

Abstract
Spinal Muscular Atrophy (SMA) is an autosomal recessive disease causing degeneration of lower motor neurons and muscle atrophy. One therapeutic avenue for SMA is targeting signaling pathways in muscle to ameliorate atrophy. Muscle Atrophy F-box, MAFbx, and Muscle RING Finger 1, MuRF1, are muscle-specific ubiquitin ligases upregulated in skeletal and cardiac muscle during atrophy. Homozygous knock-out of MAFbx or MuRF1 causes muscle sparing in adult mice subjected to atrophy by denervation. We wished to determine whether blockage of the major muscle atrophy pathways by deletion of MAFbx or MuRF1 in a mouse model of SMA would improve the phenotype. Deletion of MAFbx in the Δ7 SMA mouse model had no effect on the weight and the survival of the mice while deletion of MuRF1 was deleterious. MAFbx−/−–SMA mice showed a significant alteration in fiber size distribution tending towards larger fibers. In skeletal and cardiac tissue MAFbx and MuRF1 transcripts were upregulated whereas MuRF2 and MuRF3 levels were unchanged in Δ7 SMA mice. We conclude that deletion of the muscle ubiquitin ligases does not improve the phenotype of a Δ7 SMA mouse. Furthermore, it seems unlikely that the beneficial effect of HDAC inhibitors is mediated through inhibition of MAFbx and MuRF1.
Keywords: Spinal muscular atrophy (SMA), muscle ubiquitin ligases, Muscle Atrophy F-box, MAFbx (Atrogin1), and Muscle RING Finger 1, MuRF1 (Trim63)
INTRODUCTION
Spinal Muscular Atrophy (SMA) is an autosomal recessive disorder characterized by loss of motor neurons and atrophy of the muscle [1]. The incidence of SMA is 1 in 10,000 live births, with a carrier frequency of 1 in 40–60 [2, 3]. In humans, the SMA-determining region on chromosome 5q contains two genes Survival Motor Neuron 1 (SMN1) and SMN2. SMA is caused by deletion or mutation in the SMN1 gene and retention of the SMN2 gene [1, 4]. The SMN2 gene contains a single nucleotide change (C→T) in exon 7 that affects a modulator of splicing which results in the exclusion of exon 7 in most transcripts [5–8]. SMN lacking exon 7 does not oligomerize efficiently and thus gets rapidly degraded leading to low levels of SMN in SMA [9–11]. Low levels of SMN lead to the selective death of motor neurons and subsequent muscle atrophy. SMN’s only known function is the assembly of Sm proteins onto snRNAs to form snRNPs [1]. Thus, it has been predicted that low levels of SMN result in the alteration of splicing in genes that are critical for motor neuron function. Alternatively, SMN has been found in axons and while there is no detectable defect in axonal growth in mouse models of SMA, it has been suggested that SMN is important in mRNA transport [12]. To date, the specific function of SMN that is critical for the development of SMA has not been determined [1].
SMA results from a reduction in the level of functional SMN protein, whereas complete absence of SMN is embryonic lethal in any organism [4, 15–17]. The copy number of SMN2 varies in the human population. In general, a greater number of SMN2 copies has been shown to result in a milder SMA phenotype in SMA patients [13, 14]. To model SMA in mice, the murine Smn gene was knocked out and the human SMN2 gene was introduced to provide low levels of SMN protein [18, 19]. The presence of two copies of SMN2 on an Smn null background results in mice with SMA that live 5 days [18]. The addition of the SMNΔ7 transgene into these mice extended survival to 14 days creating the Δ7 SMA mouse model (Smn−/−; SMN2+/+; SMNΔ7+/+) [20].
In SMA patients, the loss of strength in proximal muscles is the phenotypic presentation of the disease. Thus it has been proposed that preventing muscle atrophy could ameliorate the symptoms of muscle weakness in SMA patients [21]. Clinical trials with creatine, phenylbutryate, gabapentin and thyrotropin releasing hormone in type II and III SMA patients showed no significant effect on the disease course [22]. Delivery of follistatin, a negative regulator of muscle growth, improved the muscle mass but had no increase in maximium survival in SMA mice [23]. Moreover, transgenic over-expression of follistatin resulted in increased muscle mass with no improvement in motor function or survival in SMA mice [24]. To date the most effective therapies in SMA mice increase SMN expression by reintroduction of SMN with viral vectors or by blocking negative regulators of splicing in SMN2 with antisense oligonucleotides [25–28]. Increasing SMN levels systemically or in the central nervous system specifically rescues muscle weakness and increases survival in SMA mouse models [26]. Previous studies have shown that early introduction of any SMN-inducing therapy is needed for maximum effect on survival and phenotypic improvement in mice [27, 29, 30].
Muscle Atrophy F-box, MAFbx (also called Atrogin1), and Muscle RING Finger 1, MuRF1 (also called Trim63), are two muscle specific E3 ubiquitin ligases that are required for muscle atrophy [31]. Ubiquitin ligases target the sarcomeric, contractile, signaling, metabolic and transcriptional muscle proteins to the ubiquitin proteasome system (UPS) [32]. The UPS degrades muscle proteins thus maintaining both regular turnover and muscle mass. Upon receiving a signal for atrophy, the ubiquitin ligases are upregulated causing increased breakdown of muscle proteins, tipping the balance towards decrease in muscle mass [33–35]. MAFbx also down-regulates protein synthesis in muscles [34]. The known substrates of MAFbx are MyoD [36] and calcineurin [37]. A second muscle ubiquitin ligase, MuRF1 targets myosin light-chain, MyLC1 and MyLC2, myosin heavy chain (MyHC), myosin-binding protein-C (MyBP-C) [38] and cardiac troponin I [39]. MuRF1 may also have a role in post-transcriptional modification and titin turn over [40]. Homozygous deletion of either MAFbx or MuRF1 results in sparing of muscle mass in mice subjected to atrophy by denervation [31]. The deletion of muscle ubiquitin ligase results in increased muscle weight, and the maintenance of mean fiber size, and fiber size variability [31]. Thus, deletion of MAFbx or MuRF1 has been shown to protect against muscle atrophy in mice.
We proposed that deletion of the ubiquitin ligases in the Δ7 SMA mouse model could ameliorate atrophy in the SMA mouse and result in increased weight and survival. Using MAFbx−/− or MuRF1−/− transgenic mice we deleted the ubiquitin ligases in the Δ7 SMA mouse. We found that loss of MAFbx did not improve the weight or survival of SMA mice, although there was a minimal increase in muscle fiber size. Furthermore, deletion of MuRF1 in the Δ7 SMA mouse actually decreased survival. It has been suggested that HDAC inhibitors act to benefit SMA mice by inhibition of the upregulation of MAFbx and MuRF1 [41]. We measured the expression of MAFbx, MuRF1, MuRF2 and MuRF3, and found increased expression of MAFbx and MuRF1 at postnatal day 14 (PND14), while the expression of MuRF2 and MuRF3 was unchanged in SMA mice. It appears unlikely that HDAC inhibitors act by blocking the upregulation of ubiquitin ligases given that deletion of MAFbx or MuRF1 did not improve survival in Δ7 SMA mice.
MATERIALS AND METHODS
Mouse strains and breeding
The MAFbx−/− and MuRF1−/− mice used for the experiments have been described previously [31]. They were crossed to Δ7 SMA mice (Smn+/−, SMN2+/+, SMNΔ7+/+) [20]. Mouse genotypes were as follows. MAFbx−/−-SMA : MAFbx−/−, Smn−/−, SMN2+/+, SMNΔ7+/+; MuRF1−/−-SMA : MuRF1−/−, Smn−/−, SMN2+/+, SMNΔ7+/+; SMA : MAFbx+/+, MuRF1+/+, Smn−/−, SMN2+/+, SMNΔ7+/+; and MAFbx−/−-Smn+/− : MAFbx−/−, Smn+/−, SMN2+/+, SMNΔ7+/+; MuRF1−/−-Smn+/− : MuRF1−/−, Smn+/−, SMN2+/+, SMNΔ7+/+; Smn+/− Control : MAFbx+/+, MuRF1+/+, Smn+/−, SMN2+/+, SMNΔ7+/+ and Smn+/+ Control : MAFbx+/+, MuRF1+/+, Smn+/+, SMN2+/+, SMNΔ7+/+ as controls. The breeding and maintenance of mice was in accordance to The Institutional Animal Care and Use Committee (IACUC) regulations of The Ohio State University.
Genotyping and weighing
Genomic DNA was isolated for tail clips and PCR amplified using the following primers - MAFbx K/O: 5′-CTTCCTCGTGCTTTACGGTATC and 5′-AGCACAGATATGGTACCTTCC; MAFbx WT: 5′-CTGCAACAAGGAGGTATACAGT and 5′-CATGCAGGTGTACATGCAAGTAG; MuRF1 K/O: 5′-TGGCTACCCGTGATATTGCTG and 5′-CGTTCGAGGGTTAAGAAAGTCTAG; MuRF1 WT: 5′-CGTTCGAGGGTTAAGAAAGTCTAG and 5′-GCACTCCTGCTTGTAGATGTC. The PCR conditions were 95 °C for 5 min, followed by 95 °C for 1 min, 57 °C for 1 min, 72 °C for 1 min, for a total of 35 cycles. The genotyping of SMA mice has been described previously [20]. Pups were weighed from the day of birth (PND01) until weaning (PND21).
Muscle fiber analysis
14 μm thin transverse sections were isolated from the frozen gastrocnemius muscle of PND08 pups and stained with hematoxylin and eosin (H&E) as previously described [42]. The myofiber cross-sections were viewed with a Nikon Eclipse 800 microscope (Nikon Corporation, Japan) and imaged using a Nikon FDX-35 digital camera. The cross-sectional area was measured using SPOT Advanced (v3.5.9) software (Diagnostic Instruments, Inc., MI).
Statistical Analyses
Survival (Kaplan-Meier curve) and statistical analyses (Mann-Whitney Rank Sum Test, Shapiro-Wilk Normality Test and Equal variance test) were done were performed using SigmaPlot v11 (Systat Software Inc., CA).
Digital droplet PCR (ddPCR)
Fresh tissue was flash frozen in liquid nitrogen. RNA was isolated from Trizol (Invitrogen) homogenized tissue and purified using the RNeasy kit (Qiagen). 2.0 μg of RNA was used for the RT-PCR reaction performed using AMV reverse transcriptase (Affymetrix/USB). The quantification of transcripts was done using digital droplet PCR (Bio-Rad). Approximately ten to fourteen thousand droplets, containing the template with the primers and probe, were generated. The measured fluorescence after PCR amplification was used to calculate the concentration of a transcript using Poisson statistics [25] by the software QuantaSoft (Bio-Rad). Relative levels of a transcript were determined with reference to cyclophilin expression. The sequences of the primers and probes were as follows. MAFbx: 5′-TCCTTATGCACACTGGTGCA, 5′-CTCAGCCTCTGCATGATGTTC, Probe-FAM-CAACATTAACATGTGGGTGT-MGB; MuRF1: 5′-AGCTGAGTAACTGCATCTCCATGC, 5′-TTCTGCTCCAGGATGGCGTA, Probe-FAM-CGAGTGCAGACGATCA-MGB; MuRF3: 5′-CACTTGGAGGGCTCCTCAAAG, 5′-AGAGCCTTGCTCCATGCTCTC, Probe-FAM- TGTCGAAGGTGGAGCTG-MGB; Cyclophilin: 5′-GTCAACCCCACCGTGTTCTT, 5′-TTGGAACTTTGTCTGCAAACA, Probe-VIC-CTTGGGCCGCGTCT-MGB. For MuRF2 isomers p50A and p60A, a common reverse primer in exon10 was used; 5′-AGAAGGGGCCTCAAATCCAATC and the forward primers and probes were p50A5′-GAAAGCTGCAGAGCCCTCTCAG, p60A5′-TAGGGCCTCTGGGCATTGAG; p50A Probe-FAM-TCTCCAGAACCGTTTT-MGB, p60A Probe-FAM-CAGTGAGTGGTAAGGAGTC-MGB.
RESULTS
Generation of the transgenic mouse lines
The SMA model mouse described in Le et al. [20] has a homozygous deletion of mouse Smn, and carries two copies of the human SMN2 transgene and the human cDNA expressing SMN but lacking exon 7 (Smn−/−, SMN2+/+, SMNΔ7+/+). Smn+/− mice were crossed to MAFbx and MuRF1 homozygous K/O mice to obtain MAFbx−/− and MuRF1−/− in the SMA background. Smn+/− mice with a homozygous K/O of MAFbx and MuRF1 are phenotypically normal and no different from the Smn+/− control mice. Thus, deletion of MAFbx and MuRF1 has no phenotypic effect in the absence of an atrophy signal.
Weight and survival analyses
Body weight was measured to determine if deletion of the muscle ubiquitin ligases MAFbx and MuRF1 result in muscle sparing in the Δ7 SMA mouse. Homozygous deletion of MAFbx in SMA animals (n=17), resulted in the same pattern of weight gain and loss to that of SMA pups that retain WT copies of MAFbx (n=11) (Fig. 1A). Furthermore, the deletion of MAFbx and MuRF1 in the SMA background did not improve the survival of the mice. There was no improvement in the mean survival time of MAFbx−/−-SMA (14.4±0.4 days, n=17) compared to SMA animals (17.0±0.8days, n=11) (Fig. 1C). The mean survival time of MuRF1−/−-SMA (2.6±0.8 days, n=15) was in fact much less than that of SMA mice (Fig. 1C). The controls, MAFbx−/−-Smn+/−, MuRF1−/−-Smn+/− and Smn+/− Control survived beyond 21 days (n=15 for each). Thus, deletion of MAFbx failed to improve the survival and deletion of MuRF1 significantly decreased the survival of Δ7 SMA mice.
Figure 1.
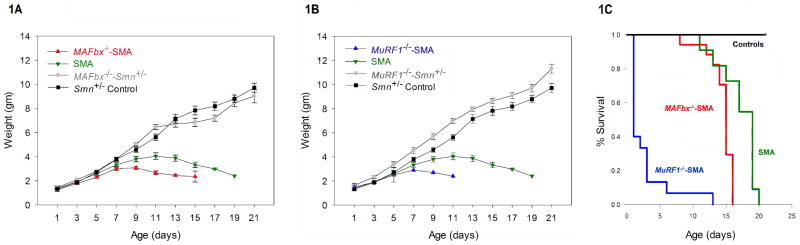
Weight and survival analyses of MAFbx−/−-SMA and MuRF1−/−-SMA animals. (A) MAFbx−/−-SMA (red) mice showed a pattern of weight gain similar to SMA (green) until PND03. (B) MuRF1−/−-SMA (blue) mice showed early death and one mouse survived till PND12. The weights of MAFbx−/−-Smn+/− and MuRF1−/−-Smn+/− controls (gray) were similar to the Smn+/− control (black). (C) The mean survival time of MuRF1−/−-SMA (blue) was 2.6±0.8 days (n=15), MAFbx−/−-SMA (red) was 14.4±0.4 days (n=17) compared to SMA animals (n=11) (green) with 17.0±0.8 days. Thus, deletion of neither MAFbx nor MuRF1 improved the survival in SMA. MAFbx−/−-Smn+/− and MuRF1−/−-Smn+/− (gray) and the Smn+/− control (black) survived for >21 days (n=15). (Log-Rank P = <0.001) (error bars = SEM)
Analyses of muscle morphology
Next, we studied if muscle fiber size was preserved upon deletion of the ubiquitin ligases in the SMA background. The muscle morphology was examined by H&E staining of the gastrocnemius muscle of MAFbx−/−-SMA (Fig. 2A) and SMA (Fig. 2B) mice at PND08. The mean fiber size distribution was measured for each group (n=2070 fibers per group) (Fig. 1C). The muscle fiber size in MAFbx−/−–SMA mice (mean 315±68 μm2, median 225 μm2) was determined to be greater than the fiber size in SMA mice (mean 211±1 μm2, median 204 μm2). The fiber size distribution in MAFbx−/−–SMA reveals a significant increase in large fibers as compared to SMA (P<0.001). MAFbx−/−-Smn+/− and Smn+/− Controls had similar fiber sizes (mean 323±2 μm2, median 312 μm2 and mean 272±2 μm2, median 258 μm2 respectively). The increase in number of large fibers upon deletion of MAFbx in SMA would be expected as the loss of MAFbx will prevent severe atrophy. It should however be noted that median fiber size of MAFbx−/−–SMA is still less than the median fiber size in Smn+/− Control (204 μm2 vs 258μm2). Thus, while deletion of MAFbx in the SMA background does not improve total body weight or survival, it does result in an increase in gastrocnemius fiber size in SMA animals.
Figure 2.

Gastrocnemius muscle fiber of PND08 (A) MAFbx−/−–SMA (B) SMA pups (at 20x magnification) after H&E staining and (C) their corresponding muscle fiber area frequency distributions. MAFbx−/−–SMA: Mean fiber size - 315±68 μm2, median size - 225 μm2 which was higher than that of SMA (mean fiber size 211±1 μm2, median 204 μm2). The difference in the median values between SMA and MAFbx−/−–SMA was significant (Mann-Whitney Rank Sum Test, P<0.001). The fiber size distribution of both SMA and MAFbx−/−–SMA vary significantly from a normal distribution (Shapiro-Wilk Normality Test, P<0.001). For the other controls: MAFbx−/−-Smn+/−: mean fiber size - 323±2 μm2, median size - 312 μm2 while Smn+/− Control: mean fiber size - 272±2 μm2 and median - 258 μm2. (n=2070 for each group)
Expression of the muscle ubiquitin ligases in SMA animals
To investigate why deletion of MAFbx and MuRF1 in the SMA background did not improve weight or survival, we studied the early postnatal expression of the muscle ubiquitin ligases in the Δ7 SMA mouse. Previous studies have shown that in the skeletal muscle, MuRF1 is present embryonically but is upregulated only postnatally [43], yet deletion of MAFbx and MuRF1 have been studied only in adult models of induced atrophy [34]. Two other members of the muscle-specific tripartite-motif (TRIM) family of ubiquitin ligases are MuRF2 and MuRF3. Of the two isoforms of MuRF2 with known timing of expression, p50A and p60A, p50A dominates in embryonic stages with a switch to the p60A isoform postnatally [32, 43]. MuRF1 and MuRF3 are expressed only postnatally [32, 43]. We performed ddPCR to determine if the ubiquitin ligases are upregulated in the Δ7 SMA mouse from PND02 to PND14. Finally, because studies in SMA have shown early heart failure, arrhythmia and cardiac defects [44–46], we examined the levels of the ubiquitin ligases in both skeletal and cardiac muscle.
The expression of MAFbx, MuRF1, MuRF2p50A, MuRF2p60A and MuRF3 were quantified in SMA and Smn+/+ Control (Smn+/+, SMN2+/+, SMNΔ7+/+) mice at PND02, PND05, PND08 and PND14 by digital droplet PCR (ddPCR) (Fig. 3 and 4). Cyclophilin expression was used as an internal control to calculate the relative fluorescence units (RFU). In the skeletal muscle, we found low expression of both MAFbx and MuRF1 levels between SMA and Smn+/+Control mice at time points before PND14 (Fig. 3A). At PND14, MAFbx and MuRF1 expression in SMA mice increase dramatically as compared to Smn+/+ Control: MAFbx increased to 11.8 fold (mean RFU in SMA - 1.04±0.28, mean RFU in Smn+/+ Control - 0.08±0.02, *P=0.04) and MuRF1 increased to 3.9 fold (mean RFU in SMA - 1.8±0.18, mean RFU in Smn+/+ Control - 0.46±0.13, **P=0.01). Similarly, the levels of MAFbx and MuRF1 show an increase at PND14 in SMA in the cardiac tissue (Fig. 3B). MAFbx increased to 2.7 fold and MuRF1 increased to 1.97 fold of Smn+/+ Control’s expression, however the increase did not reach statistical significance (MAFbx: mean RFU in SMA - 1.22±0.32, mean RFU in Smn+/+ Control - 0.45±0.16, MuRF1: mean RFU in SMA - 1.34±0.26, mean RFU in Smn+/+ Control - 0.68±0.12). Thus, the expression of MAFbx and MuRF1 are increased in the SMA model. Finally, we measured the levels of MuRF2 and MuRF3 to determine if they increase postnatally in the Δ7 SMA mouse (Figure 4). We found no significant difference in expression levels of the MuRF2 isoforms (Fig. 4A, B) or MuRF3 (Fig. 4C, D) in the Δ7 SMA mouse in the skeletal and cardiac muscle. Therefore, MuRF2 and MuRF3 appear to be expressed normally at the RNA level in the early postnatal period (PND02-14) in the Δ7 SMA mouse.
Figure 3.
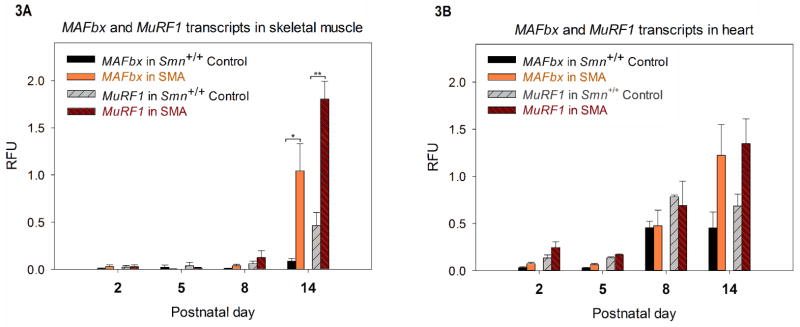
Quantification of MAFbx and MuRF1 transcripts (by ddPCR) at PND01, PND 05, PND08 and PND14 of Smn+/+ Control and SMA animals. (n=3 for each group at each time point) An increase in the mean relative fluorescence units (RFU) for MAFbx and MuRF1 was observed at PND14 in SMA. (A) MAFbx and MuRF1 transcripts in skeletal muscle: MAFbx in Smn+/+ Control and SMA were 0.08±0.02 and 1.04±0.28 respectively at PND14, while for MuRF1 transcripts the mean RFU for Smn+/+ Control was 0.46±0.13 SMA while that for SMA was 1.80±0.18 at PND14. (*P=0.04, **P=0.01) (B) MAFbx and MuRF1 transcripts in heart: For MAFbx, the mean RFU for Smn+/+ Control was 0.45±0.16 v/s 1.22±0.32 for SMA at PND14. For MuRF1, the mean RFU for Smn+/+ Control and SMA were respectively 0.68±0.12 and 1.34±0.26 at PND14. (error bars = SEM)
Figure 4.

Quantification of MuRF2p50A, MuRF2p60A and MuRF3 transcripts (by ddPCR) at PND01, PND 05, PND08 and PND14 of Smn+/+ Control and SMA animals. (n=3 for each group at each time point) There is no significant difference in MuRF2 and MuRF3 transcripts between control and SMA at any time point. MuRF2 transcripts in (A) skeletal muscle and (B) heart. MuRF3 transcripts in (C) skeletal muscle and (D) heart. (error bars = SEM)
DISCUSSION
The prominent feature of SMA in humans is motor neuron loss and subsequent atrophy of muscles [47]. In type 0 SMA which has marked symptoms at birth, the muscle fibers are universally small and atrophied. In type 1 and type 2 SMA there is large grouping of atrophied muscle fibers, indicative of denervation [47]. In type 3 SMA there is less pronounced atrophy of muscle, at least at the stages examined to date and the grouping of fibers is less dramatic with relatively small numbers of atrophied fibers and some predominance of angulated fibers [47]. It is believed that the hypertrophied fibers in type 1 and the normal size fibers are innervated by motor neurons as a result of sprouting and the hypertrophy may be due to these fibers taking over the function of the atrophied fibers [47]. In EMG studies on type 3 cases, there is evidence of sprouting of the surviving motor neurons [48]. Large CMAPs are often found in type 3 patients for a particular muscle [49].
The genes MAFbx (Atrogin 1) and MuRF1 (Trim63) are upregulated upon a signal for atrophy of muscle [31]. Indeed, their upregulation has been observed in 13 distinct models of skeletal muscle injury (denervation, immobilization, hindlimb suspension, lipopolysaccharide injection, sepsis, glucocorticoid dexamethasone, cachectic cytokine interleukin-1 (IL-1) or nutritional deprivation) [34]. In all the studies adult mice, as opposed to neonatal mice, have been examined. Removal of either MAFbx (Atrogin 1) or MuRF1 (Trim63) significantly blocks atrophy from occurring as these E3 ligases are responsible for mediating muscle protein breakdown through the ubiquitin proteasome system. As the genes (MAFbx or MuRF1) have been shown to play a major role in atrophy, we investigated whether removal of these genes in SMA reduced the atrophy of muscle. In the present study we found that deletion of MuRF1 in SMA mice significantly decreased the survival whereas deletion of MAFbx had no impact on weight or survival. The loss of MAFbx in SMA significantly altered the fiber size distribution leading to an increased number of large fibers. Furthermore, the decrease in fiber size occurs before the marked upregulation in MAFbx and MuRF1 expression. This indicates that an alternative pathway is used in SMA to produce small fibers. While MAFbx and MuRF1 play a role at a later stage (PND14), their role in early stages of atrophy is unclear.
Previous studies have shown that the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) improves body weight and survival in the Δ7 SMA mouse model [41, 50]. In addition, the authors found a significant increase in the number and size of myofibers [50]. While TSA does increase SMN expression it also blocks HDAC4 activity, which can change the expression of genes important for the development of atrophy [41, 50]. Furthermore, elevation of MuRF1 and MAFbx was reported in both SMA mice and human muscle tissue from SMA patients [41]. The authors thus proposed that TSA improves SMA muscle pathology by inhibiting the muscle atrophy pathway via downregulation of the muscle ubiquitin ligases [41], and that blocking the atrophy pathway may improve SMA. Interestingly, the activation of MAFbx and MuRF1 occur relatively late (PND11) and well after the effective window for TSA to have a benefit. The first dose of TSA was administered at PND05 [41]. Moreover, there is a marked reduction in muscle fiber size in SMA animals prior to the high expression of MAFbx and MuRF1 indicating that at least the early fiber size reduction is independent of these two genes. In the current paper we show that loss of either MAFbx or MuRF1 in the Δ7 SMA mouse model does not improve the survival. The fiber size distribution in SMA mice with MAFbx deletion tends towards larger fibers, but there is no increase in total body weight or survival. Based on the present work, TSA’s beneficial role in SMA is due to a pathway other than the downregulation of MAFbx and MuRF1.
To analyze the expression pattern of the muscle ubiquitin ligases in a neonatal Δ7 SMA mouse, we quantified the transcripts of MAFbx, MuRF1, MuRF2 and MuRF3 in muscle tissue. MuRF2 isoform p50A is predominantly expressed prenatally with a shift to expression of isoform p60A postnatally, while MuRF1 and MuRF3 levels are upregulated postnatally [32, 43]. The muscle ubiquitin ligases are expressed selectively in the skeletal and cardiac muscle tissue [31]. Since cardiac defects have been implied in the pathology of SMA [44–46], we measured the mRNA levels of the muscle ubiquitin ligases in the cardiac tissue of the Δ7 SMA mouse. Our findings indicate an 11.8 fold increase in the levels of MAFbx and a 3.9 fold increase in the levels of MuRF1 in the skeletal muscle of Δ7 SMA mice at PND14. The levels of MuRF2 and MuRF3 are comparable in control and SMA animals. Thus MAFbx and MuRF1 are upregulated at PND14 yet we find no increase in weight or survival when these genes are deleted in the SMA mice. It could be that the increased expression is too late in the lifespan of the Δ7 SMA mouse to have any effect. It is also possible that other muscle ubiquitin ligases, unknown as of now, might be responsible for atrophy in SMA.
In summary, though the molecular mechanisms of atrophy in the Δ7 SMA mouse seem to involve MAFbx and MuRF1 at late stages (PND14), the genetic deletion of these ubiquitin ligases did not improve the weight or survival of the mice. While significant changes have been found in the muscle of SMA mice, it is unclear if restoration of SMN only to muscle rescues these defects. It is possible that modifying muscle function may have a greater impact in milder forms of SMA, but to date treatments clearly directed to muscle have shown limited efficacy in mouse models of SMA.
Acknowledgments
We thank Regeneron Pharmaceuticals, Tarrytown, NY, for providing us with the MAFbx and MuRF1 knock-out mice. This work was supported by NICHD RO1 HD 060586 grant to AHMB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burghes AH, Beattie CJ. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nature Reviews Neuroscience. 2009;10:597–609. doi: 10.1038/nrn2670. Epub 2009 Jul 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. American journal of medical genetics Part A. 2010;152A:1608–16. doi: 10.1002/ajmg.a.33474. [DOI] [PubMed] [Google Scholar]
- 3.Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. European journal of human genetics: EJHG. 2012;20:27–32. doi: 10.1038/ejhg.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 5.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Human molecular genetics. 1999;8:1177–83. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 6.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6307–11. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nature genetics. 2002;30:377–84. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 8.Gennarelli M, Lucarelli M, Capon F, et al. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochemical and biophysical research communications. 1995;213:342–8. doi: 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 9.Lorson CL, Strasswimmer J, Yao JM, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nature genetics. 1998;19:63–6. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 10.Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Human molecular genetics. 2000;9:259–65. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 11.Burnett BG, Munoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Molecular and cellular biology. 2009;29:1107–15. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGovern VL, Gavrilina TO, Beattie CE, Burghes AH. Embryonic motor axon development in the severe SMA mouse. Human molecular genetics. 2008;17:2900–9. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burghes AH. When is a deletion not a deletion? When it is converted American journal of human genetics. 1997;61:9–15. doi: 10.1086/513913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAndrew PE, Parsons DW, Simard LR, et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. American journal of human genetics. 1997;60:1411–22. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9920–5. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Human molecular genetics. 1997;6:1205–14. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 17.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nature genetics. 1997;16:265–9. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 18.Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Human molecular genetics. 2000;9:333–9. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 19.Hsieh-Li HM, Chang JG, Jong YJ, et al. A mouse model for spinal muscular atrophy. Nature genetics. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 20.Le TT, Pham LT, Butchbach ME, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Human molecular genetics. 2005;14:845–57. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 21.Engvall E, Wewer UM. The new frontier in muscular dystrophy research: booster genes. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2003;17:1579–84. doi: 10.1096/fj.02-1215rev. [DOI] [PubMed] [Google Scholar]
- 22.Bosboom W, Vrancken AF, van den Berg LH, Wokke JH, Iannaccone ST. Drug treatment for spinal muscular atrophy types II and III. The Cochrane database of systematic reviews. 2009:CD006282. doi: 10.1002/14651858.CD006282.pub2. [DOI] [PubMed] [Google Scholar]
- 23.Rose FF, Jr, Mattis VB, Rindt H, Lorson CL. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Human molecular genetics. 2009;18:997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumner CJ, Wee CD, Warsing LC, et al. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Human molecular genetics. 2009;18:3145–52. doi: 10.1093/hmg/ddp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porensky PN, Mitrpant C, McGovern VL, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Human molecular genetics. 2012;21:1625–38. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Science translational medicine. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foust KD, Wang X, McGovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nature biotechnology. 2010;28:271–4. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Dominguez E, Marais T, Chatauret N, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Human molecular genetics. 2011;20:681–93. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 29.Le TT, McGovern VL, Alwine IE, et al. Temporal requirement for high SMN expression in SMA mice. Human molecular genetics. 2011;20:3578–91. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lutz CM, Kariya S, Patruni S, et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. The Journal of clinical investigation. 2011;121:3029–41. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 32.Perera S, Holt MR, Mankoo BS, Gautel M. Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Developmental biology. 2011;351:46–61. doi: 10.1016/j.ydbio.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. The Journal of nutrition. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- 34.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. The international journal of biochemistry & cell biology. 2005;37:1974–84. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 35.Glass DJ. Signaling pathways perturbing muscle mass. Current opinion in clinical nutrition and metabolic care. 2010;13:225–9. doi: 10.1097/mco.0b013e32833862df. [DOI] [PubMed] [Google Scholar]
- 36.Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. The Journal of biological chemistry. 2005;280:2847–56. doi: 10.1074/jbc.M411346200. [DOI] [PubMed] [Google Scholar]
- 37.Li HH, Kedar V, Zhang C, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. The Journal of clinical investigation. 2004;114:1058–71. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen S, Brault JJ, Gygi SP, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. The Journal of cell biology. 2009;185:1083–95. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:18135–40. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. The Journal of cell biology. 2002;157:125–36. doi: 10.1083/jcb.200108089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bricceno KV, Sampognaro PJ, Van Meerbeke JP, Sumner CJ, Fischbeck KH, Burnett BG. Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy mice. Human molecular genetics. 2012;21:4448–59. doi: 10.1093/hmg/dds286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gavrilina TO, McGovern VL, Workman E, et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Human molecular genetics. 2008;17:1063–75. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perera S, Mankoo B, Gautel M. Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. Journal of muscle research and cell motility. 2012;33:107–22. doi: 10.1007/s10974-012-9288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bevan AK, Hutchinson KR, Foust KD, et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Human molecular genetics. 2010;19:3895–905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Human molecular genetics. 2010;19:3906–18. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Human molecular genetics. 2010;19:4059–71. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- 47.Dubowitz V, Sewry CA, Fitzsimons RB. Muscle biopsy: a practical approach. 2. Eastbourne: Baillière Tindall; 1985. p. xiv.p. 720. [Google Scholar]
- 48.Hausmanowa-Petrusewicz I, Karwanska A. Electromyographic findings in different forms of infantile and juvenile proximal spinal muscular atrophy. Muscle & nerve. 1986;9:37–46. doi: 10.1002/mus.880090106. [DOI] [PubMed] [Google Scholar]
- 49.Lewelt A, Krosschell KJ, Scott C, et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle & nerve. 2010;42:703–8. doi: 10.1002/mus.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Avila AM, Burnett BG, Taye AA, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. The Journal of clinical investigation. 2007;117:659–71. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]