

Abstract
Here we report the design and synthesis of a panel of stapled peptides containing a distance-matching biphenyl cross-linker based upon a peptide capsid assembly inhibitor reported previously. Compared with the linear peptide, the biphenyl-stapled peptides exhibited significantly enhanced cell penetration and potent antiviral activity in the cell-based infection assays. Isothermal titration calorimetry and surface plasmon resonance experiments revealed that the most active stapled CAI peptide binds to the C-terminal domain of HIV capsid protein as well as envelop glycoprotein gp120 with low micromolar binding affinities, and as a result, inhibits both the HIV-1 virus entry and the virus assembly.
Keywords: peptides, protein-protein interaction, HIV capsid, virus assembly, virus entry
Helix-mediated protein-protein interactions are attractive drug targets because they mediate diverse fundamentally important biological processes including cell growth, apoptosis, and virus assembly.1 For example, a phage-based selection has led to the discovery of a peptide capsid assembly inhibitor (CAI; sequence = ITFEDLLDYYGP) that binds to the C-terminal domain (CTD) of the HIV capsid protein p24 with a Kd of 15 μM.2 Since p24 is responsible for the oligomerization and assembly of protein envelope surrounding virus genome, the binding of helical CAI to CTD inhibits the assembly of immature HIV-1.3 Due to the lack of cell penetration, however, CAI peptide failed to show antiviral activities in the cell-based assays. To overcome this problem, we previously reported the use of hydrocarbon stapling chemistry4 to convert linear CAI peptide into a cell-penetrating stapled peptide that showed potent and broad antiviral activities.5 In an effort to use simpler stapling chemistries to generate cell-permeable CAI, herein we report the synthesis of stapled CAI peptides using a cysteine-based biaryl cross-linking chemistry and characterization of their antiviral activities in the cell-based single-cycle and multi-cycle infection assays. Furthermore, we found these biaryl-stapled CAI peptides exhibited dual activities in inhibiting both the virus assembly and the virus entry by binding to the C-terminal domain of the capsid protein and the envelop glycoprotein gp120, respectively.
We recently reported the design of distance-matching biaryl cross-linkers suitable for stapling peptides containing cysteines at i,i+7 positions.6 The applications of this stapling chemistry to bioactive peptides have led to increased biological activity, improved cell penetration, and enhanced serum stability.7 To apply biaryl stapling chemistry to CAI, we needed to replace two solvent-exposed residues in CAI with two cysteines. Inspection of the crystal structure of the CAI–CTD complex revealed that the CAI peptide inserts into the deep hydrophobic groove of CTD as an amphipathic α-helix, with the hydrophobic face composed of Phe-3, Leu-6, Leu-7, Tyr-9 and Tyr-10 making van der Waals interaction with the CTD hydrophobic grove (Figure 1). To preserve the hydrophobic contact, we replaced the solvent exposed Glu-4 and Gly-11 with D- and L-cysteine, respectively, to prepare for the installation of the biphenyl cross-linker, Bph (Figure 1b). Previously, we showed that the substitution of Pro-12 with Glu at the C terminus was beneficial as Glu forms a salt bridge with the Arg173 in the CTD. Since CAI is comprised of more than 50% hydrophobic residues, we further appended a Ser-Gly-Ser tail at the C terminus in order to enhance water solubility. A series of Bph-stapled CAI peptides were thus prepared (Table 1, see the Supplemental Information for details). Substitutions were then made at the selected positions in order to probe the importance of the individual residues. The identities of the stapled CAI peptides were confirmed by mass spectrometry (Table S1).
Figure 1.

(a) Crystal structure of CAI bound to CTD (PDB code: 2BUO). CTD is rendered in surface model while CAI is shown in ribbon. The side chains of hydrophobic residues F3, L6, L7, Y9 and Y10 are shown in blue tube while the solvent exposed hydrophilic resides are shown in green tube. (b) Helical wheel diagram of CAI viewed from the N- to C-terminus. The residues suitable for stapling are colored in red, and the chemical structure of Bph is shown.
Table 1.
Sequences of CAI peptides and their antiviral activities in multi-cycle (MT-2) and single-cycle (TZM-b1) assays a
| peptide | sequence | MT-2 cells IC50 (μM) | TZM-b1 cells IC50, (μM) |
|---|---|---|---|
| CAI | ITFEDLLDYYGP | >135 | >135 |
| NYAD-36b | ISFR8ELLDYYS5ESGSc | 1.5 ± 0.17 | 2.0 ± 0.4 |
| 1 | ISFc′ELLDYYC′ESGS | 2.8 ± 0.2 | 1.9 ± 0.2 |
| 2 | ISFc′QLLDYYC′ESGSd | 5.5 ± 1.4 | 3.1 ± 0.3 |
| 3 | ISFc′ELLNYYC′ESGS | 8.8 ± 0.5 | 1.8 ± 0.1 |
| 4 | ISFc′QLLNYYC′ESGS | 5.7 ± 0.1 | 1.6 ± 0.2 |
| 5 | ISFc′ELADYYC′ESGS | 4.1 ± 0.3 | 1.4 ± 0.2 |
| 6 | ISFc′ELLDYAC′ESGS | 14.0 ± 1.1 | 17.3 ± 1.4 |
The antiviral assays were performed three times to obtain average IC50 ± SD.
Activity data were drawn from reference 8.
Hydrocarbon stapling occurs between R8 and S5: R8 = (R)-2-(7′-octenyl)alanine; S5 = (S)-2-(4′-pentenyl)alanine.
c′ and C′ denote Bph-linked D-cysteine and L-cysteine, respectively.
NA, no activity; ND, not determined
To examine whether Bph-stapling leads to increased antiviral activity, we evaluated the stapled CAI peptides in both multi-cycle infection assay and single-cycle neutralization assay using MT-2 and TZM-bl cells, respectively. All stapled peptides exhibited low-micromolar potency (IC50 < 10 μM) with the exception of stapled peptide 6 in which Tyr-10 is replaced by Ala (Table 1). Because Tyr-10 is a key hydrophobic residue involved in the binding to CTD, it is expected that substitution with Ala will result in reduced binding and significant reduction in antiviral activity. Compared to peptide 1, peptides 2–5 with substitutions of Glu-5 by Gln, Leu-7 by Ala, and/or Asp-8 by Asn resulted in modest decreases in antiviral activities (Table 1), likely due to their peripheral locations with respect to the binding interface (Figure 1b). In contrast, linear CAI peptide was inactive in both multi- and single-cycle assays, indicating that Bph-stapling is both necessary and sufficient in generating potent antiviral activities. Among the peptides tested, peptide 1 gave the highest antiviral activity in both assays (Table 1). Importantly, we did not observe any toxicity at the peptide concentrations used in the cell-based assays (CC50 > 100 μM for majority of the stapled peptides in both cell lines, see Table S2 for details), indicating that the reduction in p24 levels (multi-cycle) or luciferase production (single-cycle) in cells treated with the stapled peptides is due to viral inhibition and not to cytotoxicity.
Subsequently, we tested stapled peptide 1 that shows the highest antiviral activity and selectivity index (SI = CC50/IC50) against a panel of pseudo viruses pseudotyped with ENVs from primary HIV-1 isolates subtype A, A/D, A/G, B, C and D reference viruses in the single-cycle assay. These Env-pseudotyped viruses exhibit a neutralization phenotype that is typical of most primary HIV-1 isolates.9 As shown in Table 2, peptide 1 is active against all subtypes of HIV-1 viruses (IC50 = 1.0~17.9 μM). In particular, peptide 1 was most effective against the viruses from subtype B with IC50 values in the range of 1.6 ~ 4.6 μM. Since all the clones share the same CTD but differ in the envelops (ENVs), the discrete sensitivity is likely due to the difference in interacting with the envelope of HIV-1. However, in the absence of the knowledge about the binding site for the stapled peptide, we analyzed the entire gp160 sequences of all the clones and could not identify the specific patterns underlining higher sensitivity of some clones over the others.
Table 2.
Neutralization activity of stapled peptide 1 in single-cycle assay against a panel of HIV-1 ENV pseudotyped reference viruses
| NIH # | ENV clone | Subtype | IC50 (μM) |
|---|---|---|---|
| 11889 | QB726.70M.ENV.B3 | A | ~15.4 |
| 11891 | QF495.23M.ENV.A3 | A | 1.4 ± 0.06 |
| 11892 | QF495.23M.ENV.B2 | A | ~10 |
| 11896 | QH343.21M.ENV.A10 | A | ~15.4 |
| 11901 | QA790.204I.ENV.A4 | AD | 7.0 ± 0.3 |
| 11904 | QA790.204I.ENV.E2 | AD | ~7.7 |
| 11601 | CRF02_AG (Clone 263) | AG | 4.4 ± 0.25 |
| 11591 | CRF02_AG (Clone 211) | AG | 4.0 ± 0.7 |
| 11035 | pREJO4541 clone 67 (SVPB16) | B | 3.4 ± 0.5 |
| 11036 | pRHPA4259 clone 7 (SVPB14) | B | 1.6 ± 0.3 |
| 11038 | pCAAN5342 clone A2 (SVPB19) | B | 4.6 ± 0.5 |
| 11022 | PVO, clone 4 (SVPB11) | B | 3.5 ± 0.1 |
| 11058 | SC422661, clone B (SVPB8) | B | 3.7 ± 0.6 |
| 11306 | Du156, clone 12 (SVPC3) | C | 17.9 ± 1.7 |
| 11311 | ZM233M.PB6, SVPC9 | C | 3.6 ± 0.5 |
| 11315 | ZM135M.PL10a, SVPC15 | C | 1.0 ± 0.2 |
| 11507 | HIV-25925-2, clone 22 | C | 1.1 ± 0.2 |
| 11508 | HIV-26191-2, clone 48 | C | 6.3 ± 0.1 |
| 11912 | QA013.70I.ENV.M12 | D | ~15.4 |
| 11913 | QA465.59M.ENV.A1 | D | ~15.4 |
| 11914 | QA465.59M.ENV.D1 | D | ~7.7 |
| 11916 | QD435.100M.ENV.B5 | D | ~15.4 |
| 11917 | QD435.100M.ENV.A4 | D | 1.0 ± 0.3 |
| 11918 | QD435.100M.ENV.E1 | D | ~15.4 |
To confirm that Bph stapling increases cell-penetration, we further prepared a FITC-labeled stapled peptide 1, and compared its uptake by HEK293T cells to FITC-labeled CAI. Figure 2 shows that cells treated with FITC-1 showed strong intracellular fluorescence compared to cells treated with FITC-CAI, indicating that Bph-stapling is responsible for the cellular uptake of the peptide. The increased cell penetration is likely due to the increased hydrophobicity as the far-UV CD measurement did not show increased helicity for stapled peptide 1 compared to linear CAI peptide (data not shown).
Figure 2.
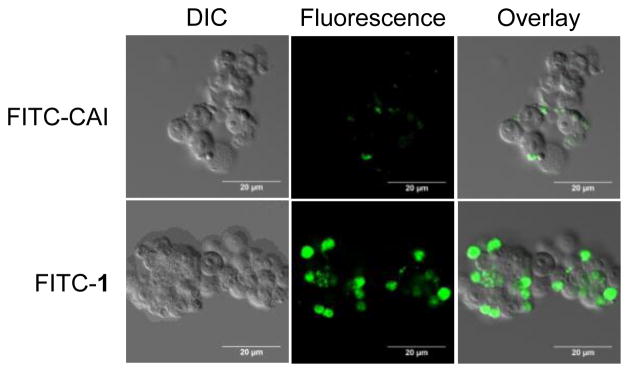
Comparison of the cell-penetration property of CAI and stapled peptide 1 in HEK293T cells. Representative confocal microscopy images of HEK293T cells incubated with 5 μM of FITC-labeled peptide at 37°C for 2 h. DIC = differential interference contrast; scale bar = 20 μm.
Since peptide 1 exhibited the highest activity in inhibiting the virus assembly as indicated in multi-cycle assay, we measured the binding affinity of peptide 1 toward the C-terminal domain (CTD) of the HIV capsid protein using isothermal titration calorimetry (ITC) (Figure 3). The integrated binding isotherm curve was fitted to the single site binding model to yield the peptide 1:CTD binding stoichiometry of 1.35 ± 0.185 along with the thermodynamic parameters ΔH = 1.431 ± 0.259 kcal/mol, ΔS = 18.4 cal/mol·K, and dissociation constant Kd = 17.5 ± 6.2 μM. In contrast, the ITC measurement with the liner CAI peptide gave the CAI:CTD binding stoichiometry of 0.98 ± 0.05, ΔH = −9.45 ± 0.64 kcal/mol, ΔS = −7.91 cal/mol·K, and Kd = 8.3 ± 0.71 μM. Therefore, despite a two-fold reduction in binding affinity toward CTD compared to linear CAI, stapled peptide 1 exhibited greater inhibition of the virus assembly, which can be attributed to its significantly improved cell-penetration (Figure 2).
Figure 3.

Isothermal titration calorimetric analyses of the CTDM184A-W185A–peptide interactions. (a) 625 μM CTDM184A/W185A was titrated in 10 μL aliquots into 20 μM peptide 1. (b) 350 μM CTDM184A/W185A was titrated in 10 μL aliquots into 15 μM CAI. Integrated binding isotherms of the titration (bottom) are fitted to a single-site binding equation.
Since stapled CAI peptides showed higher potency in the single-cycle virus neutralization assay than in the multi-cycle virus infection assay, we suspected that these peptides may also inhibit HIV-1 virus entry in addition to virus assembly. To probe this possibility, we examined whether peptide 1 can bind to HIV-1 envelope glycoprotein gp120 as many of the inhibitors that target the early stage of virus life cycle bind to the HIV-1 envelope glycoprotein.8,10 We employed the surface plasmon resonance (SPR) technique to determine the binding affinity of peptide 1 towards the Yu2gp120 protein (Figure 4). By fitting the resulting sensorgrams to the simple 1:1 (Langmuir) binding model, the association rate constant (ka) was determined to be 7.44 × 102 M−1 s−1 while the dissociation rate constant was determined to be 5.53 × 10−3 s−1. The dissociation constant, Kd, was then calculated to be 7.4 μM, which is surprisingly lower than the dissociation constant against CTD (Kd = 17.5 ± 6.2 μM) as measured by ITC (Figure 3), suggesting that the stapled CAI peptide can impede HIV-1 virus entry through an unexpected binding to the envelop glycoprotein. Since gp120 does not share sequence or structure homology with the CTD of p24, it remains to be determined to which region of gp120 the stapled peptide 1 binds. Similar inhibition of HIV-1 virus entry was also observed with the hydrocarbon stapled CAI peptides such as NYAD-36 (Table 1).8
Figure 4.

Kinetic analysis of stapled peptide 1/gp120 interaction by surface plasmon resonance (SPR). Kinetic data were collected for different concentrations of peptide 1 to a gp120 surface. Yu2 full-length gp120 was immobilized on a CM5 sensor chip, and 2-fold dilutions of peptide 1 were allowed to pass over the sensor chip.
In summary, we have successfully extended the cysteine-based biaryl stapling chemistry to the design of cell-permeable, stapled CAI peptides that showed potent and broad antiviral activities. ITC and SPR-based mechanistic studies indicated that the most potent stapled CAI peptide inhibits HIV-1 virus entry by binding to the envelop glycoprotein gp120 as well as the virus assembly by binding to the C-terminal domain of the HIV-1 capsid protein. Since the stapled peptide 1 only showed modest binding affinities toward CTD (Kd = 17.5 μM) and gp120 (Kd = 7.4 μM), the enhanced activity observed in the cell-based assays (IC50 = 1–2 μM) suggests that the binding of these two targets has synergistic effect on inhibiting HIV virus replication. This unique dual-binding mode is particularly attractive for designing potent and selective antiviral therapies targeting the multiple steps of the HIV-1 life cycle.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Pardee Foundation and the Oishei Foundation (to Q.L.) and the National Institutes of Health (AI 091604 to A.K.D.) for financial support.
Footnotes
Supplemental tables and experimental procedures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Yin H, Hamilton AD. Angew Chem Int Ed. 2005;44:4130. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]; (b) Verdine GL, Walensky LD. Clin Cancer Res. 2007;13:7264. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 2.Sticht J, Humbert M, Findlow S, Bodem J, Müller B, Dietrich U, Werner J, Kräusslich HG. Nat Struct Mol Biol. 2005;12:671. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- 3.Ternois F, Sticht J, Duquerroy S, Krausslich H-G, Rey FA. Nat Struct Mol Biol. 2005;12:678. doi: 10.1038/nsmb967. [DOI] [PubMed] [Google Scholar]
- 4.(a) Blackwell HE, Grubbs RH. Angew Chem Int Ed. 1998;37:3281. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; (b) Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891. [Google Scholar]
- 5.Zhang H, Bhattacharya S, Tong X, Waheed AA, Hong A, Heck S, Curreli F, Goger M, Cowburn D, Freed EO, Debnath AK. J Mol Biol. 2008;378:565. doi: 10.1016/j.jmb.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muppidi A, Wang Z, Li X, Chen J, Lin Q. Chem Commun. 2011;47:9396. doi: 10.1039/c1cc13320a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Muppidi A, Li X, Chen J, Lin Q. Bioorg Med Chem Lett. 2011;21:7412. doi: 10.1016/j.bmcl.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang HG, Lin Q. J Am Chem Soc. 2012;134:14734. doi: 10.1021/ja306864v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H, Curreli F, Waheed AA, Mercredi PY, Mehta M, Bhargava P, Scacalossi D, Tong X, Lee S, Cooper A, Summers MF, Freed EO, Debnath AK. Retrovirology. 2013;10:136. doi: 10.1186/1742-4690-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Mascola JR, D’Souza P, Gilbert P, Hahn BH, Haigwood NL, Morris L, Petropoulos CJ, Polonis VR, Sarzotti M, Montefiori DC. J Virol. 2005;79:10103. doi: 10.1128/JVI.79.16.10103-10107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. J, Virol. 2005;79:10108. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Virology. 2005;339:213. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]; (b) Madani N, Schön A, Princiotto AM, Lalonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Kwon YD, Huang CC, Wyatt R, Kwong PD, Freire E, Smith AB, 3rd, Sodroski J. Structure. 2008;16:1689. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. Proc Natl Acad Sci USA. 2012;109:5663. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang T, Zhang Z, Wallace OB, Deshpande M, Fang H, Yang Z, Zadjura LM, Tweedie DL, Huang S, Zhao F, Ranadive S, Robinson BS, Gong YF, Ricarrdi K, Spicer TP, Deminie C, Rose R, Wang HG, Blair WS, Shi PY, Lin PF, Colonno RJ, Meanwell NA. J Med Chem. 2003;46:4236. doi: 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.