

Abstract
During apoptotic cell death, cellular stress signals converge at the mitochondria to induce mitochondrial outer membrane permeabilization (MOMP) through BCL-2 family proteins and their effectors. BCL-2 proteins function through protein-protein interactions, the mechanisms and structural aspects of which are only just being uncovered. Recently, the elucidation of the dynamic features underlying their function has highlighted their structural plasticity and the consequent complex thermodynamic landscape governing their protein-protein interactions. These studies show that canonical interactions involve a conserved, hydrophobic groove, whereas noncanonical interactions function allosterically outside the groove. Here, we review the latest structural advances in understanding the interactions and functions of mammalian BCL-2 family members and discuss new opportunities to modulate these proteins in health and disease.
Keywords: B cell lymphoma-2 (BCL-2) family proteins, mitochondrial outer membrane permeabilization (MOMP), mitochondrial apoptosis
BCL-2 family proteins drive apoptosis
The most common form of programmed cell death in biology and disease is the mitochondrial pathway of apoptosis [1]. During apoptosis, cellular stress signals converge at the mitochondria to induce mitochondrial outer membrane permeabilization (MOMP): typically the “point of no return” during a cell’s controlled self-destruction (Figure 1) [2]. Cellular stress may be extrinsic, initiated by the engagement of death receptors at the plasma membrane, or intrinsic, such as chemotherapy-induced DNA damage. Through release of cytochrome c (cyt c) [3], MOMP triggers a caspase activation cascade, which in turn activates the downstream machinery to orchestrate the apoptotic cellular dismantling and clearance [4, 5]. MOMP is orchestrated by the BCL-2 family of proteins (Figure 1) [6].
Figure 1. The BCL-2 family protein-mediated mitochondrial pathway of apoptosis.
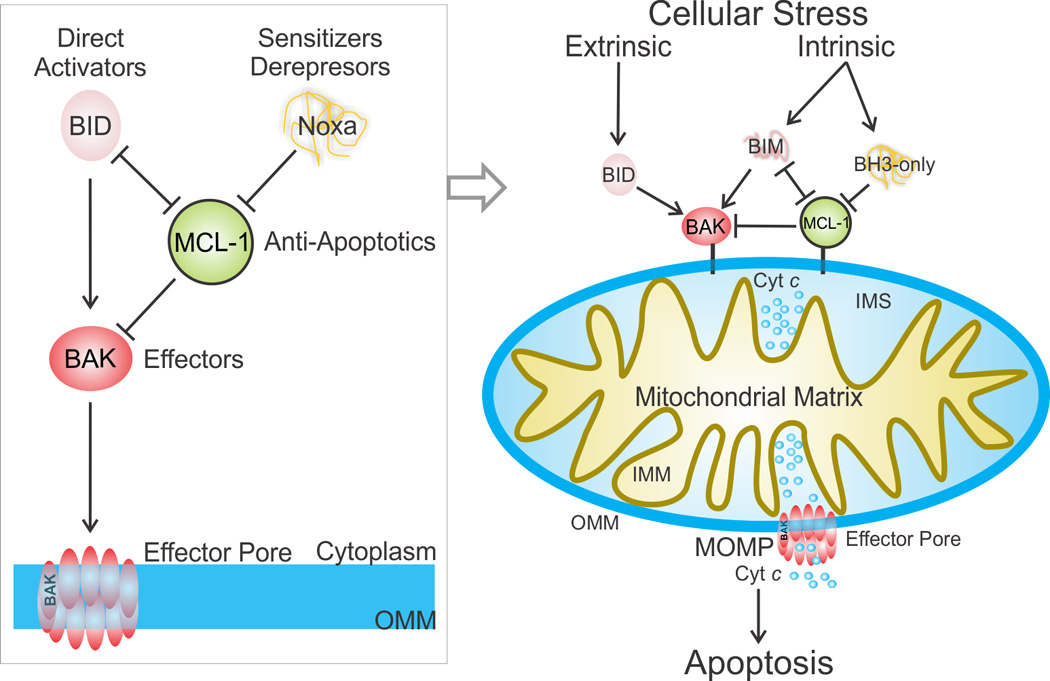
Cell extrinsic and intrinsic cellular stress engages mitochondrial apoptosis through BCL-2 family proteins, which regulate mitochondrial integrity, as shown on the right side of the figure. The extrinsic stress pathway is mounted by ligand binding of death receptors at the plasma membrane resulting in caspase 8-mediated activation of BID (not shown). The intrinsic pathway is poorly characterized in different cell types upstream of BH3-only proteins, and depending on the stress its engagement results in heterogeneous upregulation of BH3-only activities. For example, BIM is upregulated in response to kinase inhibitors and microtubule stabilizing drugs [103]. When the stress activates enough of the pro-apoptotic BH3-only proteins to overcome the anti-apoptotic response, the effectors BAK and BAX are directly activated by BID, BIM, and potentially other BH3-only proteins, and form pores in the outer mitochondrial membrane (OMM) releasing cytochrome c (cyt c), a process known as the mitochondrial outer membrane permeabilization (MOMP). Cyt c triggers the activation of the downstream caspase cascade leading to apoptosis (not shown). The unified model of BCL-2 family protein function in MOMP is depicted at the left. IMM, inner mitochondrial membrane. IMS, inter membrane space. See also Box 1 for a detailed description of BCL-2 family proteins.
Since the discovery of BCL-2 family proteins in the late 1980s, their mechanisms of action and interplay in apoptosis have been elucidated in exquisite molecular detail [6–8]. These proteins function through protein-protein interactions, which often involve allosteric control of their conformations and dynamics, that provide a molecular basis for understanding the signaling that evokes MOMP.(described later). Recently, the elucidation of the dynamic features underlying the function of these proteins has highlighted their structural plasticity and the consequent complex thermodynamic landscape governing their protein-protein interactions, which now include sparsely populated or “invisible” conformational states. Clarification of the molecular mechanisms of action of BCL-2 family proteins has fueled drug discovery efforts for their pharmacologic modulation, and related targeted therapies show great promise in the clinic [9]. Here we review the latest structure-based mechanisms furthering our understanding of mammalian BCL-2 family protein-protein interactions and allostery in MOMP and apoptosis and discuss new opportunities to modulate these proteins in health and disease.
A unified model of BCL-2 family protein-protein interactions
The mammalian B cell lymphoma-2 (BCL-2) protein family contains both pro-apoptotic and anti-apoptotic members. The family members BCL-2 antagonist killer 1 (BAK), BCL-2-associated X protein (BAX), and possibly BCL-2-related ovarian killer (BOK)–termed “effectors”—mediate MOMP, and the anti-apoptotic family members BCL-2, BCL-xL, BCL-w, Myeloid cell leukemia 1 (MCL-1), and BCL-2-related gene A1 (A1) inhibit it. The subfamily of BH3-only proteins (sharing only the third BCL-2 homology [BH] domain) function to regulate the other two classes by directly activating the pro-apoptotic effectors (e.g. BCL-2 interacting domain death agonist (BID), BCL-2 interacting mediator of cell death (BIM), and p53-upregulated modulator of apoptosis (PUMA)), or by blocking the activity of the anti-apoptotic proteins without directly engaging the pro-apoptotic effectors (e.g. BCL-2 antagonist of cell death (BAD), BCL-2 interacting killer (BIK), BCL-2 modifying feactor (BMF), harakiri (HRK), and Noxa) [6, 10]. The latter are known as derepressors or sensitizers because they disrupt existing anti-apoptotic complexes or occupy binding sites in anti-apoptotic proteins, respectively, without directly causing MOMP. Homology analyses revealed that effector and anti-apoptotic BCL-2 proteins share 4 BH regions (BH1–4), resulting in high structural homology throughout their globular folds that we refer to as the BCL-2 core (Box 1). With the exception of BID [11], BH3-only proteins lack a globular fold and are intrinsically disordered [12].
Box 1. BCL-2 family proteins uncut.
The BCL-2 family of proteins control mitochondrial apoptosis through a tightly regulated system of checks and balances mediated by direct binding interactions between pro-apoptotic and anti-apoptotic members [6, 7, 104]. The three classes of pro-apoptotic proteins are the effectors, the direct activators and the deprepressors/sensitizers (Figure I). All activators share only the BCL-2 homology region 3 (BH3-only) and with the exception of BID are intrinsically disordered. The effectors and anti-apoptotic proteins share 4 BCL-2 homology regions, BH1–BH4, numbered chronologically based on their identification (see scheme). Anti-apoptotic BCL-2 proteins and the effectors BAK [22] and BAX [54] are characterized by a common fold referred to as the BCL-2 core. It consists of a bundle of eight α-helices that forms a solvent-exposed hydrophobic groove between helices α2-α5 capped by a short C terminal helix α8 [65, 105]. This groove contains residues from the BH1–BH3. We dubbed this groove the BC groove because it binds the BH3 region of binding partners and the C-terminal transmembrane targeting region, as elucidated in the structures of BAX and BCL-w [6]. The activator protein BID [11] displays a similar architecture. Note that the BH3-only proteins BID, BIM, and PUMA exhibit features of both direct activators and derepressors/sensitizers, depending upon experimental setting.
The BC groove of all BCL-2 cores, except that of BID, function by binding BH3 domains of interacting partners similar to the original complex of BCL-xL–BAK BH3. A BH3 domain engages hydrophobic pockets in the BC groove with up to seven hydrophobic residues adopting a helical conformation even when derived from intrinsically disordered family members [65]. These domains exhibit a homologous central segment spanning 3–4 α-helical turns containing conserved Leu and Asp amino acids. When engaged in complexes, the conserved Leu is deeply buried within the hydrophobic BC groove of the anti-apoptotic globular binding partner, and the Asp forms a salt bridge with a conserved Arg located at the N terminus of helix α5 [65]. Outside this region, the BH3 sequence is more divergent and undoubtedly plays a role in the specificity and selectivity for BC grooves within the different multi-BH region family members. The derepressor/sensitizer BCL-2 proteins behave structurally very similarly to the direct activator BIM with a disordered to helical transition of the BH3 upon engagement of anti-apoptotic BCL-2 proteins [6]. The difference is that they are unable to engage and directly activate the effectors.
The anti-apoptotic BCL-2 family proteins function by antagonizing the pro-apoptotic direct activator–effector MOMP axes through protein-protein interactions [13]. Depending on the cellular stress, anti-apoptotic BCL-2 proteins may inhibit direct activator BH3-only proteins (Mode 1), activated effector BCL-2 proteins (Mode 2), or act by both mechanisms. The relative extent of either inhibitory mode is largely governed by the relative affinities associated with the different BCL-2 family protein partnerships (discussed later). The pro-apoptotic role of derepressor/sensitizer BH3-only proteins is to antagonize the anti-apoptotic BCL-2 proteins by competing for a common site engaged by virtually all pro-apoptotic proteins, thereby relieving inhibition of direct activator–effector MOMP interactions [6]. Based on the above classification, we offer a unified model for BCL-2 family function in MOMP (Figure 1, Box 1).
Plasticity of BCL-2 family proteins
An emerging feature of protein-protein interactions between BCL-2 family members is the functional role of structural plasticity, which may be categorized into ‘canonical’ and ‘non-canonical’ interactions based on the engagement or lack thereof of the BH3 and C terminus binding (BC) groove. This is a conserved, hydrophobic groove on the surface of the BCL-2 core, demarcated on one face of the globular domain by the BH1–BH3 regions (Box 1). Canonical interactions at the BC groove involve binding of BH3 ligands to form hetero-dimeric or homo-oligomeric complexes. Non-canonical interactions outside the BC groove can allosterically activate or inhibit effectors, mediate effector oligomerization, modulate the affinity of BH3-only proteins for the BC groove of anti-apoptotic binding partners, or control interactions with binding partners outside the BCL-2 family.
Canonical BC groove mechanisms
Anti-apoptotic and effector BCL-2 family proteins canonically bind the BH3 regions of pro-apoptotic proteins at the BC groove, as originally illustrated for the anti-apoptotic complex of BCL-xL and a BAK BH3 peptide [14] (Box 1) and recently for the pro-apoptotic complexes of BAK [15] or BAX [16] and a BID BH3 peptide. The similarities and differences in the BC grooves and their interactions with BH3 ligands correlate with the biological functions of BCL-2 family proteins. Structural features of the BC groove dictate the specificities of effectors for direct activators, and of anti-apoptotic BCL-2 proteins for different BH3 ligands (discussed later). Moreover, the BC groove of effectors may mediate one of several types of oligomeric interactions relevant to MOMP.
BC groove-mediated effector activation
BCL-2 family proteins, including the effector BAK, constitutively target the outer mitochondrial membrane (OMM) through a hydrophobic helix C-terminal to the BCL-2 core (labeled TM in Figure 2, and in Figure I of Box 1). By contrast, BAX is primarily localized in the cytosol and requires a preliminary step during activation to help target it to the OMM [17]. Currently, the preferred model for this step involves engagement of a non-canonical site on a surface of the BCL-2 core of BAX that is opposite to the BC groove, which induces OMM targeting by the C-terminal helix α9 (described later) [18]. To perhaps antagonize this preliminary step, in healthy cells BAX is constantly retro-translocated from the mitochondria to the cytosol by anti-apoptotic BCL-2 family proteins; consequently, it resides primarily in the cytosol [19].
Figure 2. Mechanisms of effector-mediated MOMP.
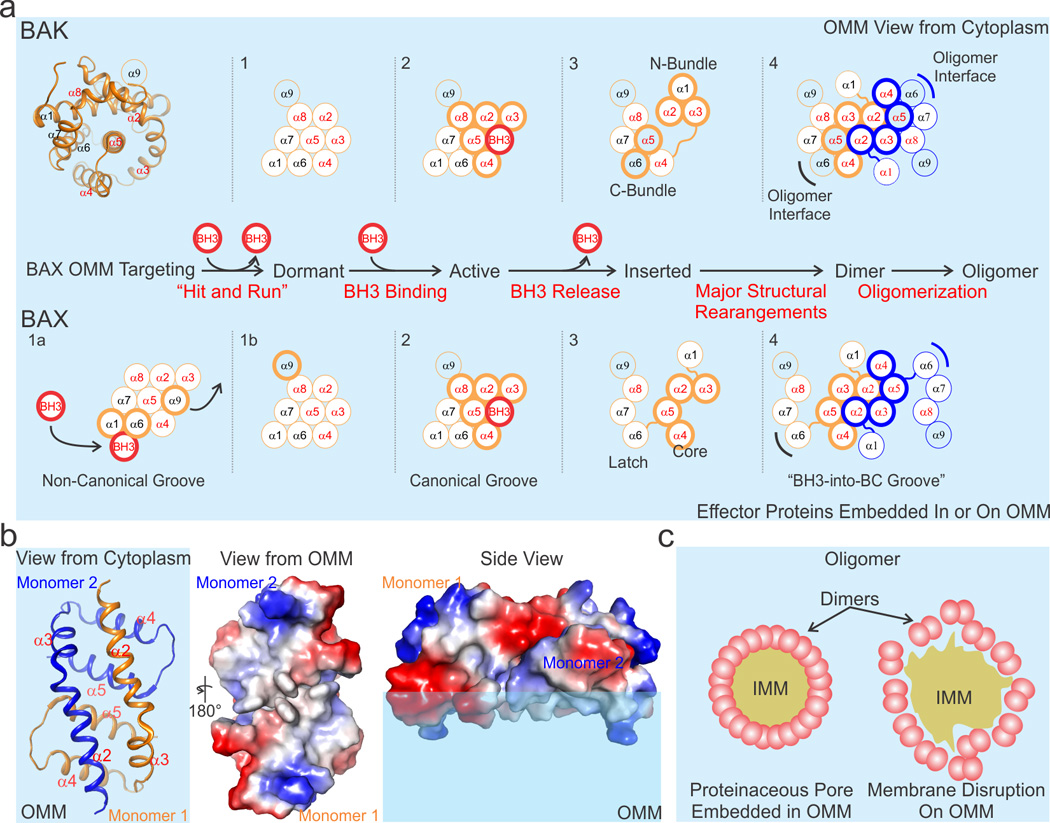
a) The effectors undergo a multistep activation mechanism culminating in oligomerization as illustrated in the flow diagram. At least four and five distinct conformations (numbered 1–4 and separated by dotted lines) have been associated with BAK and BAX during MOMP, respectively. The cartoon representation of the structure of the BCL-2 core of BAK (helices α1–α8) at the top left orients the initial model for both BAK and BAX by centering the core on helix α5. BAK is constitutively targeted to the OMM by α9 (conformation 1), and BAX resides primarily in the cytosol (conformation 1a). During apoptosis, direct activator BH3s (red circle) allosterically engage the non-canonical groove of BAX, releasing α9 from the BC groove (the helices that comprise the BC groove are labeled in red font, i.e. α2–α5, α8) on the opposite face and facilitating α9 OMM targeting (conformation 1b). Conformations 1 of BAK and 1b of BAX are considered dormant, as the OMM insertion of helix α9 does not lead to MOMP. Both effectors are directly activated by BH3 engagement at the canonical BC groove (conformation 2). Possible mechanisms for effector conformational changes associated with monomer insertion in OMM include a N- and C-terminal bundle separation for BAK, and a latch and core separation for BAX (conformation 3). Both effectors form seemingly symmetric dimers by a BH3 interaction of one monomer to the canonical BC groove of another (conformation 4). A second unstable interface may be mediated by α6 (indicated by black and blue brackets). Helices actively involved in the step-wise mechanism are shown with a thick outline. Inserted helices are colored light blue like the OMM. OMM is represented by the light blue background. b) A low-resolution crystal structure of the “BH3-into-groove” dimer has suggested that a hydrophobic region (shown in white) may be formed (view from the OMM ) that has the potential to destabilize the outer leaflet of the OMM (side view). Electropositive, electronegative, and neutral (hydrophobic) patches are blue, red, and white, respectively. c) Effector oligomers (pink), made up of conformation 4 dimers in panel a, may form a well-organized proteinaceous pore that perforates the OMM (left), or they may loosely associate and, through the disruption of the OMM outer leaflet, induce destabilization and OMM rupture (right). IMM, inner mitochondrial membrane.
BOX1 Figure I. The structure–function cheat sheet of BCL-2 family proteins.
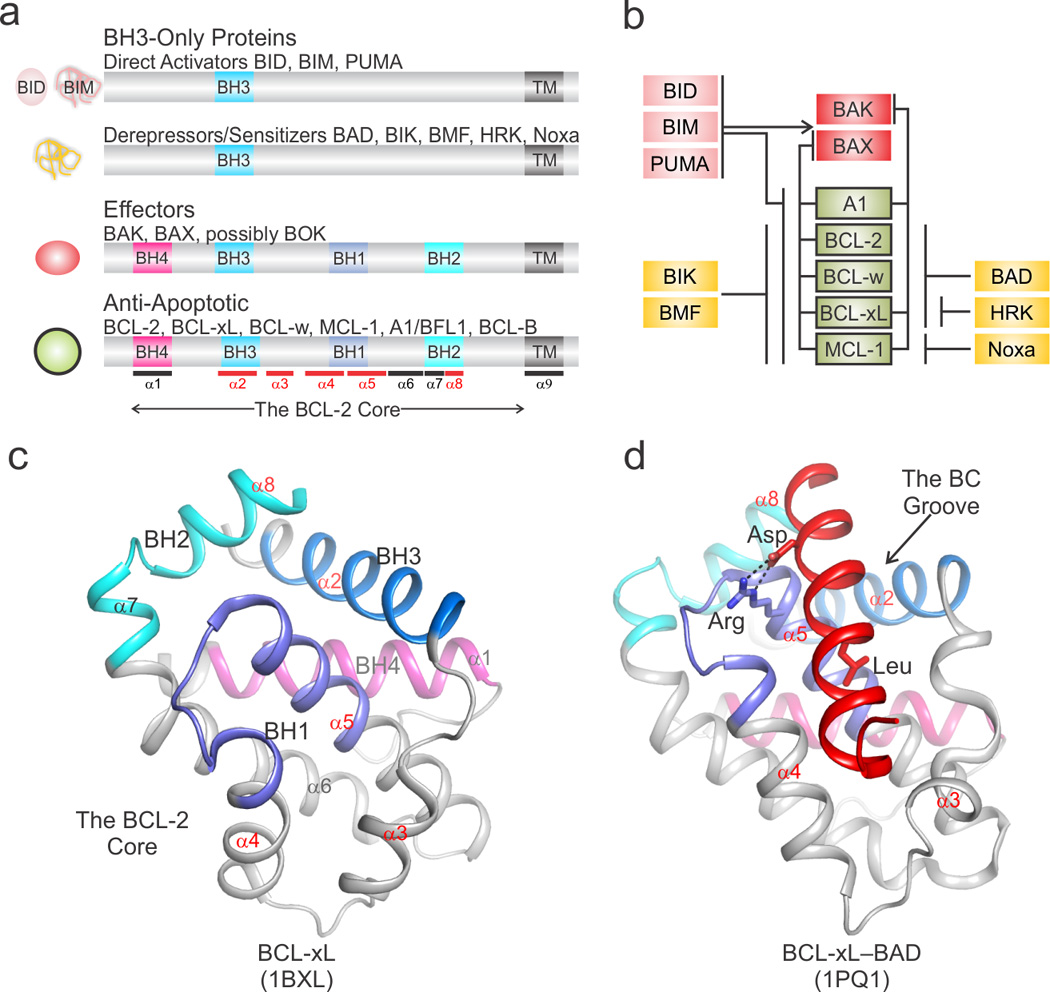
a) Schematic representation of BCL-2 family proteins identifying BCL-2 homology (BH) and transmembrane-targeting (TM) regions with respect to the BCL-2 core. At the left, the cartoons illustrate the structured, globular (rounded) or unstructured, intrinsically disordered (noodle-like) nature of the particular class of BCL-2 family proteins. The approximate position of α-helices in the BCL-2 core is marked at the bottom. Helices that delineate the BC groove are highlighted in red. b) The functional interaction network between pro- and anti-apoptotic BCL-2 family members is color coded as in panel a. c) The structure of the BCL-2 core of BCL-xL identifies the BH regions and α-helices colored as in panel a. d) A representative structure highlighting the BH3-into-BC groove interaction for the complex between BCL-xL and the BH3 region of BAD (shown in red). The conserved Leu and Asp side chains of BAD BH3 are engaged in hydrophobic and electrostatic interactions, respectively, with complementary sites in the BCL-2 core. PDB identifiers are shown at the bottom.
Although the details of effector activation may differ, BAK and BAX follow a similar activation mechanism requiring engagement of the BC groove by direct activator BH3 helices. This was determined using solution NMR spectroscopy between the BCL-2 core of BAK and BID BH3 [15], but had been previously suggested by several studies [20–24]. In the complex with BID BH3, the BC groove of BAK undergoes a transition from an occluded conformation in its unbound state to an open conformation to accommodate binding of the activator BID BH3 with moderate affinity (Kd ~ 10 µM; Figure 2). Similarly, a complex between the BCL-2 core of BAX and BID BH3, captured in a domain-swapped BAX homodimer, revealed a subtle apo-to-bound conformational transition at the BC groove of BAX (Figure 2) [16]. How BH3-only proteins other than BID and BIM engage the canonical BC groove of the effectors remains to be elucidated structurally, but PUMA, BMF and Noxa were proposed to act as weak direct activators [20, 23, 25–28]. These observations illustrate the plasticity of BCL-2 family proteins.
BC groove effector oligomerization
Though stable direct activator–effector complexes form between truncated proteins in the absence of the OMM [15, 16], these may reflect “trapped” states and the effector structure is likely to be different in the presence of the OMM [29]. As we currently lack a high-resolution structure of activated effectors engaging the OMM, our current model for this process was compiled from extensive biochemical evidence and one low-resolution crystal structure of a truncated, BC groove-containing form of BAX (Figure 2). This model predicts that in the presence of the OMM, conformational changes destabilize the activated BCL-2 core revealing effector conformations with an energy landscape reminiscent of partially disordered proteins (summarized in Figure 2). The conformational transitions have been characterized through stabilization with antibodies recognizing N-terminal and BH3 epitopes [30–32], binding of the exposed effector BH3 region with anti-apoptotic BCL-2 proteins, limited proteolysis to map stable fragments of on-activation-pathway effector intermediates [13], and extensive mutagenesis to accompany related studies. For BAK, the evidence suggests permanent N-terminus exposure and transient BH3 exposure. Following the initial conformational changes within a monomer, a “BH3-into-BC groove” symmetric homodimer forms for both effectors and buries the transiently exposed BH3 [30, 33–35]. The BH3-into-BC groove homodimer, captured in the minimal BH3-into-groove crystal structure of truncated BAX, resembles a canonical BH3–BC groove interaction (Figure 2) [16]. We cannot predict exactly how regions of this minimal BH3-into-groove homodimer, determined in the absence of membranes, may alter the OMM, or how it assembles in the context of the full-length protein. A previous study suggested that a similar minimal core induces apoptosis when targeted to the OMM using helix α9 [36] and a recent study detailing BAK oligomerization by electron paramagnetic resonance indicated that BAK may form a similar BH3-into-groove homodimer [37]. Intriguingly, one face of the BH3-into-groove dimer is enriched in hydrophobic residues and may displace lipids in the outer bilayer, thus destabilizing the local OMM. Downstream of the BH3-into-groove interface, biochemical evidence suggests that helix α6 on the opposite face of the BC groove may mediate a non-canonical oligomerization interface, but high-resolution details are not available [38, 39]. Additionally, membrane topology-mapping studies suggest that helices α5, α6 and α9 of BAX are inserted into the membrane, raising the possibility of multiple intermediate conformations governing effector-mediated MOMP [40]. Although the BH3-into-groove dimer supports a non-proteinaceous, lipidic MOMP pore [16], gel filtration chromatography studies have suggested that a proteinaceous effector oligomer of about 20 monomers may resemble the putative MOMP pore (Figure 2) [23, 33, 41], although much larger oligomers have been postulated from confocal microscopy studies of MOMP [42]. Irrespective of the actual size of the effector oligomers, one possibility is that they consolidate local perturbations of the OMM lipid structure to create a large lipidic pore, ultimately responsible for MOMP and the release of inter-membrane proteins.
Disorderly engagement of the BCL-xL BC groove by PUMA
Though extensive allosteric plasticity is critical for the activation of effector BCL-2 proteins, interplay between binding events, protein conformations and conformational dynamics appear to also play regulatory roles in anti-apoptotic BCL-2 proteins. The BH3-only protein PUMA, a transcriptional target of p53, directly regulates the transcription-independent, cytosolic, pro-apoptotic function of p53 [43]. PUMA, uniquely among the BH3-only proteins, is capable of binding BCL-xL, releasing cytosolic p53 from an inhibitory complex with BCL-xL through a dynamic allosteric mechanism [44]. A critical Trp residue within the BH3 region of PUMA engages a His residue in helix α3 of BCL-xL to induce unfolding of this region of BCL-xL, thus disrupting a significant portion of the BCL-xL interface with p53. In this regulatory context, the plasticity of BCL-xL α3 is exploited to allow simultaneous interaction of BCL-xL with p53 and BH3 binding partners. Only the BH3 region of PUMA, in an exquisitely specific fashion, triggers extensive unfolding of this segment and thus prevents p53 engagement. The PUMA-unleashed p53 then acts as a non-canonical (because it does not contain a BH3 region) direct activator of BAX [45] or BAK [46].
More generally, a repositioning of helix α3 away from helix α4 constitutes the main conformational rearrangement that leads to the opening of the BC groove upon BH3 ligand binding to anti-apoptotic BCL-2 proteins [6]. The plasticity of this segment appears, therefore, to play a role in determining the specificity and affinity of anti-apoptotic BCL-2 proteins to different BH3 partners. Additionally, as in the case of the BCL-xL interaction with PUMA, the plasticity of this segment contributes to their functional regulation. It is fascinating that regulatory structural rearrangements occur within the same two helices within the BC groove of both the anti-apoptotic and effector BCL-2 family proteins, despite the different structural and functional outcomes of these interactions. Arguably, the overall fold of multi-domain BCL-2 proteins may have evolved to confer a ‘built-in’ malleability within this region that allows for diverging modes of regulation and therefore functional outcomes. The fact that extensive structural rearrangements, though incompletely understood at present, are essential for the critical function of MOMP may have imposed structural plasticity in an ancestral molecule and this feature, through evolutionary diversification, persists within many BCL-2 protein family members.
Molecular basis of BH3 specificity for BC groove
Structure-function studies have elucidated the molecular basis for recognition of BH3 regions of pro-apoptotic proteins by the BCL-2 core of anti-apoptotic [6, 47] and effector BCL-2 family proteins [15, 16]. The minimal (4–5 helix turns) and extended (>6 helix turns) BH3 region binds similarly to the BC groove of BCL-2 family proteins for all tested pairs. Both hydrophobic and hydrophilic/electrostatic contacts define the selectivity and specificity of BH3–BC groove complexes (Figure 4). This mode of interaction provides an explanation for 1) the selectivity of anti-apoptotic BCL-2 family proteins for derepressors/sensitizers, illustrated here by the BAD BH3–BCL-xL and Noxa BH3–MCL-1 complexes [48, 49] whereby BAD BH3 is a low affinity ligand for MCL-1 and Noxa BH3 for BCL-xL; 2) the specificity of these anti-apoptotic proteins for the effector proteins, illustrated by the BAX BH3–BCL-2 complex [50], whereas BAK BH3 does not bind BCL-2; and 3) the promiscuity of direct activator BH3-only proteins for anti-apoptotic and effector BCL-2 family proteins, illustrated by complexes between BID BH3 and MCL-1, BAK and BAX (Figure 4) [15, 16, 51], wherea derepressors/sensitizers do not engage the effectors. Structural clashes inferred from the available BH3–BCL-2 core complex structures provide a structural explanation for the low-affinity pairs. The role of the clashes in determining specificity was verified by switching clashes to permissible interactions by mutagenesis, which in most cases conferred binding in otherwise non-interacting BH3–BCL-2 core pairs. Moreover, in several instances the minimal BH3 regions required for binding have been identified. Interestingly, the effectors BAK and BAX require, respectively, a C-terminal and N-terminal extension to the overlapping BID BH3 as a minimal region for their activation (Figure 4). As discussed later, this difference may be exploited to design effector targeting compounds. With the available structures of BH3–BCL-2 core complexes, we can, in principle, predict permissible interactions or clashes for all possible pairs. This type of analysis recently revealed why BAD and NOXA BH3, which clash with the BC grooves of BAK and BAX, are not direct activators of the effectors. Mutagenesis to eliminate the clashes and convert the BAD and NOXA BH3 regions into direct activators, confirmed this analysis (Figure 4).
Figure 4. BC groove specificity for BH3-only ligands.
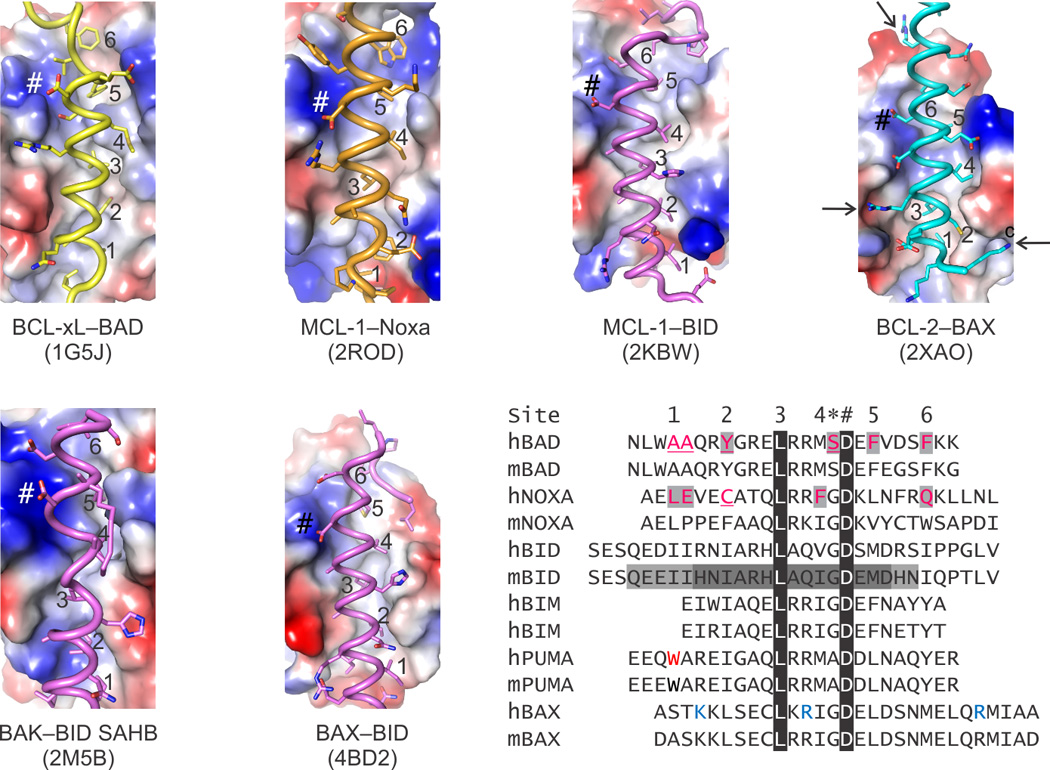
The electrostatic potential on the surface of BC grooves reveals the similarities and differences between folded BCL-2 family proteins, which ultimately dictate the specificity of the binding pairs as summarized in Box 1. Sequence homology highlights features shared by BH3 ligands, including up to seven hydrophobic residues that engage six pockets along the BC groove (labeled 1–6 in the sequence alignment and in the structures) and a conserved Asp always found in a salt bridge (labeled *) preceded by a small residue (labeled #). Peripheral to these positions, which are marked in each structure panel, there are additional interactions, many electrostatic in nature, that contribute to the specificity. For instance, in the BCL-2–BAX complex three positively charged Lys and Arg residues (blue in the alignment and identified by arrows in the structure) were shown to be essential for the affinity of BAX for BCL-2. In addition to the specificity, the BC grooves have evolved to be selective only for certain BH3s; others typically produce structural clashes when modeled based on the known complexes. Up to four residues in BAD BH3 and Noxa BH3, predicted to clash, had to be replaced to directly activate BAK and BAX. Residues highlighted and underlined in human BAD and NOXA were replaced by those found in human BID BH3 for BAK and BAX activation, respectively. Interestingly, specificity of BID BH3 for BAK and BAX was conferred by minimal BH3 regions that required, respectively, a C- and N-terminal extension to a common core (sequence highlighted by dark gray). Black highlights show 100% sequence conservation. Electropositive, electronegative, and neutral (hydrophobic) patches are blue, red and white, respectively.
Non-canonical BC groove mechanisms
SAHBed in the BAX
The initial steps of BAX activation implicate a transient “hit and run” mechanism originally proposed by the Korsmeyer laboratory studying the BID–BAK axis [24]. Based on observations that oligomeric BAK did not remain associated with activator BID, this hit-and-run model postulated that BID dissociates after inducing the active BAK conformation. At the molecular level, interactions by activator BH3 domains occur first at a non-canonical site [18, 52, 53], and then at the BC groove [16]. In BAX, the C-terminal helix α9 uniquely occupies the BC groove generally exploited for BH3 domain interactions [54] and it is displaced during activation to target BAX to the OMM. Specifically, the BH3 domain of BIM [18, 55] and probably that of BID [27, 56] can interact with an interface formed between helices α1 and α6 of BAX (Figure 2a) on the face opposite to the BC groove and, through a presumed allosteric mechanism, induce the displacement of helix α9. Recently, a similar activation mechanism has also been reported for the BH3 domain of PUMA [57]. The weak and transient nature of non-canonical native activator BH3–BAX complexes have evaded structural analyses. A breakthrough to aid these analyses was the use of covalently stabilized, or “stapled”, α-helical peptides of BCL-2 proteins (SAHBs), which were adopted to minimize the entropy loss associated with folding-upon-binding into α-helices,. This in turn increased the affinities of BH3–effector interactions compared with their endogenous counterparts [58].
p53: the non-canonical effector activator
How proteins other than the known direct activator BH3-only proteins, BID, BIM and PUMA exploit effector activation remains to be characterized. A likely candidate is p53, which activates BAX- and BAK-dependent MOMP. p53-dependent direct activation of BAX [45] and BAK [46] is not mediated by a BH3 region in p53, because p53 does not contain such region, and may therefore be achieved through non-canonical mechanisms. Although awaiting structural characterization, ample biochemical and genetic evidence supports this alternative mechanism of effector-mediated MOMP. BAX activation by p53 was abrogated upon ablation of the proline rich region between the transcriptional activation and DNA binding domains of p53 [45]. This p53 segment may activate BAX directly or favor a suitable spatial organization of neighboring domains to trigger BAX activation via a transient interaction [45]. The interaction between BAK and p53 is primarily meditated by the DNA binding domain of p53 [59, 60].
While p53-mediated direct activation of BAX [37] and BAK [38] has been experimentally observed in cells and in cell-free conditions without a requirement for p53-induced transcription [61], it should be noted that mutants that discriminate these activities of cytosolic p53 from transcriptionally active p53 have not yet been identified. Furthermore, the specific cellular signals that regulate the relative contributions of “cytosolic” or “nuclear/transcriptional” p53 to the induction of apoptosis remain unknown [62]. The relative extent to which the cytosolic activity of p53 function physiologically therefore remains controversial [63].
Allosteric regulation of the PUMA–BCL-xL–p53 complex
The complex between BCL-xL and p53 constitutes another example of a non-canonical interaction with a BCL-2 core. The DNA binding domain (DBD) of p53, characterized by a positively charged surface, interacts with a negatively charged lobe of BCL-xL comprising the C terminus of helix α1 and the two short loops connecting helices α3 and α4 and helices α5 and α6 (Figure 3) [59, 64, 65]. Similar features appear to characterize the interaction between the DBD of p53 and BCL-2, which is structurally similar to BCL-xL [66].
Figure 3. Allosteric regulation of BCL-xL–p53 complex formation by the BH3-only protein PUMA.
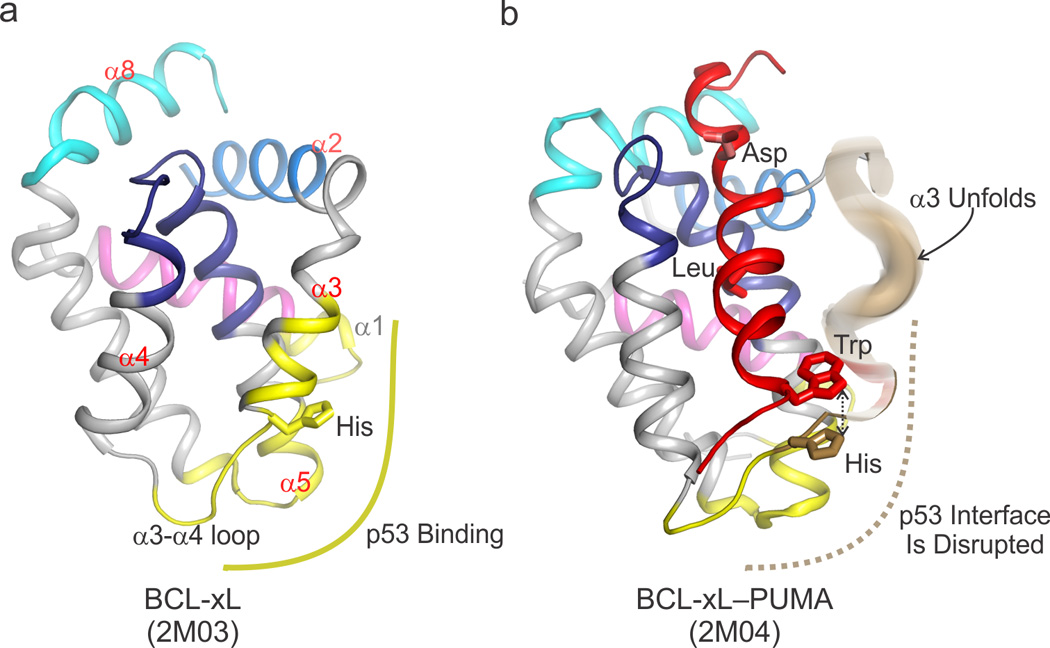
a) The basic DNA binding domain of p53 binds an acidic lobe of BCL-xL (shown in yellow) comprising the C terminal segment of α1, α3, the loop between α3 and α4, and the C terminus of α5. This non-canonical binding site lies outside the BC groove of BCL-xL (the helices that comprise the BC groove are labeled in red font, i.e. α2–α5, α8) The four BH regions are colored according to the scheme in Figure I (Box1). b) Upon binding of PUMA BH3 (colored red) to the BC groove of BCL-xL, a π-stacking interaction between a tryptophan in PUMA and a histidine in BCL-xL induces unfolding of its α3 (colored brown) and disrupts the interface with p53. Since a tryptophan residue is only found at that particular position in PUMA, and no other BH3 domains, this regulatory mechanism uniquely confers an additional level of signaling complexity to the PUMA–BCL-xL interaction that is absent in other pairs of interacting BCL-2 family proteins.
The structural plasticity displayed in various binding contexts by helix α3 of BCL-xL is exploited in formation of the complex between BCL-xL and the p53 DBD. Specifically, the BCL-xL helix α3 segment adopts conformations that both optimize its contact interface with p53, yet simultaneously are compatible with the binding of BCL-xL to BH3-only proteins other than PUMA [64].
The intrinsically disordered N-terminal transcriptional activation domain of p53 has also been shown to weakly interact with the BC groove of BCL-xL [67, 68] and MCL-1 in a non-canonical fashion. The first and second transcriptional activation domains of p53 engage the BC groove as helices in an orientation opposite to that of canonical BH3 partners [67–69]. Given the weak nature of these complexes, their specific biological function remains unclear.
Viral solutions to effector inhibition
Several viruses inhibit BAK- and BAX-dependent apoptosis to prevent premature self-clearance of infected cells and favor their own replication [70, 71]. The anti-apoptotic functions of viral proteins rely on blocking BAK or BAX activation by direct interaction with these proteins [72, 73]. Although most inhibitors follow canonical BH3–into–BC groove inhibitory mechanisms and can inhibit BH3 regions of BCL-2 family activators and effectors [74], some viral inhibitors engage different sites on the effectors, thereby preventing their activation. The atypical complex between BAX and the viral mitochondria-localized inhibitor of apoptosis protein (vMIA) involve a short amphipathic, positively charged helix that engages a negatively charged lobe of BAX comprising the loops connecting helices α3 and α4 and helices α5 and α6 [75]. Intriguingly, this interaction is reminiscent of BCL-xL–p53 interaction (described earlier), suggesting the possibility of a conserved noncanonical interaction mode exploited in patho-physiology.
In summary, the difference between canonical and non-canonical interactions may be generally explained by the extensive hydrophobic nature of the former, where both the BH3 ligand and BC groove provide broad surface complementarity expanded on the periphery by hydrophilic interactions that define specificity. On the other hand, in non-canonical complexes the BCL-2 core may assume the role of a versatile protein-protein interaction domain by exploiting surfaces outside the BC groove.
Drugging BCL-2 family proteins
BC groove drugs
Since the early 2000s, significant progress has been made toward the pharmaceutical targeting of BCL-2 family proteins [9, 76]. Small molecule BH3-mimetic inhibitors of anti-apoptotic family members including BCL-2, BCL-xL, and BCL-w [77–79] were designed to prevent anti-apoptotic BCL-2 proteins from sequestering pro-apoptotic family members, thereby acting as mimetics of derepressors/sensitizers (Figure 5). The original BH3 mimetic ABT-737 was generated based on screening and binding characterization of weak binding small molecule “fragments” using NMR spectroscopy. Larger molecules were then synthesized by structure-based drug design from these fragments [80]. ABT-737 engages multiple sites within the BC groove of BCL-xL or BCL-2 and was later improved to a bioavailable analogue, ABT-263 (navitoclax) (Figure 5) [81–83]. Clinical studies with ABT-737 and ABT-263 as single agents in chronic lymphocytic leukemia have been encouraging [84]. Moreover, these compounds synergize in combination with experimental drugs that cause additional cellular stress and have shown efficacy for a broad spectrum of BCL-2 and BCL-xL–dependent malignancies [85, 86]. A notable side effect has been the depletion of blood platelets, or thrombocytopenia, which has been associated with targeting of the BCL-xL–BAK axis, which is essential to platelet survival [83]. To circumvent this specific toxicity, a potent BCL-2–specific inhibitor, ABT-199, was developed by further refining the ABT-263 scaffold using a structure-guided approach (Figure 5). ABT-199 does not cause side effects related to thrombocytopenia [87, 88] and showed therapeutic promise against diverse forms of cancer [89, 90]. A BCL-xL–specific compound, WEHI-539, which targets similar hydrophobic pockets as the ABT compounds, was recently described [91]. The evolution of BH3 mimetics of BCL-2 family proteins highlights how detailed structural knowledge of the various anti-apoptotic proteins and subtle variations in the specificities of their interaction with BH3 partners were harnessed for the development of specific small molecule inhibitors [92]. Furthermore, the toxicity of ABT-737 and its elimination in ABT-199 provide an example of how the selective modulation of BCL-2 family proteins by small-molecules may find valuable applications as discovery tools for selectively revealing the in vivo functions of these proteins.
Figure 5. Targeting apoptosis via the BCL-2 family in biology and disease.
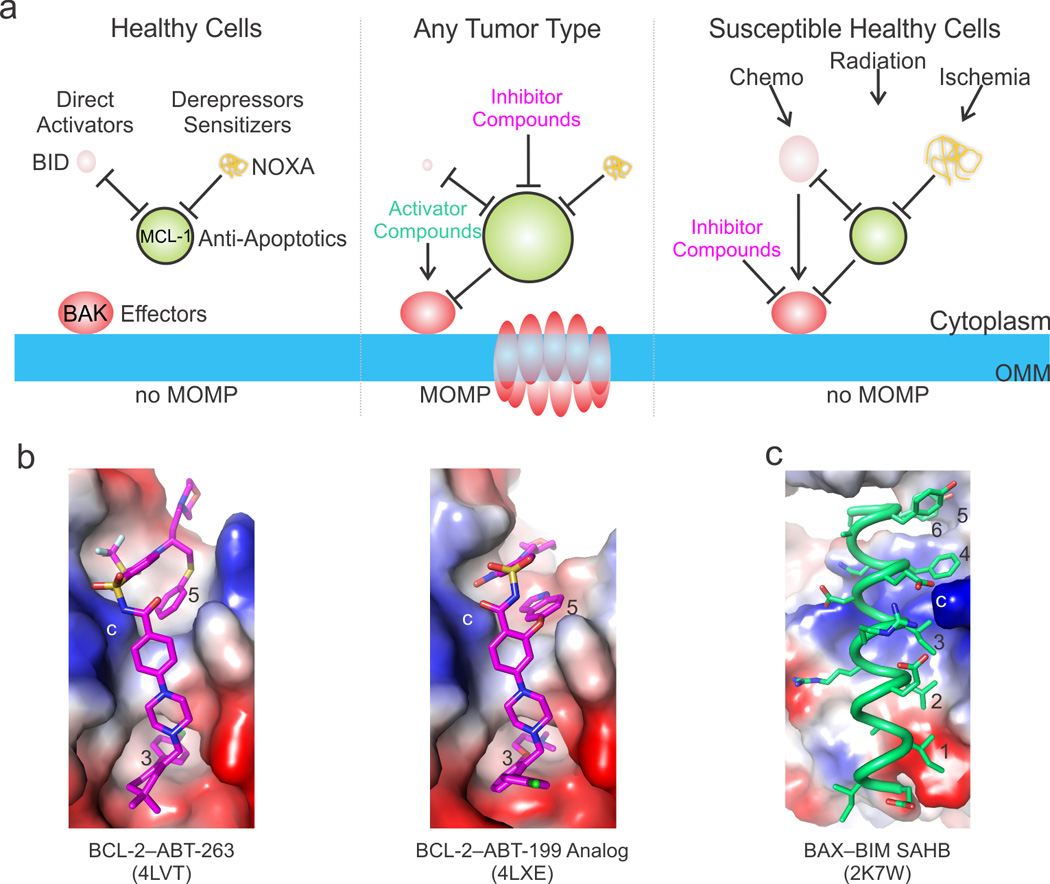
a) Targeted apoptotic therapy addresses the upregulation of anti-apoptotic and the downregulation of activator BCL-2 family proteins in cancer and the aberrant overactivation of the pathways during pathological insults and toxicity related stress. We illustrate this be varying the size of the BCL-2 family protein cartoons to reflect the fluctuations in intracellular levels. For example, in healthy cells the BH3-only protein levels are low and their pro-apoptotic activities are kept in check by anti-apoptotic BCL-2 proteins, overall preventing the direct activator–effector axes from being engaged (left panel). In tumors, the BH3-only proteins are down-regulated and the anti-apoptotic BCL-2 proteins are upregulated, together preventing MOMP. Compounds that mimic the activities of BH3-only proteins can be used to activate the effectors and inhibit the anti-apoptotic BCL-2 proteins (middle panel). On the other hand, compounds that inhibit the effectors may be used to block excessive cell death in healthy tissues susceptible to chemotherapy and radiation therapy or in healthy or diseased tissues prone to ischemic cell death. b) The most promising therapeutics have been designed at Abbvie for the anti-apoptotic BCL-2 family proteins, including ABT-263, which targets both BCL-2 and BCL-xL, and the BCL-2-specific compound ABT-199. Both compounds take advantage of scaffolds that engage the BC groove from the third hydrophobic contact (occupied by a conserved Leu in BH3 ligands) to beyond the fifth. These compounds have also been engineered to explore the electrostatic landscape of the BC groove, including a similar charge interaction to the conserved Arg–Asp (labeled *). c) BH3 mimetics for the non-canonical interaction described for the stabilized alpha helix of BCL-2 protein BIM (SAHB) at a groove on BAX distantly resembling the BC groove have also been described, and either SAHBs or their small molecule mimetics may trigger BAX. Importantly, both BAK- and BAX-mediated MOMP require activation by engagement of their BC grooves, which provides additional opportunities for modulating the mitochondrial pathway with activators or inhibitors. Positive, negative, and neutral (hydrophobic) patches are blue, red, and white, respectively.
Drugging allosteric conformational transitions
The allosteric conformational transitions associated with BCL-2 family effector activation have provided new opportunities for drug discovery. The effectors may, in principle, be triggered by compounds that mimic the direct activator BH3 domains during scenarios in which the endogenous direct activators are expressed at low levels, as documented for several types of tumors (Figure 5) [93, 94], or the mitochondria are not primed for apoptosis [95]. Alternatively, susceptible tissues, such as the gut and lung epithelia, may benefit by protection against MOMP during general chemotherapy and radiation therapy protocols (Figure 5). Inhibition of effector activation may also be beneficial in limiting the consequences of ischemic cell death during stroke [96]. Effector-mediated MOMP may be blocked with inhibitory BH3 mimetics targeting their BC groove or with molecules targeting non-canonical sites (e.g. vMIA mimetics). Small molecule activators and inhibitors could theoretically be designed for all of the hotspots associated with the intermediates of the effector activation mechanism. However, in the absence of structures of effector intermediates in membranes, the best targeting site for activation and inhibition remains their BC groove [15, 16]. Pertinent to BAX modulation, the non-canonical trigger site opposite the BC groove may provide an additional targeting site [18, 97]. This, however, has repercussions for the BAX targeting strategy either as a single agent or as a combination of two different agents that engage this site and the BC groove exclusively. This challenge may be circumvented with BH3 mimetics, such as the BIM, BAX and PUMA SAHBs, which have the capacity to engage both sites [18, 55, 57, 58]. A major drawback of this strategy, however, may be that SAHBs are relatively plasma membrane impermeable and therefore may not reach their in vivo targets in a wide variety of tumors [98]. Nonetheless, advances in the delivery of SAHBs to the target tumor sites may revolutionize their use as drugs [58]. In the near future, we ultimately envision modulation of BCL-2 family-mediated apoptosis in pathophysiology by a combination of a compound that mimics direct activator BH3–effector interactions with another that mimics derepressor/sensitizer BH3–anti-apoptotic BCL-2 family protein interactions—the apoptotic “double whammy.”
Concluding remarks
Most of the key members of the mammalian BCL-2 family proteins, including the effectors BAK and BAX, the promiscuous direct activators BID and BIM, the anti-apoptotic BCL-2, BCL-xL and MCL-1 and the classical derepressor/sensitizer BAD as reviewed here, are now well understood genetically, biochemically and structurally,. Remaining unknowns in this regard include elucidating the definitive function of BH3-only proteins claimed to act as borderline direct activators, including PUMA, Noxa, and others, and how non–BCL-2 family proteins can activate the effectors. A major knowledge gap is the lack of mechanistic understanding of the molecular events involving protein-protein and protein-lipid interactions that mediate MOMP. Multidisciplinary research focusing on BCL-2 family protein interactions in the presence of membranes, involving reductionist approaches using reconstituted membrane systems and others involving measurements in live cells [29, 99], seeks to fill this gap. In particular, how the BCL-2 family effectors, induce MOMP remains to be addressed. Moreover, how the interactions of full-length BH3-only proteins with globular BCL-2 family members compare with those of their truncated counterparts, typically encompassing BH3 peptides, needs further exploration and verification. Targeting strategies for MCL-1, the anti-apoptotic family member conferring resistance to cell death in cancer [100], are also lacking. Genetic ablation of this family member results in the most severe phenotypes in mice [101], thus there are adverse toxicity considerations. These concerns are compounded because MCL-1 has a BC groove that remains refractory to drug discovery efforts to identify highly specific inhibitors, mainly due to its lack of plasticity [102]. Nonetheless, anti-cancer drug discovery efforts to directly target effectors holds the promise of ultimately directly manipulating the BCL-2 family proteins with small molecules and bypassing the potentially deleterious effects of their indirect activation through chemotherapy and radiation therapy. Beyond understanding and manipulating the key members through reductionist mechanistic investigations, which inarguably have been tremendously illuminating, the next phase in BCL-2 family research will aim to decode their interactions with the proteome, lipidome and metabolome, which has the potential to not only fine tune their activity but also to reveal uncharacterized critical regulators, such as additional direct activators and non-canonical modulators.
Highlights.
Binding-induced conformational transitions within BCL-2 family proteins regulate apoptosis.
Canonically and non-canonically ligand binding occurs at the globular BCL-2 cores.
Allosteric activation of BCL-2 effectors initiates mitochondrial outer membrane permeabilization (MOMP).
PUMA-induced unfolding of BCL-xL releases p53 to trigger BCL-2 effectors and MOMP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hotchkiss RS, et al. Cell death. N. Engl. J. Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 3.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 4.Enari M, et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki J, et al. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–406. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 6.Chipuk JE, et al. The BCL-2 family reunion. Mol. Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gross A, et al. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 8.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell. Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 9.Lessene G, et al. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 10.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends. Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou JJ, et al. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- 12.Hinds MG, et al. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- 13.Llambi F, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sattler M, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 15.Moldoveanu T, et al. BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 2013;20:589–597. doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czabotar PE, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 17.Walensky LD. Direct BAKtivation. Nat. Struct. Mol. Biol. 2013;20:536–538. doi: 10.1038/nsmb.2579. [DOI] [PubMed] [Google Scholar]
- 18.Gavathiotis E, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edlich F, et al. Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145:104–116. doi: 10.1016/j.cell.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai H, et al. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J. Cell Biol. 2011;194:39–48. doi: 10.1083/jcb.201102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leshchiner ES, et al. Direct activation of full-length proapoptotic BAK. Proc. Natl. Acad. Sci. 2013;110:E986–E995. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moldoveanu T, et al. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol. Cell. 2006;24:677–688. doi: 10.1016/j.molcel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Kim H, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei MC, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 25.Dai H, et al. Evaluation of the BH3-Only Protein Puma as a Direct Bak Activator. J. Biol. Chem. 2013 Nov 21; doi: 10.1074/jbc.M113.505701. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du H, et al. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J. Biol. Chem. 2011;286:491–501. doi: 10.1074/jbc.M110.167148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ren D, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–1393. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim H, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 29.Shamas-Din A, et al. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013;5:a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dewson G, et al. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell. 2008;30:369–380. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths GJ, et al. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 1999;144:903–914. doi: 10.1083/jcb.144.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 33.Bleicken S, et al. Molecular details of Bax activation, oligomerization, and membrane insertion. J. Biol. Chem. 2010;285:6636–6647. doi: 10.1074/jbc.M109.081539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dewson G, et al. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 2012;19:661–670. doi: 10.1038/cdd.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh KJ, et al. Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J. Biol. Chem. 2010;285:28924–28937. doi: 10.1074/jbc.M110.135293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.George NM, et al. A three-helix homo-oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev. 2007;21:1937–1948. doi: 10.1101/gad.1553607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aluvila S, et al. Organization of the Mitochondrial Apoptotic BAK Pore: Oligomerization of the BAK Homodimers. J. Biol. Chem. 2013 Dec 11; doi: 10.1074/jbc.M113.526806. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dewson G, et al. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell. 2009;36:696–703. doi: 10.1016/j.molcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Ma S, et al. Assembly of the Bak apoptotic pore: A critical role for the Bak alpha6 helix in the multimerization of homodimers during apoptosis. J. Biol. Chem. 2013;288:26027–26038. doi: 10.1074/jbc.M113.490094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Annis MG, et al. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Q, Gehring K. Heterodimerization of BAK and MCL-1 activated by detergent micelles. J. Biol. Chem. 2010;285:41202–41210. doi: 10.1074/jbc.M110.144857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou L, Chang DC. Dynamics and structure of the Bax-Bak complex responsible for releasing mitochondrial proteins during apoptosis. J Cell Sci. 2008;121:2186–2196. doi: 10.1242/jcs.024703. [DOI] [PubMed] [Google Scholar]
- 43.Chipuk JE, et al. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 44.Follis AV, et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 2013;9:163–168. doi: 10.1038/nchembio.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chipuk JE, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 46.Leu JI, et al. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell. Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 47.Petros AM, et al. Defining the p53 DNA-binding domain/Bcl-x(L)-binding interface using NMR. FEBS Lett. 2004;559:171–174. doi: 10.1016/S0014-5793(04)00059-6. [DOI] [PubMed] [Google Scholar]
- 48.Day CL, et al. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J. Mol. Biol. 2008;380:958–971. doi: 10.1016/j.jmb.2008.05.071. [DOI] [PubMed] [Google Scholar]
- 49.Petros AM, et al. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ku B, et al. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011;21:627–641. doi: 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Q, et al. Apoptotic regulation by MCL-1 through heterodimerization. J. Biol. Chem. 2010;285:19615–19624. doi: 10.1074/jbc.M110.105452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cartron PF, et al. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol. Cell. 2004;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 53.Gallenne T, et al. Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J. Cell Biol. 2009;185:279–290. doi: 10.1083/jcb.200809153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki M, et al. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 55.Gavathiotis E, et al. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korsmeyer SJ, et al. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 57.Edwards AL, et al. Multimodal Interaction with BCL-2 Family Proteins Underlies the Proapoptotic Activity of PUMA BH3. Chem. Biol. 20:888–902. doi: 10.1016/j.chembiol.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LaBelle JL, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sot B, et al. Comparative biophysical characterization of p53 with the pro-apoptotic BAK and the anti-apoptotic BCL-xL. J. Biol. Chem. 2007;282:29193–29200. doi: 10.1074/jbc.M705544200. [DOI] [PubMed] [Google Scholar]
- 60.Pietsch EC, et al. Oligomerization of BAK by p53 utilizes conserved residues of the p53 DNA binding domain. J. Biol. Chem. 2008;283:21294–21304. doi: 10.1074/jbc.M710539200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chipuk JE, et al. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–381. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 62.Schuler M, Green DR. Transcription, apoptosis and p53: catch-22. Trends Genet. 2005;21:182–187. doi: 10.1016/j.tig.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 63.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hagn F, et al. Bcl-xL changes conformation upon binding to wild-type but not mutant p53 DNA binding domain. J. Biol. Chem. 2010;285:3439–3450. doi: 10.1074/jbc.M109.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petros AM, et al. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 66.Tomita Y, et al. WT p53, but not tumor-derived mutants, bind to Bcl-2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006;281:8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 67.Bharatham N, et al. Molecular basis of Bcl-X(L)-p53 interaction: insights from molecular dynamics simulations. PLoS One. 2012;6:e26014. doi: 10.1371/journal.pone.0026014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu H, et al. The MDM2-binding region in the transactivation domain of p53 also acts as a Bcl-X(L)-binding motif. Biochemistry. 2009;48:12159–12168. doi: 10.1021/bi901188s. [DOI] [PubMed] [Google Scholar]
- 69.Ha JH, et al. Dual-site interactions of p53 protein transactivation domain with anti-apoptotic Bcl-2 family proteins reveal a highly convergent mechanism of divergent p53 pathways. J. Biol. Chem. 2013;288:7387–7398. doi: 10.1074/jbc.M112.400754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arnoult D, et al. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. 2004;101:7988–7993. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Handke W, et al. Live or let die: manipulation of cellular suicide programs by murine cytomegalovirus. Med. Microbiol. Immunol. 2012;201:475–486. doi: 10.1007/s00430-012-0264-z. [DOI] [PubMed] [Google Scholar]
- 72.Cam M, et al. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ. 2010;17:655–665. doi: 10.1038/cdd.2009.147. [DOI] [PubMed] [Google Scholar]
- 73.Handke W, et al. Viral inhibition of BAK promotes murine cytomegalovirus dissemination to salivary glands. J. Virol. 2013;87:3592–3596. doi: 10.1128/JVI.02657-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kvansakul M, Hinds MG. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013;4:e909. doi: 10.1038/cddis.2013.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma J, et al. Structural mechanism of Bax inhibition by cytomegalovirus protein vMIA. Proc. Natl. Acad. Sci. 2013;109:20901–20906. doi: 10.1073/pnas.1217094110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer. 2005;5:876–885. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 77.Lee EF, et al. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007;14:1711–1713. doi: 10.1038/sj.cdd.4402178. [DOI] [PubMed] [Google Scholar]
- 78.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 79.van Delft MF, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shuker SB, et al. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 81.Ackler S, et al. ABT-263 and rapamycin act cooperatively to kill lymphoma cells in vitro and in vivo. Mol. Cancer Ther. 2008;7:3265–3274. doi: 10.1158/1535-7163.MCT-08-0268. [DOI] [PubMed] [Google Scholar]
- 82.Shoemaker AR, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin. Cancer Res. 2008;14:3268–3277. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- 83.Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 84.Balakrishnan K, Gandhi V. Bcl-2 antagonists: a proof of concept for CLL therapy. Invest. New Drugs. 2013;31:1384–1394. doi: 10.1007/s10637-013-0002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ackler S, et al. Navitoclax (ABT-263) and bendamustine +/− rituximab induce enhanced killing of non-Hodgkin's lymphoma tumours in vivo. Br. J. Pharmacol. 2012;167:881–891. doi: 10.1111/j.1476-5381.2012.02048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen J, et al. The Bcl-2/Bcl-xL/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol. Cancer Ther. 2011;10:2340–2349. doi: 10.1158/1535-7163.MCT-11-0415. [DOI] [PubMed] [Google Scholar]
- 87.Souers AJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 88.Vogler M, et al. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br. J. Haematol. 2013 doi: 10.1111/bjh.12457. [DOI] [PubMed] [Google Scholar]
- 89.Touzeau C, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2013 Jul 17; doi: 10.1038/leu.2013.216. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vaillant F, et al. Targeting BCL-2 with the BH3 Mimetic ABT-199 in Estrogen Receptor-Positive Breast Cancer. Cancer Cell. 2013;24:120–129. doi: 10.1016/j.ccr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 91.Lessene G, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 92.Petros AM, et al. Discovery of a potent and selective Bcl-2 inhibitor using SAR by NMR. Bioorg Med Chem Lett. 2010;20:6587–6591. doi: 10.1016/j.bmcl.2010.09.033. [DOI] [PubMed] [Google Scholar]
- 93.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ng KP, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012;18:521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 95.Sarosiek KA, et al. Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol. 2013;51:751–765. doi: 10.1016/j.tcb.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Galluzzi L, et al. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim. Biophys. Acta. 2009;1787:402–413. doi: 10.1016/j.bbabio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 97.Gavathiotis E, et al. Direct and selective small-molecule activation of proapoptotic BAX. Nat. Chem. Biol. 2012;8:639–645. doi: 10.1038/nchembio.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Higueruelo AP, et al. Protein-protein interactions as druggable targets: recent technological advances. Curr. Opin. Pharmacol. 2013;13:791–796. doi: 10.1016/j.coph.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 99.Kale J, et al. Shedding light on apoptosis at subcellular membranes. Cell. 2012;151:1179–1184. doi: 10.1016/j.cell.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 100.Varadarajan S, et al. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ. 2013;20:1475–1484. doi: 10.1038/cdd.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang X, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27:1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Friberg A, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Happo L, et al. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 2012;125:1081–1087. doi: 10.1242/jcs.090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shamas-Din A, et al. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 105.Muchmore SW, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]