

Highlights
-
•
EPAC1 promotes AP1-dependent transcription in HUVECs.
-
•
Delineation of the minimal, EPAC1-responsive SOCS3 promoter in human cells.
-
•
Phosphorylated c-Jun (Ser 63) is constitutively associated with the SOCS3 promoter.
-
•
Phosphorylation of c-Jun on Ser63 requires activation of PKA, but not EPAC1.
-
•
c-Jun is required for SOCS3 induction by cyclic AMP in MEFs.
Keywords: Cyclic AMP, Transcription, SOCS3, EPAC1, c-Jun, AP1
Abstract
The cyclic AMP sensor, EPAC1, activates AP1-mediated transcription in HUVECs. Correspondingly, induction of the SOCS3 minimal promoter by EPAC1 requires a single AP1 site that constitutively binds phosphorylated (Ser63) c-Jun in DNA-pull-down assays. c-Jun (Ser63) becomes further phosphorylated following cyclic AMP stimulation and specific activation of protein kinase A (PKA), but not through selective activation of EPAC1. Moreover, despite a requirement for c-Jun for SOCS3 induction in fibroblasts, phospho-null c-Jun (Ser63/73Ala) had little effect on SOCS3 induction by cyclic AMP in HUVECs. AP1 activation and SOCS3 induction by EPAC1 in HUVECs therefore occur independently of c-Jun phosphorylation on Ser63.
1. Introduction
Elevations in intracellular cyclic AMP in response to activation of adenosine and prostaglandin receptors in human umbilical vein endothelial cells (HUVECs) leads to inhibition of interleukin 6 (IL-6) signalling complex activation of STAT3 transcription factors and the mitogen-activated protein (MAP) kinase, ERK [1]. This inhibition occurs independently of the classical route for cyclic AMP signalling, through protein kinase A (PKA), but is rather dependent on the induction of the suppressor of cytokine signalling 3 (SOCS3) gene in response to activation of exchange protein activated by cyclic AMP (EPAC) 1 [1]. SOCS3 is an E3 ubiquitin ligase component that targets IL6 signalling components for proteolytic degradation [2], whereas EPAC1 is a specific guanine nucleotide exchange factor (GEF) for the Ras GTPase homologues Rap1 and Rap2, which EPAC1 activates independently of PKA [3]. Cyclic AMP binding sites in EPAC proteins facilitate their direct activation by cyclic AMP, thereby relieving auto-inhibitory influences of the cyclic nucleotide binding domain (CNBD) toward the catalytic GEF domain [3]. Recent research now implicates EPAC1 in the regulation of multiple inflammatory processes in vascular endothelial cells, like HUVECs, including regulation of endothelial cell–cell junction stability [4] and activation of integrins [5], reduction in endothelial permeability and down-regulation of IL-6-mediated inflammatory processes [1]. The involvement of EPAC1 in the activation of multiple protective mechanisms in one cell type highlights the importance of EPAC1 for the proper functioning of VECs and presents an intriguing model by which distinct cellular processes interact to initiate a co-ordinated program of protective, anti-inflammatory events.
Given the importance of EPAC1 in limiting pro-inflammatory IL6 signalling, we have begun to further delineate the molecular basis of how elevations in intracellular cyclic AMP positively control the expression of the SOCS3 gene. Our initial findings implicated mobilisation of CCAAT enhancer binding protein (C/EBP) transcription factors, C/EBPβ and C/EBPδ, which are sufficient to induce SOCS3 gene induction in HUVECs following EPAC1 activation [6]. Although EPAC1 activation is sufficient to induce SOCS3 expression in HUVECs, maximal SOCS3 gene expression also requires supporting activity from the MAP kinases, ERK and JNK [7,8]. In this case ERK is activated by cyclic AMP, independently of both PKA and EPAC1 [7], and this is required for the phosphorylation and activation of multiple transcription factors associated with the SOCS3 promoter, including C/EBPβ (Thr235), STAT3 (Ser727) and SP3 (Ser73) [9]. JNK activation is also promoted by cyclic AMP and principally leads to activation of the AP1 transcriptional complex component c-Jun through phosphorylation on Ser63 [8]. In light of recent reports implicating EPAC proteins as regulators of JNK activity in diverse cell types [10,11], here we address whether EPAC1 is involved in a similar regulation of AP1 activity through c-Jun phosphorylation in HUVECs.
2. Materials and methods
2.1. Materials
Primary antibodies to total c-Jun, total STAT3, phospho-STAT3 (Tyr705), phospho-c-Jun (Ser63) and anti-β-tubulin were purchased from New England Biolabs. Anti-SOCS3 antibody was from Santa Cruz Biotechnology. Secondary antibodies, anti-rabbit, anti-goat and anti-mouse IgG conjugated with horse radish peroxidase (HRP), were purchased from GE Healthcare. Forskolin, rolipram and MG132 were purchased from Merck/Calbiochem. 8-(4-Chlorophenylthio)-2′-O-methyladenosine-3′, 5′-cyclic monophosphate (007) and N6-Benzoyladenosine-3′, 5′-cyclic monophosphate (6-Bnz) were purchased from Biolog, Bremen, Germany. Recombinant human interleukin 6 (IL-6) and soluble IL6 receptor (sIL-6Rα) were bought from R&D Systems Europe Ltd. (Abingdon, UK).
2.2. Plasmids
The AP1 reporter construct (pMN34, containing 7 AP1 repeats) was provided by Professor Walter Kolch (University College Dublin, Republic of Ireland). Mouse SOCS3 promoter constructs were a generous gift from Professor Johannes Bode (Heinrich-Heine University, Dusseldorf, Germany) with permission from Professor Shlomo Melmed (Cedars-Sinai Medical Centre, Los Angeles, USA). These included pGL3-SOCS3-159 Luc, which contains the promoter region −159 to +929 of the murine SOCS3 gene fused to the coding region of firefly luciferase as described previously [12], as well as promoter truncates -107Luc, -79Luc, -68Luc, and -49Luc and pGL3-SOCS3-107Luc constructs mutated to disrupt the putative AP1 site as described [9]. Plasmids (pcDNA3) expressing wild type human c-Jun or phospho-null mutant, c-Jun (Ser63/73Ala), were generous gifts from Professor Christopher Hughes (University of California Irvine, USA).
2.3. Luciferase assays
Human umbilical vein endothelial cells (HUVECs) were grown in human endothelial cell growth medium 2 (PromoCell Heidelberg, Germany) at 37 °C in humidified 5% (v/v) CO2. Cultures of 80–90% confluent HUVECs in 12-well culture clusters were transfected with 0.125 μg Renilla Luciferase reporter construct (pGL4.74) plus 1.125 μg of murine SOCS3-Luc promoter constructs using Lipofectamine 2000 (Life Technologies, UK). Transfected cells were then incubated with treatments overnight at 37 °C and luciferase assays carried out the next day using the Promega Dual Luciferase Reporter Assay System, according to the manufacturer’s protocols. Luciferase activities were measured using a BMG Labtech luminometer. Mouse embryo fibroblasts (MEFs) from wild type and c-Jun−/− (KO) mice were provided by Professor David Gillespie (Beatson Institute of Cancer Research, Glasgow, Scotland) and cultured as described [13].
2.4. c-Jun transfection and immunoblotting
HUVECs were grown to 80–90% confluence and then transfected with human c-Jun or c-Jun (Ser63/73Ala) using Lipofectamine 2000 according to the manufacturer’s instructions. Cell lysates were then prepared in sample buffer (50 mM Tris–HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 1% (v/v) β-Mercaptoethanol, 12.5 mM EDTA, 0.02% (w/v) bromophenol blue, 100 mM DTT). Protein samples were then separated by SDS–PAGE on 10% (w/v) gels and then transferred to nitrocellulose membranes. These were blocked for 1 h at room temperature in 5% (w/v) BSA and then immunoblotted with antibodies specific for SOCS3, tubulin, total c-Jun, STAT3 and phospho-STAT3 (Tyr705) and visualised using ECL chemiluminescence protocols (GE Healthcare).
2.5. DNA pull-down assays
Biotin-modified oligonucleotides were synthesised (Yorkshire Biosciences) that corresponded to proximal promoter sequences from the human SOCS3 gene that include putative AP1 and STAT transcription factor binding sites (Fwd 5′-CGAGTAGTGACTAAACATTACAAGA and Rev 5′-TCTTGTAATGTTTAGTCACTACTCG) or with the AP1 site disrupted (Fwd 5′-CGAGTAAAGCTTAAACATTACAAGA and Rev 5′-TCTTGTAATGTTTAAGCTTTACTCG). Double-stranded oligonucleotides were prepared by mixing equal amounts of forward (biotin-labelled) and reverse oligonucleotide at 100 °C for 1 h and then slow cooling to room temperature (∼30 min). Nuclear extracts were isolated from stimulated HUVECs using a Nuclear Extract Kit (Active Motif). Protein concentrations in nuclear extracts were normalised following Bradford assay, and then DNA pull-down experiments using biotinylated probes were performed as previously described [14]. Oligonucleotides were recovered using streptavidin-agarose beads, which were then boiled in Laemmli sample buffer and analysed by immunoblotting.
2.6. Statistics
Data was analysed using one-way analysis of variance (ANOVA) with a Tukey–Kramer post-test.
3. Results
We previously found that EPAC1 activation by cyclic AMP in human umbilical vein endothelial cells (HUVECs) leads to mobilisation of C/EBP transcription factors and up-regulation of the gene encoding suppressor of cytokine signalling 3 (SOCS3) [6]. Since SOCS3 induction occurs independently of the classical route of cyclic AMP-mediated transcription, namely the phosphorylation of CREB by PKA [15], this represents a novel mechanism for cyclic AMP-mediated transcription. Intriguingly, we recently found that elevations in cyclic AMP in HUVECs also leads to the activation of JNK MAP kinase, which also appears to be a requirement for SOCS3 transcription [8]. However the mechanisms underlying this remain unclear. Normally JNK activates the oncoprotein c-Jun through phosphorylation on Ser 63 and Ser73, leading to activation of the activator protein-1 (AP1) transcription factor complex as a homodimer, or as a heterodimer with Fos [16]. With this in mind we investigate here whether c-Jun and AP1 contribute to the regulation of SOCS3 induction in response to cyclic AMP in HUVECs.
In order to test whether cyclic AMP and EPAC1 activation leads to activation of AP1 transcription factors, we transfected HUVECs with a luciferase gene reporter construct expressing multiple copies of the AP1 transcription factor binding motif, and then stimulated cells with the EPAC-specific cyclic AMP analogue, 8-pCPT-2 -O-Me-cAMP (007), the PKA-specific agonist, 6-Bnz (10 μM and 500 μM), or a combination of the adenylate cyclase activator forskolin and the cyclic AMP-specific phosphodiesterases inhibitor, rolipram, (F/R). As a positive control cells were also stimulated with the phorbol ester, phorbol 12-myristate 13-acetate (PMA), which is a well-known activator of AP1 [17]. In these experiments 007 served as a specific activator of the EPAC1 isoform, since EPAC2 is not expressed in HUVECs [7]. Moreover, we chose to stimulate cells with forskolin in combination with the PDE4 inhibitor, rolipram, rather than a general PDE inhibitor, such as IBMX, because PDE4s have been shown to be intimately linked with the regulation of EPAC1 in HUVECs [18]. We found that similar to PMA, both 007 (10 μM) and F/R provoked a significant induction of AP1 activity with, surprisingly, 10 μM 007 provoking an activation of AP1 which was significantly greater than that induced by F/R treatment (Fig. 1a). In contrast, stimulation with 10 μM 6-Bnz did not promote a significant increase in AP-1 activity and it was only until a concentration of 500 μM 6-Bnz was applied that a significant effect was observed, which suggests an off-target effect of 6-Bnz at high concentrations. Together, these results suggest that AP1 activity is exquisitely sensitive to EPAC1, and not PKA activation in HUVECs.
Fig. 1.

EPAC1 activation promotes AP1-dependent transcription of the SOCS3 gene in HUVECs. (a) HUVECs were transfected with an AP1 luciferase reporter construct and then stimulated with the EPAC-specific agonist 007 (10 μM), the PKA-specific agonist 6-Bnz (at either 10 μM or 500 μM, as indicated), a combination of the adenylate cyclase activator forskolin (10 μM) and the cyclic AMP-specific phosphodiesterase inhibitor rolipram (10 μM), or the protein kinase C activator, PMA (100 nM). Cell extracts were then prepared and luciferase activities measured as described in Section 2. Significant differences relative to diluent-treated cells are indicated, ∗∗∗P < 0.001 (n = 3), as are significant increases in 007-treated cells compared to those treatment with 6-Bnz or F/R, ###P < 0.001. (b) HUVECs were transfected with luciferase constructs containing deletions and truncations of the murine SOCS3 promoter. Cells were then stimulated for 16 h with either F/R or 007 (lower panel). Cells extracts were then assayed for luciferase activity. Significant differences relative to diluent-treated cells are indicated, ∗∗∗P < 0.001, as are significant decreases in luciferase activity relative to F/R-treated cells, ###P < 0.001 (n = 3). (c) HUVECs were transfected with the minimal murine SOCS3 promoter or the minimal promoter with disruptive deletions in the putative AP1 site. Cells were then stimulated with either 6-Bnz (500 μM) or 007 (10 μM). Cell extracts were then assayed for luciferase activity and significant differences relative to diluent-treated cells are indicated ∗∗∗P < 0.001; as are significant differences relative to 007-treated cells transfected with minimal promoter vs. with AP1-mutated constructs, ###P < 0.001 (n = 3).
We have previously shown that transcriptional activation of SOCS3 in COS1 cells in response to PMA treatment requires an AP1, a distal STAT, a proximal STAT and an SP3 transcription factor binding site, all of which are found within the minimal SOCS3 promoter [9]. We therefore transfected HUVECs with luciferase reporter constructs containing deletions of the murine SOCS3 promoter where the individual AP1, STAT and SP3 DNA binding sites had been removed sequentially (Fig. 1b). Transfected cells were then stimulated with F/R or 007 and luciferase activities determined. We found that F/R treatment provoked activation of minimal promoter construct containing all four transcription factor binding sites. However, the combined deletion of the AP1 and distal STAT sites abolished the response to F/R treatment (Fig. 1b), and also to 007 treatment (Fig. 1b, lower panel). Moreover, individual ablation of the AP1 site alone was sufficient to ablate the response of the minimal SOCS3 promoter to both 007 and F/R treatment (Fig. 1b, lower panel). In contrast, we found that induction of the minimal SOCS3 promoter by high concentrations of 6-Bnz (500 μM) was not affected by deletion of the AP1 site, indicating that at these concentrations 6-Bnz acts independently of AP1 to induce SOCS3 expression (Fig. 1c). We also found that, unlike 007, lower concentrations of 6-Bnz (10 μM) were unable to induce activation of the SOCS3 minimal promoter (results not shown). Together, these results suggest that induction of SOCS3 transcriptional activity following EPAC1 activation, but not PKA activation, requires a distinct AP1 site within the SOCS3 minimal promoter in HUVECs. It should be noted that in Fig. 1 007 produces an activation of the AP1 reporter that is greater than that induced by F/R. In contrast, in Fig. 1b (lower panel), F/R is more effective than 007 at inducing induction of the SOCS3 minimal promoter. This is probably due to the fact that F/R activates a range of transcription factors to induce SOCS3 gene expression [8,9], through the activation of ERK and JNK MAP kinase pathways [8,9], whereas SOCS induction in response to 007 appears only to require induction of AP1 activity. This may help explain why the SOCS3 promoter is more responsive to F/R treatment and why AP1 activity is more sensitive to stimulation with 007 than with F/R.
Given that the transcription factor c-Jun is a constituent of AP1 homodimers and heterodimers, we next determined whether c-Jun associates with the putative AP1 binding site in the SOCS3 minimal promoter and whether it becomes activated following EPAC1 or PKA activation. We therefore synthesised two biotinylated DNA probes corresponding to the section of the SOCS3 promoter containing the putative AP1 and distal STAT binding sites (SOCS3 WT; Fig. 2). We also synthesised a corresponding DNA probe where the AP1 site had been disrupted by targeted substitution of nucleotides in the consensus AP1 binding site (SOCS3 ΔAP1; Fig. 2). The two biotinylated probes were then used in DNA pull-down assays with nuclear extracts from HUVECs that had been stimulated with either 007 (10 μM), the PKA-specific cyclic AMP analogue, 6-Bnz (10 μM) or F/R (Fig. 2). DNA precipitates were then immunoblotted with antibodies that recognise c-Jun phosphorylated on the JNK-target site, Ser 63 (pc-Jun (Ser 63)), or antibodies that recognise either total c-Jun or STAT3 proteins. Results indicated that STAT3 associated with both the SOCS3 WT DNA and SOCS3 ΔAP1 probes, and levels remained unchanged following stimulation (Fig. 2). In contrast, whereas c-Jun was seen to associate with the SOCS3 WT in a partially active form, as indicated by the lower mobility of probe-associated c-Jun protein and positive staining with the pc-Jun (Ser63) antibody, little pc-Jun (Ser63) immunoreactivity was seen to associate with the SOCS3 ΔAP1 probe (Fig. 2). Despite this, a corresponding reduction in total c-Jun immunoreactivity associated with the SOCS3 ΔAP1 probe meant that the ratio of pc-Jun (Ser 63):total c-Jun was relatively unchanged (Fig. 2; lower panel). However, the appearance of a high molecular weight form of total c-Jun associated with the SOCS3 ΔAP1 probe (Fig. 2; upper panel), which might represent an inactivated, hyper-phosphorylated form of c-Jun [8], indicates that although the relative level of Ser63 phosphorylation is unchanged following disruption of the SOCS3 AP1 site, most of the probe-associated c-Jun is in an inactive form. Together, these results suggest that the putative AP1 binding site in the SOCS3 minimal promoter has the ability to interact with active c-Jun. Moreover, activation of EPAC1 with 007 had essentially no effect on the amount of active, Ser63-phosphoryted c-Jun associated with the SOCS3 AP1 site. However activation of PKA with 6-Bnz-cAMP (10 μM), or activation of both EPAC1 and PKA with F/R, lead to an increase in the amount of pc-Jun (Ser63) associated with the SOCS3 WT probe (Fig. 2). This suggests that cyclic AMP-stimulated phosphorylation and activation of c-Jun associated with the SOCS3 promoter requires PKA, but not EPAC1.
Fig. 2.
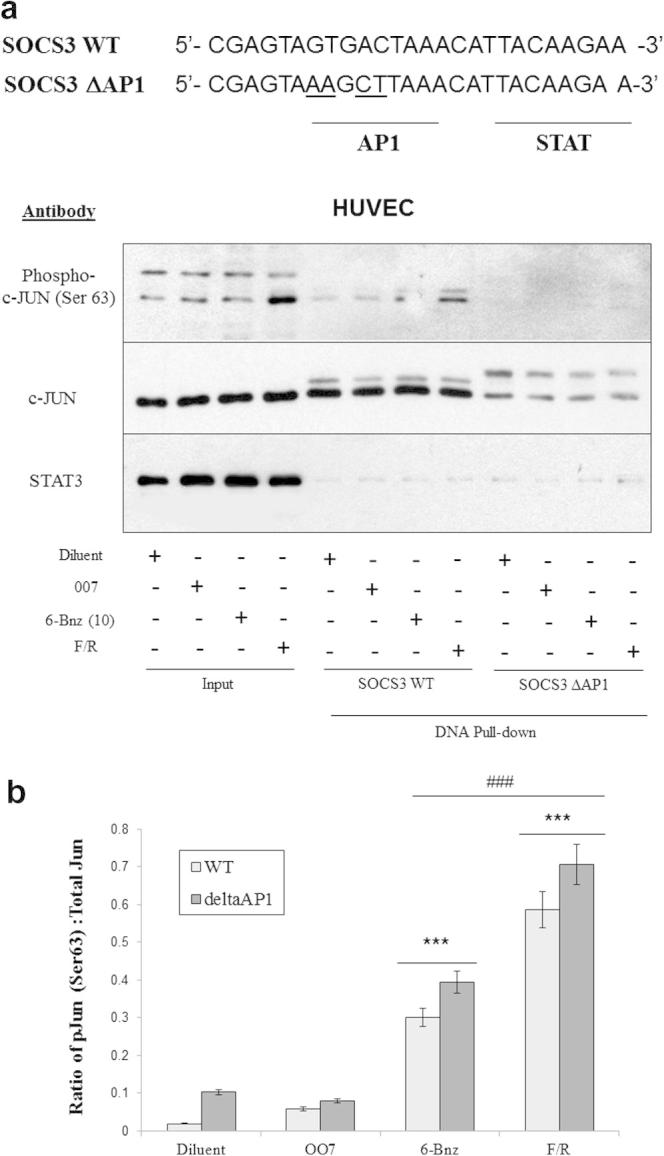
Activation of PKA, but not EPAC1, promotes AP1-associated c-Jun activation in HUVECs. HUVECs were stimulated for 5 h with either 007 (10 μM), the PKA-activator, 6-Bnz (10 μM), or F/R. Nuclear extracts were then prepared and precipitated with the biotinylated oligonucleotides detailed in the upper panel. Cell extracts (input) and precipitated proteins were then Western blotted with the indicated antibodies. Results are representative of an experiment carried out on three separate occasions and densitometic analysis of multiple immunoblots is displayed as a histogram in the lower panel as the ratio of pJun (Ser 63) to total Jun immunoreactivity. Significant differences relative to diluent-treated cells are indicated, ∗∗∗P < 0.001, as is the significant decrease in immunoreactivity in samples from 6-Bnz-treated cells relative to F/R-treated cells, ###P < 0.001 (n = 3).
These results raise the question as to the requirement for c-Jun in the control of SOCS3 induction in response to elevations in intracellular cyclic AMP. We previously demonstrated that the transcription factors C/EBPβ and C/EBPδ are both required and sufficient for induction of the SOCS3 gene in HUVECs following EPAC1 activation [19]. We therefore examined whether the same is true for c-Jun. We therefore transfected HUVECs with cDNAs encoding wild type human c-Jun (c-Jun WT) or human c-Jun, where Ser63 and Ser73 had been mutated to alanine (c-Jun AA; Fig. 3a). Cells were then stimulated with F/R, in the presence or absence of the proteasome inhibitor, MG132, to inhibit proteolytic digestion of SOCS3 protein following its synthesis, thereby stabilising its expression, as described [1]. We found that F/R provoked a robust induction of SOCS3 protein in the presence of MG132, which was unaffected by transfection of cells with either c-Jun WT or c-Jun AA (Fig. 3a). In contrast, we did find that over-expression of both c-Jun WT or c-Jun AA enhanced the ability of F/R to suppress IL6-promoted STAT3 phosphorylation in HUVECs (Fig. 3b). Finally, to test whether c-Jun is required for cyclic AMP-induced SOCS3 expression we used mouse embryonic fibroblasts (MEFs) from wild type mice and animals where both alleles for c-Jun had been deleted (KO; Fig. 3c). We found that in the presence of MG132, F/R stimulation of WT cells led to a robust induction of SOCS3 expression, which was completely ablated in c-Jun KO MEFs (Fig. 3c). Moreover, transfection of c-Jun KO MEFS with either c-Jun WT or c-Jun AA cDNAs rescued the ability of F/R to induce SOCS3 expression (Fig. 3c). Together these results indicate that c-Jun is required, but alone insufficient, to induce SOCS3 gene expression in response to elevations in intracellular cyclic AMP.
Fig. 3.

c-Jun is required, but not sufficient, to promote SOCS3 induction by cyclic AMP. (a) HUVECs were transfected with vectors expressing human c-Jun or c-Jun where Ser63 and Ser73 had been converted to alanine (Ser63/73Ala). Cells were then stimulated with F/R (5 h) in the presence or absence of the proteasome inhibitor MG132 to stabilise SOCS3 expression. Cell extracts were then immunoblotted with the indicated antibodies and denstitometric values are displayed as a histogram in the lower panel. Significant increases in SOCS3 immunoreactivity relative to diluent-treated cells are shown, ∗P < 0.05 and ∗∗∗P < 0.001, as is the significant in increase in immunoreactivity in F/R + MG132 cells relative to cells treated with MG132 alone (###P < 0.001, n = 3). (b) HUVECs were transfected with wild type human c-Jun or mutant c-Jun (Ser63/73Ala), and then stimulated with combinations of F/R and IL6 for 5 h as indicated. Cell extracts were then immunoblotted with antibodies that recognise c-Jun, total STAT3 and STAT3 phosphorylated on Tyr705. Densitometric analysis of immunoblots was carried out and results are presented as a histogram in the lower panel. Significant increases in pSTAT3 (Tyr 705) immunoreactivity relative to diluent-treated cells are shown, ∗∗∗P < 0.001, as are significant decreases in immunoreactivity in c-Jun transfected cells, relative to mock-transfected cells (###P < 0.001, n = 3). (c) Wild type or c-Jun knockout mouse embryonic fibroblasts (MEFs) were transfected with or without (mock) c-Jun or c-Jun (Ser63/73Ala) and then stimulated in the presence or absence of F/R (5 h) plus MG132, as indicted. Cells extracts were then prepared and immunoblotted with the indicated antibodies. Densitometric values from immunoblots are displayed in the lower panel where significant increases in SOCS3 immunoreactivity relative to diluent-treated cells are shown, ∗∗∗P < 0.001 (n = 3).
4. Discussion
The principle findings of this study are that EPAC1 induces activation of AP1-mediated transcription and that AP1 is a requirement for the induction of the SOCS3 gene by EPAC1 in HUVECs. Our work also shows that the principle AP1 component, c-Jun, although required for SOCS3 induction by cyclic AMP in MEFs (Fig. 3c), is not sufficient for SOCS3 induction in HUVECs (Fig. 3a). This is in contrast to C/EBP transcription factors that are absolutely required and sufficient for SOCS3 induction in response to EPAC1 activation in HUVECs [6]. This suggests that c-Jun may therefore play a supporting role in controlling the activation of the SOCS3 promoter in response to C/EBP activation. In this respect, it has been reported that c-Jun can form heterodimers with C/EBP isoforms that promote the transcription of the PU.1 promoter during monopoiesis [20]. Whether similar Jun/C/EBP dimers regulate SOCS3 induction in HUVECs remains to be determined.
With regards to cyclic AMP-dependent c-Jun phosphorylation on the SOCS3 promoter, this appears to be regulated through PKA-, rather than EPAC1-dependent mechanisms, since the PKA-selective agonist 6-Bnz (10 μM), but not 007, was able to promote Ser63 phosphorylation in DNA-pull-down assays (Fig. 2). Moreover, although PKA can promote phosphorylation of Ser63, this phosphorylation appears to be dispensable for the induction of SOCS3 expression in both HUVECs (Fig. 3a) and MEFs (Fig. 3c). Accordingly, AP1 activation and SOCS3 promoter induction was only promoted by 500 μM 6-Bnz in HUVECs (Fig. 1a and c). Together, this indicates a PKA-independent mode of action of cyclic AMP on AP1-dependent SOCS3 induction and suggests that for EPAC1 to induce SOCS3 expression it must either activate c-Jun through Ser63-independent mechanisms or promote dimerization of c-Jun with another active component of the AP-1 complex, such as c-Fos [21] or C/EBP isoforms [20]. Intriguingly, although c-Jun is not sufficient to induce SOCS3 expression in HUVECs, it is sufficient to suppress IL6 signalling to STAT3 activation (Fig. 3b). This suggests that c-Jun may play a wider, SOCS3-independent, role in the control of anti-inflammatory signalling in HUVECs. Overall we describe new mechanisms by which EPAC1 can regulate transcription in HUVECs through the control of AP-1 transcription factors, in a PKA-independent manner. This control appears not to involve the classical route of JNK-mediated phosphorylation of c-Jun and may therefore involve cooperation between c-Jun and other transcription factors, such as C/EBPs. This remains to be formally tested.
Acknowledgements
This work was supported by a British Heart Foundation project Grant (ref PG/10/26/28303) awarded to S.J.Y.
References
- 1.Sands W.A., Woolson H.D., Milne G.R., Rutherford C., Palmer T.M. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol. Cell. Biol. 2006;26:6333–6346. doi: 10.1128/MCB.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piessevaux J., Lavens D., Peelman F., Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev. 2008;19:371–381. doi: 10.1016/j.cytogfr.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Bos J.L. Epac proteins: multi-purpose cAMP targets. Trends Biochem. Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt M., Sand C., Jakobs K.H., Michel M.C., Weernink P.A. Epac and the cardiovascular system. Curr. Opin. Pharmacol. 2007;7:193–200. doi: 10.1016/j.coph.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Netherton S.J., Sutton J.A., Wilson L.S., Carter R.L., Maurice D.H. Both protein kinase A and exchange protein activated by cAMP coordinate adhesion of human vascular endothelial cells. Circ. Res. 2007;101:768–776. doi: 10.1161/CIRCRESAHA.106.146159. [DOI] [PubMed] [Google Scholar]
- 6.Yarwood S.J., Borland G., Sands W.A., Palmer T.M. Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J. Biol. Chem. 2008;283:6843–6853. doi: 10.1074/jbc.M710342200. [DOI] [PubMed] [Google Scholar]
- 7.Woolson H.D., Thomson V.S., Rutherford C., Yarwood S.J., Palmer T.M. Selective inhibition of cytokine-activated extracellular signal-regulated kinase by cyclic AMP via Epac1-dependent induction of suppressor of cytokine signalling-3. Cell. Signal. 2009;21:1706–1715. doi: 10.1016/j.cellsig.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Wiejak J., Dunlop J., Stoyle C., Lappin G., McIlroy A., Pediani J.D., Gao S., Yarwood S.J. The protein kinase C inhibitor, Ro-31-7459, is a potent activator of ERK and JNK MAP kinases in HUVECs and yet inhibits cyclic AMP-stimulated SOCS-3 gene induction through inactivation of the transcription factor c-Jun. Cell. Signal. 2012;24:1690–1699. doi: 10.1016/j.cellsig.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiejak J., Dunlop J., Gao S., Borland G., Yarwood S.J. Extracellular signal-regulated kinase mitogen-activated protein kinase-dependent SOCS-3 gene induction requires c-Jun, signal transducer and activator of transcription 3, and specificity protein 3 transcription factors. Mol. Pharmacol. 2012;81:657–668. doi: 10.1124/mol.111.076976. [DOI] [PubMed] [Google Scholar]
- 10.Eid A.H., Chotani M.A., Mitra S., Miller T.J., Flavahan N.A. Cyclic AMP acts through Rap1 and JNK signaling to increase expression of cutaneous smooth muscle alpha2C-adrenoceptors. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H266–H272. doi: 10.1152/ajpheart.00084.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochbaum D., Tanos T., Ribeiro-Neto F., Altschuler D., Coso O.A. Activation of JNK by Epac is independent of its activity as a Rap guanine nucleotide exchanger. J. Biol. Chem. 2003;278:33738–33746. doi: 10.1074/jbc.M305208200. [DOI] [PubMed] [Google Scholar]
- 12.Auernhammer C.J., Bousquet C., Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc. Natl. Acad. Sci. USA. 1999;96:6964–6969. doi: 10.1073/pnas.96.12.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacLaren A., Black E.J., Clark W., Gillespie D.A. c-Jun-deficient cells undergo premature senescence as a result of spontaneous DNA damage accumulation. Mol. Cell. Biol. 2004;24:9006–9018. doi: 10.1128/MCB.24.20.9006-9018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramberg V., Tracy L.M., Samuelsson M., Nilsson L.N., Iverfeldt K. The CCAAT/enhancer binding protein (C/EBP) delta is differently regulated by fibrillar and oligomeric forms of the Alzheimer amyloid-beta peptide. J. Neuroinflammation. 2011;8:34. doi: 10.1186/1742-2094-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meinkoth J.L., Alberts A.S., Went W., Fantozzi D., Taylor S.S., Hagiwara M., Montminy M., Feramisco J.R. Signal transduction through the cAMP-dependent protein kinase. Mol. Cell. Biochem. 1993;127–128:179–186. doi: 10.1007/BF01076769. [DOI] [PubMed] [Google Scholar]
- 16.Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 17.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 18.Rampersad S.N. Cyclic AMP phosphodiesterase 4D (PDE4D) Tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J. Biol. Chem. 2010;285:33614–33622. doi: 10.1074/jbc.M110.140004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yarwood S.J., Kilgour E., Anderson N.G. Cyclic AMP potentiates growth hormone-dependent differentiation of 3T3-F442A preadipocytes: possible involvement of the transcription factor CREB. Mol. Cell. Endocrinol. 1998;138:41–50. doi: 10.1016/s0303-7207(98)00049-5. [DOI] [PubMed] [Google Scholar]
- 20.Cai D.H., Wang D., Keefer J., Yeamans C., Hensley K., Friedman A.D. C/EBP alpha:AP-1 leucine zipper heterodimers bind novel DNA elements, activate the PU.1 promoter and direct monocyte lineage commitment more potently than C/EBP alpha homodimers or AP-1. Oncogene. 2008;27:2772–2779. doi: 10.1038/sj.onc.1210940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curran T., Franza B.R., Jr. Fos and Jun: the AP-1 connection. Cell. 1988;55:395–397. doi: 10.1016/0092-8674(88)90024-4. [DOI] [PubMed] [Google Scholar]