimicrobial protection.
Although lacking both CD40L and BAFF, mouse splenic ILCs expressed APRIL, a BAFF-related molecule that enhances plasma cell survival36. By also expressing DLL1, mouse splenic ILCs would sustain the differentiation and/or survival of plasmablasts and plasma cells emerging during IgG3 responses to TI antigens. In mice, ILC depletion did not impair IgM production, suggesting that ILCs predominantly enhance MZ B cell-derived IgG3 rather than B-1 cell-derived IgM responses. Nonetheless, splenic ILCs sustained pre-immune production of IgM to bacterial PC, which points to the involvement of ILCs in IgM responses to some but not all TI antigens.
In ILC-deficient mice, impaired TI IgG3 production was associated with a reduction of NBH cells, which help MZ B cells via BAFF and APRIL10. Accordingly, human splenic ILCs enhanced the survival and B cell-helper activity of NBH cells through GM-CSF. This cytokine also induced NET-like structures, which might enable NBH cells to capture blood-borne antigens transiting through the MZ4, 10. Human splenic ILCs also released IL-8, which may contribute to the recruitment of NBH cells. Mice do not express IL-8, but their splenic ILCs may attract NBH cells via alternative factors, including GM-CSF. Remarkably also splenic innate response activator (IRA) B cells express GM-CSF49, which could explain the persistence of some NBH cells in mice lacking ILCs.
GM-CSF further derives from TH17 cells37, but the analysis of T cell-deficient mice suggested that ILCs can regulate the homeostasis of NBH cells independently of T cells. In mice, splenic ILCs sustained both IgG3+ plasmablasts and IgG3+ plasma cells, whereas mouse NBH cells predominantly helped IgG3+ plasma cells, suggesting that multiple subsets of innate immune cells orchestrate TI IgG3 responses in a hierarchical manner. Of note, IgG3 may provide protection against certain pathogens following its interaction with ficolins, a family of soluble pattern recognition receptors released by hepatocytes and various cells of the innate immune system41. In summary, our studies indicate that splenic ILCs orchestrate a stromal-innate cell network that fosters TI antibody production by MZ B cells. Harnessing this network with adjuvants may enhance protective humoral responses to poorly immunogenic TI antigens4, 50, including viral glycoproteins and bacterial carbohydrates.
ONLINE METHODS
Human samples
Mononuclear cells and neutrophils were isolated from the peripheral blood of healthy volunteers or histologically normal spleens from deceased organ donors or splenectomized trauma patients without clinical signs of infection or inflammation10. Tonsils were from individuals with follicular hyperplasia10. The use of blood and tissues was approved by the Ethical Committee for clinical investigation of Institut Hospital del Mar d'Investigacions Mèdiques (CEIC-IMIM 2011/4494/I). Tissue samples were collected by the department of Pathology of Hospital del Mar. Prior to collection, signed informed consent was obtained from the patient or his/her parent or guardians. All the blood and tissue samples were coded and relevant clinical information remained anonymous.
Mice
Rorc–/– (Rorcgfp/gfp), Cd3e–/–, Rag1–/–, Rag1–/–Il2rg–/– and Csf2–/– mice on the C57BL/6 background have been described previously29,51-54. C57BL/6 CD90.1 mice were purchased from the Jackson Laboratory. All mice were housed in specific pathogen-free conditions. Male and female mice were used at age 8-12 weeks unless specified in the text. All the experiments involving mice were performed in accordance with approved protocols from the Institutional Animal Care at RIKEN and Icahn School of Medicine at Mount Sinai.
Mouse chimeras and neutrophil depletion
4 × 107 bone marrow cells from Rag1–/– mice were mixed with 1 × 107 bone marrow cells from Rorc–/– mice and injected intravenously into 10-Gy irradiated 2-month old Rorc–/– mice. After transplantation, mice were given 500 mg/L ampicillin (Sigma) and 1 g/L neomycin (Sigma) in drinking water for 2 weeks. To deplete neutrophils, control RTK2758 monoclonal antibody (mAb) or neutrophil-depleting 1A8 mAb to Ly6G (BioXCell) were administrated intraperitoneally every 5 days at a dose of 250 μg/mouse for a month. Thy-1 disparate Rag1–/– chimeras were generated and depleted of ILCs with a 30-H12 mAb to Thy-1.2 (BioXCell) injected intraperitoneally every 3 days at a dose of 500 μg/mouse for a month as previously published43. LTF-2 mAb was used as control.
Adoptive transfers
2–3 × 104 ILCs from the small intestine of either Csf2+/+ or Csf2–/– mice were adoptively transferred into Rag1–/–Il2rg–/– mice via retro orbital injection as previously described and splenic neutrophils were analyzed 21 days later. 1 × 105 B16Csf2 melanoma cells overexpressing GM-CSF (from G. Dranoff) were injected into Csf2–/– mice and splenic neutrophils were analyzed 12–14 days later.
Immunization
Serum TNP-specific antibody titers were determined by ELISA 7 days after intraperitoneal injection of 50 μg TNP-Ficoll.
Cells
Human splenocytes or tonsillar mononuclear cells were obtained from fresh tissue samples as reported in published studies10. Human ILCs, NK cells, naïve B cells, MZ B cells, macrophages, T cells, NC cells and NBH cells were sorted by flow cytometry as described previously10 or reported below. To isolate human MRCs, splenic tissues were enzymatically digested in Hank's balanced salt solution (Lonza) containing 1 mg/ml collagenase-IV (Invitrogen) and 50 ng/ml DNAse-I (50 ng/ml) at 37 °C for 45 min, followed by separation on Ficoll-Hypaque gradient. Splenocytes were resuspended in EGM medium (Lonza) and cultured in 0.1% gelatin (Sigma) pre-coated plate. 72 h later, adherent cells were obtained with Accutase cell detachment solution as instructed by the manufacturer (Millipore). CD45– CD31– adherent cells were sorted by flow cytometry and expanded up to three passages in EGM medium. Murine OP9 and OP9-DLL1 stromal cell lines (from R. Gimeno) were tested for mycoplasma and grown in essential medium-α (Invitrogen) supplemented with 20% Fetalclone I (Hyclone).
Cultures and reagents
Human splenic ILCs (5 × 104/well) were plated in 96 U-bottom plates and cultured in complete RPMI medium with or without 50 ng/mL IL-7, 50 ng/mL IL-1β and/or 50 ng/mL IL-23 (Peprotech). Conditioned medium was obtained by culturing splenic ILCs in complete medium for 24 h. In co-culture experiments, splenic ILCs were first expanded with IL-7 and IL-1β for 5-8 days as previously published. ILCs were co-cultured with confluent human MRCs in flat-bottom plates for 72 h. To avoid cell-to-cell contact, some co-cultures were performed in a 24-transwell system (Corning). Human ILCs were also co-cultured with human MZ B cells, NC cells and/or NBH cells at a 1:10 ratio. Some co-cultures were supplemented with 500 ng/ml BAFF (Alexis), 0.25 μg/ml CpG ODN-2006 (InvivoGen), 30 μg/ml BAFF-R-Ig, 5 μg/ml CD40-Ig, 5 μg/ml, 10 μg/ml neutralizing MAB215 mAb to human GM-CSF, or 10 μg/ml isotype control IgG1 mAb (R&D system). To inhibit NOTCH2, human MZ B cells were incubated with 25, 5 or 2.5 μM N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) (Sigma), which interferes with the cleavage of NOTCH proteins by γ-secretase. In some experiments, human MRCs were stimulated with 100 ng/ml LTα1β2 and 10 ng/ml TNF (R&D Systems) or with ILCs in the presence or absence of 10 μg/ml mAbs MAB2101 to TNF or MAB1370 to LTα2β1/α1β2 (R&D Systems). Human NC cells were stimulated with 50 ng/ml GM-CSF (Peprotech) or 100 ng/ml LPS (InvivoGen).
Flow cytometry
Cells were incubated with Fc blocking reagent (Miltenyi Biotec) at 4 °C before adding appropriate cocktails of fluorochrome-labeled mAbs (Supplementary Tables 1 and 2). Dead cells were excluded with 4’-6-diamidine-2’-phenylindole (DAPI) as indicated by the manufacturer (Boehringer Mannheim). To stain intracellular transcription factors, human ILCs or NK cells were stained with mAbs to cell-specific surface molecules, permeabilized, fixed with a Transcription Factor Buffer Set as indicated by the manufacturer (eBioscience), and labeled with mAbs to human RORγt or T-bet (Supplementary Table 1). For intracellular cytokine staining, human ILCs were cultured for 4 h in complete RPMI medium with GolgiStop (BD Pharmingen) in the presence or absence of 10−7 M phorbol myristate acetate and 0.5 μg/ml ionomycin (Sigma). These cells were stained with mAbs to specific surface molecules, fixed, permeabilized using the Cytofix/Cytoperm kit (BD Pharmingen), and finally incubated with mAbs to human GM-CSF or IL-8 (Supplementary Table 1). To stain intracellular IgG3, mouse splenocytes were stained with mAbs to specific surface molecules, permeabilized, fixed with the Cytofix/Cytoperm kit (BD Pharmingen), and labeled with R40-82 mAb to mouse IgG3 (Supplementary Table 2). Cells were acquired using FACS calibur, FACS Aria II, LSRII or LSRFortessa (BD Biosciences) and further analyzed using the FlowJo software (Tree Star).
FACSorting
Human CD3–CD14–CD19–CD117+CD127+ ILCs, CD3–CD14–CD19–CD117–CD127– CD56+ NK cells, CD19+IgDhiCD27– naïve B cells, CD19+IgDloCD27+ MZ B cells, CD14+ macrophages and CD3+ T cells as well as mouse CD3+ T cells, B220+ B cells, CD11c+ DCs, CD11b+ macrophages, CD49b+ NK cells, Ly6G+ neutrophils, CD4+CD45+CD117+CD127+Lin– (CD3–B220–CD11b–CD11c– Ly6G–) ILCs and CD4–CD45+CD117+CD127+Lin– ILCs were stained with appropriate cocktails of fluorochrome-labeled mAbs (Supplementary Tables 1 and 2) and sorted using FACSAria II (BD Biosciences) after exclusion of dead cells through DAPI staining. The purity of FACSorted cells was consistently > 95%.
Viability and proliferation assays
Cell survival was measured using the Annexin-V Apoptosis Detection Kit II (BD Pharmingen). Gates and quadrants were drawn to give ≤ 1% total positive cells in samples incubated with isotype control mAbs. Cell proliferation was assessed by CFSE staining using the CellTrace CFSE Cell Proliferation Kit (Invitrogen).
Laser capture microdissection
Microdissections were perfomed on fresh human splenic tissues frozen in optimal cutting temperature (OCT) compound using the ArcturusXT Laser Capture Microdissection System following the manufacturer's instructions (Life Technologies). In brief, 10-μm tissue slices were isolated on membrane slides (Life Technologies) and fixed in cold acetone for 10 s. Immunohistochemistry with specific mAbs was followed by tissue dehydration and fixation with xylene. Groups of cells from completely dried tissues were acquired by microdissection and RNA was extracted using the Arcturus PicoPure RNA Extraction Kit (Life Technologies).
Immunofluorescence analysis
Frozen tissues and cells from humans or mice were fixed as previously published10 and stained with various combinations of Abs (Supplementary Tables 1 and 2). Biotinylated Abs were detected using horseradish peroxydase (HRP)-conjugated streptavidin followed by tetramethylrhodamine from a Tyramide Signal Amplification Kit (PerkinElmer Life Sciences) or streptavidin-Alexa Fluor conjugates. Nuclear DNA was stained with DAPI. Coverslips were applied with FluorSave reagent (Calbiochem). Images were obtained using Axioplan2 (Carl Zeiss) microscopy and further analyzed using Axiovision (Carl Zeiss) software. To perform NET formation assays, human circulating neutrophils were seeded on poly D-lysin coated glass cover slips and stimulated for 3 h with either human GM-CSF or human splenic ILC-conditioned medium. Cells were fixed with 4% paraformaldehyde and stained with mAbs NP57 or ab21595 to human elastase and DAPI.
Immunohistochemistry
5-μm thick formalin-fixed and paraffin-embedded tissue sections were stained with various mAbs (Supplementary Table 1) using EnVision™ + Dual Link System-HRP (DAB+) for single stainings and EnVision G/2 Doublestain System Rabbit/Mouse (DAB+/Permanent Red) for double stainings (Dako). Sections were counterstained with hematoxylin.
ELISA
Total human IgM, IgG and IgA were measured as previously published10. Soluble human BAFF ELISA Kit (Adipogen), LEGEND MAX Human APRIL/TNFSF13 ELISA Kit (Biolegend), Human GMCSF ELISA Development Kit, and Human IL-8 ELISA Development Kit (Peprotech) were used to measure human BAFF, APRIL, GM-CSF and IL-8. Human Th1/Th2/Th9/Th17/Th22 13plex FlowCytomix Multiplex (eBioscience) was used to measure human IL-17 and IL-22. The concentration of mouse serum IgM, IgG3 and IgA were determined with Mouse ELISA Quantification Set (Bethyl Laboratories). To detect mouse TNP-specific IgG3, ELISA plates coated with 5 μg/ml bovine serum albumin (BSA)-conjugated TNP (BioSearch Technologies) were sequentially incubated with mouse serum and HRP-conjugated goat polyclonal antibody A90-111P to mouse IgG3 (Bethyl Laboratories). To detect mouse phosphorylcholine-specific IgM, ELISA plates coated with 5 μg/ml BSA-conjugated phosphorylcholine were sequentially incubated with serum and an HRP-conjugated goat polyclonal antibody A90-101P to IgM (Bethyl Laboratories). BSA-conjugated phosphorylcholine was synthesized as follows. 1 mg of BSA was coupled to 1 mg p-aminophenylphosphorylcholine in 0.1 M 2-(N-morpholino)ethanesulfonic buffer (Sigma) at pH 4.5 supplemented with 2 mg 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride in a 1.6 ml final volume. This mixture was incubated for 2 h at 20 °C. Conjugated BSA was dialyzed against PBS at pH 7.4 and 4 °C.
qRT-PCR
To quantify human gene products, total RNA was extracted and reverse transcribed into cDNA as reported in previously published studies10. qRT-PCRs were performed as reported previously10 using specific primer pairs (Supplementary Table 3). To quantify mouse gene products, total RNA was extracted with TRIzol reagent (Gibco BRL). After DNase I (Invitrogen) treatment, random hexamers (Invitrogen) were used for first-strand cDNA synthesis. qRT-PCRs were performed in 96-well plates with a LightCycler 480 real-time PCR instrument (Roche Diagnostics) using the LightCycler 480 SYBR Green I Master kit (Roche Diagnostics) and specific primer pairs (Supplementary Table 4). Gene expression was normalized to that of glyceraldehyde 3-phosphate dehydrogenase in each sample.
Statistical analysis
Values were expressed as mean ± standard error of the mean (s.e.m.) or standard deviation (s.d.). Statistical significance was assessed with two-tailed or one-tailed unpaired Student's t-test unless specified otherwise. Mann-Whitney U-test or Wilcoxon matched-pairs signed rank test were performed on non-parametric data. Results were analyzed with Prism software (Graph Pad) and P values less than 0.05 were considered significant. In animal experiments, littermates (minimum 3 mice/group) were randomly distributed to the treatment groups so that all groups were age-matched and sex-matched. No specific randomization or blinding protocol was used and no animals were excluded from the analysis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
We thank A. Chorny, S. Casas (Icahn School of Medicine at Mount Sinai), Gregory F. Sonnenberg (University of Pennsylvania), G. Dranoff (Dana Farber Cancer Institute), Anna Mensa (Hospital Clínic of Barcelona), R. Gimeno, A. Bigas and M. López-Botet (Institut Hospital del Mar d'Investigacions Mèdiques) for providing reagents and samples and helping with discussions; E. Ramirez, E. Julià and O. Fornas (Barcelona Biomedical Research Park) for helping with cell sorting. Supported by European Advanced Grant ERC-2011-ADG-20110310, Ministerio de Ciencia e Innovación grant SAF2011-25241, and Marie Curie reintegration grant PIRG-08-GA-2010-276928 to A.C.; Juan de la Cierva post-doctoral fellowships to I.P. and G.M.; pre-doctoral fellowships from the Instituto de Salud Carlos III and Ministerio de Ciencia e Innovación to C.B, M.G and S.B.G.; an EUROPADnet HEALTH-F2-2008-201549 grant to A.C.; US National Institutes of Health grants R01 AI74378, R01 AI57653, U01 AI95613, U01 AI95776 IOF, P01 AI61093 and U19 096187 to A.C; Grant-in-Aid for Scientific Research in Priority Areas to S.F.; and Grant-in-Aid for Scientific Research (B) to S.F.
Footnotes
AUTHOR CONTRIBUTIONS
G.M. and M. Miyajima designed and performed research, discussed data and wrote the paper; S.B., A.M., I.P. and A. Chudnovskiy designed and performed research; C.M.B., L. Cassis, L. Comerma, M.G., M.C. and D.L. performed research; S.S., J.I.A., M.J. and J.Y. provided blood and tissue samples and discussed data; S.F. and M. Merad designed research, provided reagents and discussed data; A.C. designed research, discussed data and wrote the paper.
COMPETING INTEREST STATEMENT
The authors declare that they have no competing financial interests.
REFERENCES
- 1.Balázs M, Martin F, Zhou T, Kearney JF. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17:341–352. doi: 10.1016/s1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- 2.Kang YS, et al. A dominant complement fixation pathway for pneumococcal polysaccharides initiated by SIGN-R1 interacting with C1q. Cell. 2006;125:47–58. doi: 10.1016/j.cell.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 3.Castagnaro L, et al. Nkx2-5(+)islet1(+) mesenchymal precursors generate distinct spleen stromal cell subsets and participate in restoring stromal network integrity. Immunity. 2013;38:782–791. doi: 10.1016/j.immuni.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013;13:118–132. doi: 10.1038/nri3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan JS, Kousis PC, Suliman S, Visan I, Guidos CJ. Functions of notch signaling in the immune system: consensus and controversies. Annu. Rev. Immunol. 2010;28:343–365. doi: 10.1146/annurev.immunol.021908.132719. [DOI] [PubMed] [Google Scholar]
- 6.Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 2011;11:34–46. doi: 10.1038/nri2901. [DOI] [PubMed] [Google Scholar]
- 7.Genestier L, et al. TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus-independent responses. J. Immunol. 2007;178:7779–7786. doi: 10.4049/jimmunol.178.12.7779. [DOI] [PubMed] [Google Scholar]
- 8.Pone EJ, et al. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nat. Commun. 2012;3:767. doi: 10.1038/ncomms1769. doi:10.1038/ncomms1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Litinskiy MB, et al. Antigen presenting cells induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 2002;3:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puga I, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2012;13:170–180. doi: 10.1038/ni.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat. Rev. Immunol. 2013;13:75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 12.Satoh-Takayama N, et al. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J. Exp. Med. 2010;207:273–280. doi: 10.1084/jem.20092029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 14.Bernink JH, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013;14:221–229. doi: 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- 15.Neill DR, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mjosberg JM, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011;12:1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 17.Mjosberg J, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity. 2012;37:649–659. doi: 10.1016/j.immuni.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Luci C, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 2009;10:75–82. doi: 10.1038/ni.1681. [DOI] [PubMed] [Google Scholar]
- 19.Cella M, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. USA. 2010;107:10961–11096. doi: 10.1073/pnas.1005641107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JS, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eberl G, et al. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 23.Takatori H, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiss EA, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- 25.Satoh-Takayama N, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonnenberg GF, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Z, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–2373. doi: 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- 29.Tsuji M, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity. 2008;29:261–271. doi: 10.1016/j.immuni.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Cupedo T, et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009;10:66–74. doi: 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- 31.Buonocore S, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spits H, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 33.Linterman MA, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 2010;207:353–363. doi: 10.1084/jem.20091738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol. 2010;28:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 35.Avery DT, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Invest. 2003;112:286–297. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu VT, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat. Immunol. 2011;12:151–159. doi: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- 37.Codarri L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 38.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 39.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guinamard R, Okigaki M, Schlessinger J, Ravetch JV. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat. Immunol. 2000;1:31–36. doi: 10.1038/76882. [DOI] [PubMed] [Google Scholar]
- 41.Panda S, Zhang J, Tan NS, Ho B, Ding JL. Natural IgG antibodies provide innate protection against ficolin-opsonized bacteria. EMBO J. 2013;32:2905–2919. doi: 10.1038/emboj.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ha SA, et al. Regulation of B1 cell migration by signals through Toll-like receptors. J. Exp. Med. 2006;203:2541–2550. doi: 10.1084/jem.20061041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hepworth MR, et al. Innate lymphoid cells regulate CD4 T-cell responses to intestinal commensal bacteria. Nature. 2013 doi: 10.1038/nature12240. doi:10.1038/nature12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lanier LL. Shades of grey--the blurring view of innate and adaptive immunity. Nat. Rev. Immunol. 2013;13:73–74. doi: 10.1038/nri3389. [DOI] [PubMed] [Google Scholar]
- 45.Sawa S, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 46.Zindl CL, et al. The lymphotoxin LTalpha(1)beta(2) controls postnatal and adult spleen marginal sinus vascular structure and function. Immunity. 2009;30:408–420. doi: 10.1016/j.immuni.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weller S, et al. IgM+IgD+CD27+ B cells are markedly reduced in IRAK-4-, MyD88- and TIRAP- but not UNC-93B-deficient patients. Blood. 2012;120:4992–5001. doi: 10.1182/blood-2012-07-440776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeramilli VA, Knight KL. Development of CD27 marginal zone B cells requires GALT. Eur. J. Immunol. 2013 doi: 10.1002/eji.201243205. doi: 10.1002/eji.201243205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rauch PJ, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335:597–601. doi: 10.1126/science.1215173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szomolanyi-Tsuda E, Welsh RM. T-cell-independent antiviral antibody responses. Curr. Opin. Immunol. 1998;10:431–435. doi: 10.1016/s0952-7915(98)80117-9. [DOI] [PubMed] [Google Scholar]
- 51.Eberl G, Littman DR. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science. 2004;305:248–251. doi: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, et al. Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J. Immunol. 2008;180:1565–1575. doi: 10.4049/jimmunol.180.3.1565. [DOI] [PubMed] [Google Scholar]
- 53.Vonarbourg C, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greter M, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36:1031–1046. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.