

Abstract
The membrane fusion protein (MFP) component, MexA, of the MexAB-OprM multidrug efflux system of P. aeruginosa is proposed to link the inner (MexB) and outer (OprM) membrane components of this pump as a probable oligomer. A cross-linking approach confirmed the in vivo interaction of MexA and MexB, while a LexA-based assay for assessing protein-protein interaction similarly confirmed MexA multimerization. Mutations compromising the MexA contribution to antibiotic resistance but yielding wild-type levels of MexA were recovered and shown to map to two distinct regions within the N- and C-terminal halves of the protein. Most of the N-terminal mutations occurred at residues that are highly conserved in the MFP family (P68, G72, L91, A108, L110, and V129), consistent with these playing roles in a common feature of these proteins (e.g., oligomerization). In contrast, the majority of the C-terminal mutations occurred at residues poorly conserved in the MFP family (V264, N270, H279, V286, and G297), with many mapping to a region of MexA that corresponds to a region in the related MFP of Escherichia coli, AcrA, that is implicated in binding to its RND component, AcrB (C. A. Elkins and H. Nikaido, J. Bacteriol. 185:5349-5356, 2003). Given the noted specificity of MFP-RND interaction in this family of pumps, residues unique to MexA may well be important for and define the MexA interaction with its RND component, MexB. Still, all but one of the MexA mutations studied compromised MexA-MexB association, suggesting that native structure and/or proper assembly of the protein may be necessary for this.
Pseudomonas aeruginosa is an opportunistic human pathogen characterized by an innate resistance to multiple antimicrobials (21). The resistance has historically been attributed to the presence in this organism of an outer membrane (OM) of low permeability (55), but it is increasingly clear that resistance owes much to the operation of broadly specific, so-called multidrug efflux systems (58-60, 64) that work synergistically with limited OM permeability (18, 40, 60). Several multidrug efflux systems in P. aeruginosa have been described to date (61), although the major system contributing to intrinsic multidrug resistance is encoded by the mexAB-oprM operon (38). Hyperexpression of this system also occurs in so-called nalB (27, 28, 75, 88)- and nalC (75, 88)-type multidrug-resistant mutants. MexAB-OprM accommodates a broad range of structurally diverse antimicrobials, including dyes, detergents, inhibitors of fatty acid biosynthesis, organic solvents, disinfectants, and clinically relevant antibiotics (10, 34, 37, 39, 41, 42, 48, 70, 74, 74, 76), and is implicated in the export of homoserine lactones involved in quorum sensing (17, 57) and, possibly, virulence factors (22).
The MexAB-OprM efflux system, like the other tripartite Mex efflux systems in P. aeruginosa, consists of an inner membrane (IM) drug-proton antiporter of the resistance-nodulation-cell division (RND) family (MexB), an OM channel-forming component (OprM; also called OM factor [OMF]), and a periplasmic membrane fusion protein (MFP) (MexA) (58, 86). Crystal structures have not yet been reported for any of the efflux components of P. aeruginosa, although structures are available for the homologous OM (TolC [35]) and RND (AcrB [52]) components of the Mex-like AcrAB-TolC multidrug efflux system of Escherichia coli. The TolC channel is a trimer and spans both the OM (as a β-barrel) and periplasm (as a α-helical barrel) (35). Measuring 140 Å in length, the channel is open at the distal (extracellular) end and tapers almost to a close at the proximal (periplasmic) end, which likely interacts with the RND component, AcrB (35). Modeling studies suggest that OprM adopts a similar structure (43, 80). The AcrB RND component also exists as a trimer, composed of a 50-Å-thick transmembrane region and a 70-Å headpiece that protrudes into the periplasm (52). This headpiece has a funnel-like opening at the top that is connected to a central cavity at the bottom, which, in turn, opens to the periplasm via three vestibules that likely play a role in substrate recognition by and/or access to the pump (52). Indeed, several studies highlight the role played by the periplasmic portion of the RND transporters of E. coli and P. aeruginosa in substrate (i.e., drug) recognition (14, 15, 45, 53, 79, 83). A high-resolution crystal structure for an MFP efflux component is not yet available, although preliminary studies indicate that, e.g., AcrA is also likely trimeric (85) and that monomer AcrA is a highly asymmetric, elongated molecule of sufficient length to span the periplasm (4, 84).
MFPs appear to be oligomeric (possibly trimeric) and are typically anchored to the IM via a single transmembrane helix or via an N-terminal lipid tail, with the bulk of the protein in the periplasm (30, 81). Components of multidrug efflux systems, they also function as part of protein export systems (e.g., the HlyD family) (12). Current models suggest that MFPs interact with one or both of the IM-OM export components, and, indeed, an in vivo interaction of AcrA and AcrB has been confirmed (31, 85). Still, a TolC association with either of these proteins has yet to be demonstrated. Similarly, MexA appears to interact with MexB (79), although no association with OprM has been seen. Studies with the HlyBD-TolC hemolysin exporter have, however, also failed to show TolC association with the HlyBD components unless hemolysin was present and available for export (77). Apparently, the OM TolC component associates transiently with the other export components when substrate is actively being exported, suggesting that TolC or OprM recruitment by, e.g., AcrAB or MexAB may be similarly transient and dependent upon substrate (i.e., drug) entry into the export pathway. Intriguingly, the MFP component of the hemolysin transporter (HlyD), which has also been shown to bind its cognate IM component (HlyB), was also implicated in TolC binding or recruitment (77). Possibly, then, MFPs bind both IM and OM components of tripartite drug-protein export systems and, e.g., promote their assembly into a functional trans-cell envelope export systems. Consistent with this, the MFP component, CvaA, of the tripartite CvaAB-TolC colicin V exporter has been shown to interact with both CvaB and TolC (25).
The present study was undertaken both to assess MexA oligomerization and association with its cognate IM and OM efflux components and to identify regions or residues of MexA that are important for this. Here we report both MexA-MexA and MexA-MexB interactions and identify several residues in MexA whose mutation compromises function, in many instances as a result of interference with one or both of these processes.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The strains and plasmids used in this study are listed in Table 1. Bacteria cells were cultivated at 37°C in Luria broth (LB) (Miller's LB base [Difco] and 2 g of NaCl per liter of H2O) solidified with agar (LB containing 1.5% [wt/vol] agar) as needed. Plasmid pDN3 was constructed by excising the mexA gene of pRSP48 (itself constructed by cloning a mexA-containing 2-kb HindIII-BamHI fragment from pPV6 into pAK1900) on a HindIII-BamHI fragment and cloning it into HindIII-BamHI-restricted pRK415. To construct pDN25, sequences encoding a cleavable hexahistidine tag were first engineered (by PCR) onto the 3′ end of the gene cloned into expression vector pBAD18 (20). Briefly, the bulk of the mexB gene (all but the last 133 bp) was first cloned into pBAD18 on a EcoRI-KpnI fragment excised from pPV6 as described previously (73). The 3′ end of mexB was then amplified by using the previously described primer TK-1 (73) and primer RE2 (5′-AAGCTTAAGCTTtcaGTGGTGGTGGTGGTGGTGGGAGCCGCGCGGCACCAGTTGCCCCTTTTCGACGGACG-3′ [tandem HindIII sites are underlined, and sequences encoding a thrombin cleavage site [italic], hexahistidine [boldface], and a stop codon [lowercase] are also indicated]). PCR formulation and conditions were as described previously (72) except for an annealing temperature of 58°C. Following digestion with KpnI (present naturally in the mexB sequence being amplified with TK-1 [73]) and HindIII, the PCR product was cloned into pAK1900 and sequenced to ensure that no unwanted mutations had been introduced during PCR. The now-His-tagged 3′ end of the mexB gene was subsequently recovered from pAK1900 and cloned into KpnI-HindIII-restricted pBAD18 carrying the 5′end of mexB, to yield pRE35. The mexB-his gene was finally excised from pRE35 on an EcoRI-HindIII fragment and cloned first into pVLT31 (to produce pRE38) and then into pMMB206 (to produce pDN25). E. coli and P. aeruginosa strains containing pMMB206 or pDN25 were grown in the presence of 10 μg of chloramphenicol per ml. When pRK415, pMS604, or their derivatives were used, E. coli and P. aeruginosa strains were grown in the presence of 10 μg of tetracycline per ml. Plasmid pEX18Tc was maintained with tetracycline at 10 μg/ml (E. coli) or 50 μg/ml (P. aeruginosa).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Properties or genotypea | Source or reference |
|---|---|---|
| P. aeruginosa | ||
| K767 | PAO1, wild type | 47 |
| K1491 | K767 ΔmexR | 75 |
| K1119 | K767 ΔmexAB-oprM | 41 |
| K870 | Smr derivative of K767 | 65 |
| K2278 | K870 ΔmexA | This study |
| K2274 | K870 ΔmexRA | This study |
| K2275 | K767 ΔmexRAB | This study |
| E. coli | ||
| DH5α | endA hsdR17 supE44 thi-1 recA1 gyrA relA1 Δ(lacZYA-argF)U169 deoR [φ80 dlacΔ(lacZ)M15] | 3 |
| S17-1 | thi pro hsdR recA Tra+ | 71 |
| SU101 | lexA71::Tn5 (Def)sulA211 sulA::lacZ | 13 |
| Δ(lacIPOZYA)169/F′ lacIqlacZΔM15::Tn9 | ||
| Plasmids | ||
| pAK1900 | E. coli-P. aeruginosa shuttle cloning vector; MCS; Cbr/Apr | A. Kropinski, Queens University |
| pPV2 | pAK1900 carrying mexR-mexAB-oprM and flanking DNA on a 9-kb XhoI fragment | 62 |
| pPV6 | pAK1900 carrying mexR-mexAB-oprM and flanking DNA on a 5.5-kb XhoI-HindIII fragment | 62 |
| pRSP48 | pAK1900::mexA | This study |
| pEX18Tc | Gene-replacement vector; sacB Tcr | 23 |
| pDN1 | pEX18Tc:: ΔmexRA | This study |
| pDN2 | pEX18Tc:: ΔmexRAB | This study |
| pRK415 | Broad-host-range cloning vector; Tcr | 32 |
| pRSP17 | pRK415::mexAB-oprM | 73 |
| pDN3 | pRK415::mexA (WT) | This study |
| pDN4 | pRK415::mexA* (P68S) | This study |
| pDN5 | pRK415::mexA* (G72S) | This study |
| pDN6 | pRK415::mexA* (A108T) | This study |
| pDN7 | pRK415::mexA* (V129M) | This study |
| pDN8 | pRK415::mexA* (T256I) | This study |
| pDN9 | pRK415::mexA* (V264E) | This study |
| pDN10 | pRK415::mexA* (N270S) | This study |
| pDN11 | pRK415::mexA* (H279Y) | This study |
| pDN12 | pRK415::mexA* (V286A) | This study |
| pDN13 | pRK415::mexA* (G297D) | This study |
| pDN26 | pRK415::mexA* (L91P) | This study |
| pDN27 | pRK415::mexA* (L110P) | This study |
| pDN28 | pRK415::mexA* (N270S, S251P) | This study |
| pDN29 | pRK415::mexA* (G297D, L155P) | This study |
| pMS604 | pUC-like vector carrying a LexA1-87 WT-Fos zipper fusion and a ColE1 replicon; Tcr | 66 |
| pDN14 | pMS604::mexA (WT) | This study |
| pDN15 | pMS604::mexA* (P68S) | This study |
| pDN16 | pMS604::mexA* (G72S) | This study |
| pDN17 | pMS604::mexA* (A108T) | This study |
| pDN18 | pMS604::mexA* (V129M) | This study |
| pDN19 | pMS604::mexA* (T256I) | This study |
| pDN20 | pMS604::mexA* (V264E) | This study |
| pDN21 | pMS604::mexA* (N270S) | This study |
| pDN22 | pMS604::mexA* (H279Y) | This study |
| pDN23 | pMS604::mexA* (V286A) | This study |
| pDN24 | pMS604::mexA* (G297D) | This study |
| pDN28 | pMS604::mexA* (L91P) | This study |
| pDN29 | pMS604::mexA* (L110P) | This study |
| pVLT31 | Broad-host-range, low-copy-number cloning vector lacIq Tcr | 11 |
| pRE38 | pVLT31::mexB-6his | This study |
| pMMB206 | Broad-host-range, low-copy-number cloning vector; lacIq Cmr | 51 |
| pDN25 | pMMB206::mexB-6his | This study |
| pET21d(+) | His tag vector used to generate His6 C-terminal fusions to products of cloned genes; Apr | Novogen |
| pXZL05 | pET21d(+)::mexA | This study |
Smr, streptomycin resistance; Cbr, carbenicillin resistance; Tcr, tetracycline resistance; Apr, ampicillin resistance; Cmr; chlorampheicol resistance; mexB-6his, a mexB gene engineered to produce a MexB protein with six C-terminal histidine residues; MCS, multiple cloning site. Mutations in the MexA proteins encoded by the plasmids are shown in parentheses WT, wild-type MexA.
Construction of mex deletion strains.
To construct a P. aeruginosa strain lacking mexR and carrying an in-frame deletion of mexA, a mexA deletion strain was first constructed and mexR was subsequently deleted by using the previously described mexR deletion vector pRSP75 (75). Elimination of the chromosomal mexA gene was necessary to permit screening of mutated plasmid-borne mexA genes for changes affecting substrate selectivity (see below), while elimination of mexR served to ensure high-level expression of the chromosomally encoded MexB and OprM components. Together with the high-level expression of the plasmid-encoded MexA (under plac control [see below]), this served to ensure maximal production of a MexAB-OprM efflux system and thus ready assessment of its activity (by using MIC assays) and any changes in activity resulting from mutations in mexA. The mexA deletion was constructed in gene replacement vector pEX18Tc following amplification (via PCR) of ca. 1-kb portions upstream and downstream of the mexA sequences being deleted. The upstream region was amplified off plasmid pPV2 by using primers JT-2 (5′-GTCGACGTCGACGATTTCCTCAACGTCAGCAAG-3′ [the SalI site is underlined]) and JT-3 (5′-TCGGGGTACCTTGCATAGCGTTGTCCTCATG3′ [the KpnI site is underlined]) and cloned into SalI-KpnI-restricted pEX18Tc. The downstream region was amplified off pPV2 by using primers JT-4 (5′-TCGGGGTACCGCGAAAACCGACAGCAAGGGC3′ [the KpnI site is underlined]) and JT-5 (5′-ACCGGAATTCAGGTACATCACCAGGAACACG3′ the EcoRI is site underlined) and cloned into pEX18Tc carrying the upstream region and restricted with KpnI and EcoRI, to yield pDN1. Conditions for PCR were as described previously (72), except that dimethyl sulfoxide was omitted from the reaction mixture and annealing was carried out at 60°C. The ΔmexA construct was sequenced to ensure that no mutations had been introduced during PCR and subsequently mobilized into P. aeruginosa strain K870 via conjugation with pDN1-carrying E. coli S17-1 as described previously (75). Transconjugants were selected on LB agar containing tetracycline (50 μg/ml) and streptomycin (250 μg/ml) (to counterselect donor E. coli), and those harboring a chromosomal deletion of mexA were subsequently recovered on sucrose plates (LB agar containing 10% [vol/vol] sucrose) and screened for loss of MexA following immunoblotting of whole-cell extracts. Loss of MexA was also confirmed by using drug susceptibility assays, with mexA deletion strains demonstrating enhanced susceptibility to representative MexAB-OprM antimicrobial substrates (e.g., carbenicillin, chloramphenicol, and novobiocin). Successful complementation by the cloned mexA gene (on plasmid pDN3) also confirmed that no other gene had been affected. The mexR gene was then deleted from a representative ΔmexA strain, K2278, by using pRSP35 as described previously (75) to yield the ΔmexR ΔmexA strain K2274.
In constructing a strain lacking mexR and carrying an in-frame deletion of mexAB, it was necessary to retain the promoter region of mexAB-oprM to ensure continued expression of the downstream oprM once the mexR-mexAB deletion was engineered into the chromosome of P. aeruginosa. Initially, a ca. 1-kb region upstream of the deletion endpoint in mexA (6 bp into the coding region) was amplified from the chromosome of the ΔmexR strain K1491 by using primers DN1 (5′-AAGCTTAAGCTTGGCCACCTCGCGCTGTACGGGCTG-3′ [HindIII sites are underlined]) and DN2 (5′-GAGCTCGAGCTCTTGCATAGCGTTGTCCTCATG-3′ [SacI sites are underlined]), producing a fragment that carried a mexR deletion as well as the mexAB-oprM promoter. Following digestion with HindIII and SacI, this fragment was cloned into HindIII-SacI-restricted pEX18Tc. A ca. 1-kb fragment encompassing the region downstream of the deletion endpoint in mexB (300 bp upstream of the mexB stop codon) was then amplified from plasmid pRSP17 by using primers DN3 (5′-GAGCTCGAGCTCTCCAACGACGTGTTCTTCCAGGTG-3′ [SacI sites are underlined]) and DN4 (5′-GAATTCGAATTCCGCCGACGTCGTAGCTGCGC-3′ [BamHI sites are underlined]), and the SacI-BamHI-restricted product was cloned into pEX18Tc carrying the upstream fragment. The resultant ΔmexR-ΔmexAB construct, pDN2, was verified by nucleotide sequencing and mobilized into K767 as described above. Transconjugants were selected on tetracycline (50 μg/ml) and chloramphenicol (5 μg/ml) (to counterselect the E. coli donor), and strains carrying the mexR-mexAB deletion were selected on sucrose and screened for loss of MexA and MexB but overproduction of OprM by immunoblotting.
DNA manipulations.
Standard protocols were used for restriction endonuclease digestions, ligations, transformation, plasmid isolation, and agarose gel electrophoresis, as described by Sambrook and Russell (68). Genomic DNA of P. aeruginosa was extracted according to the protocol of Barcak et al. (5). E. coli cells were made competent by using the CaCl2 method (68) or the method of Inoue et al. (26). DNA sequencing was performed by Cortec DNA Services Laboratories (Kingston, Ontario, Canada) with universal and custom oligonucleotide primers.
In vitro mutagenesis of mexA.
Plasmid pDN3 carrying the mexA gene under control of the constitutive (in P. aeruginosa) plac was mutagenized by a 1-h treatment with hydroxylamine according to a previously published protocol (17a). Mutagenized plasmid DNA was then precipitated by using a standard protocol (68), washed in 70% (vol/vol) ethanol, and resuspended in 20 μl of H2O. E. coli S17-1 was electroporated with 1 to 3 μl of hydroxylamine hydrochloride-treated pDN3 and, following addition of 1 ml of LB, was permitted to recover at 37°C with shaking for 1 h in the absence of antibiotic selective agents. Electroporated cells (1 ml) were then added to 9 ml of LB containing tetracycline (10 μg/ml) and cultured overnight at 37°C with shaking. Mutagenized pDN3 was mobilized into the ΔmexA P. aeruginosa strain K2274 from E. coli S17-1 via conjugation as described previously (63) with modifications. Briefly, a 1.0-ml overnight culture of plasmid-containing E. coli S17-1 was mixed with 0.3 ml of an overnight LB culture of P. aeruginosa K2274 and pelleted in a microcentrifuge tube. Following resuspension in 150 μl of LB and spotting onto the center of an LB agar plate, cells were incubated for 4 h at 37°C. Bacterial growth was then resuspended in 1 ml of LB, and appropriate dilutions were plated on LB plates containing tetracycline (10 μg/ml) and streptomycin (250 μg/ml) (to counterselect the donor E. coli S17-1). Transconjugants carrying plasmids with inactivating mutations in mexA were then identified by their inability to complement the antibiotic resistance defect of the ΔmexA strain K2274. Initially, these were identified by their lack of growth on LB agar containing 30 μg of carbenicillin per ml (K2274 producing wild-type MexA from pDN3 grows readily on this concentration of carbenicillin). Subsequent susceptibility testing with additional representative MexAB-OprM antimicrobial substrates (e.g., novobiocin and chloramphenicol) confirmed the inability of these mutant mexA plasmids to restore antibiotic resistance to K2274. To confirm that the defect actually resided with the plasmid-borne mexA genes and was not attributable to spontaneous mutations in K2274, plasmids carrying prospective mutant mexA genes were isolated, reintroduced into K2274, and shown not to complement the multidrug-hypersusceptible phenotype of this strain.
Mutagenesis of mexA was also carried out by PCR with Taq DNA polymerase (Qiagen Inc., Mississauga, Ontario, Canada) as per the manufacturer's instructions. In this case, 100 ng of pDN3 DNA was amplified at 59°C for 25 cycles with primers JT-28 (5′-AAGCTTAAGCTTTGAATGTAAGTATTTTGCCTGC-3′ [tandem HindIII sites are underlined]) and JT-27 (5′-GAGCTCGAGCTCGATCACCCACGCGAAAATGG-3′ [tandem SacI sites are underlined]), and the resultant mexA-containing PCR products were digested with HindIII and SacI and cloned into HindIII-SacI-restricted pRK415. Following the ligation reaction, though, DNA was dialyzed against water, electroporated into E. coli S17-1, mobilized into P. aeruginosa K2274, and screened for mutations exactly as described above. In some instances site-directed mutagenesis with PfuI polymerase (Stratagene) and a protocol supplied with the enzyme was used to recreate mutations obtained by using one of the procedures described above.
MexA homodimerization assay.
To assess MexA-MexA interaction and the possible negative impact of mutations in mexA on this, wild-type and mutated (isolated as described above) mexA genes were individually engineered into the LexA fusion vector pMS604. This required amplification of the mexA genes (exclusive of sequences encoding the signal peptide and the cysteine residue that is normally acylated in the wild-type protein) from their respective pRK415 derivatives by using primers MexA-AgeI-for (5′-CGACCGGTACCGGTAGGAAAAAGCGAGGCG-3′ [tandem AgeI sites are underlined]) and MexA-604-rev (5′-GGCTCGAGCTCGAGTCAGCCCTTGCTGTC-3′ [tandem XhoI sites are underlined]) and the reaction conditions outlined above for construction of a mexA deletion. Following digestion with AgeI and XhoI, the mexA-containing PCR products were cloned into AgeI-XhoI-restricted pMS604 and sequenced, to confirm that no additional mutations were introduced during PCR. The resultant pMS604 derivatives encoding fusions of wild-type and mutant MexA to the DNA-binding domain of LexA were introduced into E. coli SU101, and production of MexA-LexA fusion proteins was confirmed by immunoblotting with MexA antibodies. Plasmid-bearing SU101 was then cultivated overnight in LB containing chloramphenicol (10 μg/ml), diluted 1:49 into the same medium, and grown to mid-log phase before being assayed for β-galactosidase activity as described previously (49). SU101 carries a lacZ gene under the control of an operator to which dimeric LexA can bind, and as such, dimerization of the LexA-binding domains promoted by, e.g., interaction of the MexA proteins fused to them leads to repression of lacZ. Loss of or reduction in β-galactosidase activity is thus a measure of MexA-MexA interaction.
MexA-MexB interaction assay.
To demonstrate the in vivo interaction of MexA and MexB, a plasmid-encoded histidine-tagged MexB was engineered into P. aeruginosa and copurification of MexA with MexB-His on Ni-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen) was assessed. In the initial assay, which is based upon a protocol developed by Thanabalu et al. (77), P. aeruginosa K1119 (ΔmexAB-oprM) harboring pDN3 (pRK415::mexA) and pDN25 (pMMB206:: mexB-6his) was cultured overnight in LB (50 ml) containing tetracycline (10 μg/ml), chloramphenicol (10 μg/ml), and IPTG (isopropyl-[beta]-d-thiogalactopyranoside) (1 mM) (to induce MexB-His). Cells, which typically produced levels of both proteins that were similar to that seen in nalB mutants (74), were harvested, resuspended in 1 ml of 50 mM Na2HPO4 (pH 8.0)-0.3 M NaCl (buffer A) containing 0 to 7 mM concentrations of the cross-linking agent dithiobis(succinimidylpropionate) (DSP) (Pierce), and incubated for 30 min at 37°C. Following quenching of DSP with Tris as described previously (77), cell envelopes were prepared (73) and solubilized in buffer A with 1% Triton X-100 (buffer B) for 20 min on ice. Soluble membrane fractions containing MexB-His were incubated for 20 min at room temperature in a microcentrifuge tube containing 0.5 ml of Ni-NTA agarose (Qiagen) equilibrated in buffer B. The Ni-NTA was centrifuged and, after the supernatant was discarded, washed twice with 10 ml of buffer B containing 30 mM imidazole. Bound MexB-His was then eluted with 0.5 ml of buffer B containing 1 M imidazole and 0.25 M Na-EDTA. Following treatment with dithiothreitol (DTT) (50 mM, 30 min, 37°C) to break cross-links (when cells were treated with DSP), samples (10 μl) were electrophoresed on sodium dodecyl sulfate (SDS)-polyacrylamide gels and immunoblotted with antibodies to MexB (to confirm recovery of MexB-His) and MexA (to assess corecovery of MexA). Control experiments were run with P. aeruginosa K1119 carrying pDN3 but lacking pDN25 (in which case pMMB206 was substituted) to ensure that any retention of MexA was MexB-His dependent (i.e., due to MexA binding to MexB-His) and not due to nonspecific binding to Ni-NTA. In later studies, P. aeruginosa strain K2275 was used and treatment with DSP was omitted, since MexA association with MexB-His was observed in this strain without treatment with this cross-linker. To examine the impact of mexA mutations on MexA association with MexB, the experiment described above was carried out with K2275 harboring pDN25 and pRK415 derivatives expressing the mutant MexA proteins
Susceptibility testing.
The antimicrobial susceptibilities of P. aeruginosa K2278 and K2274 carrying pRK415 and derivatives encoding wild-type and mutant MexA were assessed by using the twofold serial microtiter broth dilution method described previously (29) with an inoculum of 5 × 105 cells per ml. MICs were recorded as the lowest concentration of antibiotic inhibiting visible growth after an 18-h incubation at 37°C.
Purification of MexA and generation of MexA-specific polyclonal antiserum.
To facilitate purification of MexA for the purpose of raising antibodies to the protein, the mexA gene was cloned into the His tag expression vector pET21d(+) (Novogen). The mexA gene was amplified from plasmid pPV6 via PCR with primers mexa1xz (5′-TATAGGATCCGCATCGGCCGCTTTCGCT-C-3′ [the BamHI is site underlined]) and mexa2xz (5′-ATTACTCGAGGCCCTTGCTGTCGGTTTTC-3′ [the XhoI site is underlined]) and reaction conditions described previously for amplification and cloning of oprM into pET21d(+) (87). Following digestion with BamHI and XhoI, the mexA-containing PCR product was cloned into BamHI-XhoI-restricted pET21d(+), and the resultant vector, pXZL05, was subsequently introduced into E. coli DE21/DE3 harboring the pLysS vector. Expression of the MexA-His6 fusion encoded by pXZL05 and its subsequent purification was achieved as described previously (87), except that MexA-His6 was solublized from cell envelopes in 20 mM Tris-HCl (pH 8.0)-1% (vol/vol) Triton X-100 and Triton X-100 (0.1%[vol/vol]) replaced sarcosyl in the TALON (Clontech) metal affinity column buffers. Antibodies to purified MexA-His6 were subsequently raised in New Zealand White rabbits (Lucy Mutharia, Department of Microbiology, University of Guelph).
To remove background reactivity seen in immunoblots with the anti-MexA antiserum, it was adsorbed against whole-cell extracts of E. coli strain DE21/DE3(pLysS) and P. aeruginosa strain K1119. Briefly, overnight cultures (100 ml) grown in LB were harvested by centrifugation, washed twice with phosphate-buffered saline (PBS) (1.7 mM NaH2PO4, 8.1 mM Na2HPO4, 145 mM NaCl), and resuspended in 25 ml PBS. Cells were disrupted with a French pressure cell (two passages) following addition of RNase, DNase, and lysozyme as described previously (20a), and SDS was added to a final concentration of 0.4% (wt/vol). Following a 1-h incubation at 60°C and centrifugation to remove insoluble material, the resultant cell extracts were diluted 1:2 in PBS and 15-ml aliquots were incubated on individual nitrocellulose disks (6-cm diameter; Bio-Rad) for 3 h at 37°C. The nitrocellulose disks were washed three times in 10 ml of PBS containing 0.05% Tween 20 (10 min each) and subsequently incubated in PBS containing 3% (wt/vol) skim milk powder for 1 h at 37°C. After the disks were again washed three times with PBS, the anti-MexA polyclonal antiserum (10 ml) was incubated sequentially on three each of the E. coli and P. aeruginosa disks for 1 h at 37°C to produce the adsorbed, MexA-specific antiserum.
SDS-polyacrylamide gel electrophoresis and immunoblotting.
The protocols for SDS-polyacrylamide gel electrophoresis and Western immunoblotting have been described previously (73). The preparation of antibodies used to detect MexB (73) and OprM (87) have also already been described. To confirm expression of wild-type and mutant MexA in P. aeruginosa strain K2274 and of wild-type and mutant MexA-LexA fusions in E. coli strain SU101, immunoblotting with anti-MexA antiserum was carried out on whole-cell extracts prepared from overnight LB cultures as described previously (67).
RESULTS
Isolation of MexA mutants compromised for antibiotic resistance.
To identify residues of MexA that are important for function, >5,000 colonies of a ΔmexA derivative of PA01 (strain K2274) harboring hydroxylamine-mutated, plasmid-borne mexA were screened for increased susceptibility to representative MexAB-OprM antimicrobial substrates (carbenicillin, chloramphenicol, and novobiocin). While the wild-type mexA gene (on pDN3) enhanced resistance of K2274 to all three antibiotics (MIC of carbenicillin, 128 μg/ml; MIC of chloramphenicol, 64 μg/ml; and MIC of novobiocin, 512 μg/ml), several dozen colonies carrying mutated pDN3 exhibited susceptibility characteristic of K2274 lacking a mexA gene (MIC of carbenicillin, 2 μg/ml; MIC of chloramphenicol, 8 μg/ml; and MIC of novobiocin, 16 μg/ml). Of these, 12 produced wild-type levels of MexA protein (Fig. 1A and data not shown). Subsequent nucleotide sequencing confirmed single mutations in the mexA genes of 10 colonies (Table 1, plasmids pDN4 to -10, -12, -26, and -27) and double mutations in 2 others (Table 1, pDN28 and -29). When the S251P and N270S mutations of pDN28 were individually engineered into mexA, only the latter (on plasmid pDN11 [Table 1]) compromised MexA-dependent antibiotic resistance (MICs of carbenicillin, chloramphenicol, and novobiocin were the same as those for K2274 without mexA), although a MexA product was not visible in immunoblots (Fig. 1A, lane 9). Similarly, only the G297D mutation of pDN29 (engineered to produce pDN12) fully compromised MexA-dependent antibiotic resistance, and it too failed to produce detectable MexA (Fig. 1A, lane 12). These 12 inactivating mutations clustered in two regions of MexA, one in the N-terminal portion of this 383-amino-acid protein between residues P68 and V129 and the second in the C terminus between residues T256 and G297, identifying, perhaps, two distinct functional domains in this protein.
FIG. 1.

Western immunoblot of whole-cell extracts of P. aeruginosa K2274 (A) and E. coli SU101 (B), demonstrating expression of wild-type and mutant MexA proteins from mexA genes cloned onto pRK415 (A) and pMS604 (B). Lane 1, wild-type MexA; lane 2, vector control; lane 3, MexAP68S; lane 4, MexAG72S; lane 5, MexAA108T; lane 6, MexAV129 M; lane7, MexAT256I; lane 8, MexAV264E; lane9, MexAN270S; lane 10, MexAH279Y; lane 11, MexAV286D; lane 12, MexAG297D; lane 13, MexAL91P; lane 14, MexAL110P. See Table 1 for designations of the pMS604 and pRK415 derivatives carrying the indicated wild-type and mutant mexA genes.
In vivo interaction of MexA with MexB. Although preliminary cross-linking studies suggested that MexA and MexB interact in vivo, the results were not unequivocal (79). To better assess this interaction, then, a ΔmexAB-oprM strain (K1119) carrying plasmids encoding wild-type MexA (pDN3) and a wild-type but hexahistidine-tagged MexB (pDN25) was treated with a cross-linking agent (DSP) and MexB-His was subsequently recovered on Ni-NTA, with copurification of MexA used as an indication of in vivo MexA-MexB association. As seen in Fig. 2A (top panel, lane 3), very little MexA was bound to and eluted from the Ni-NTA when cells were not previously treated with DSP, although MexA levels recovered from Ni-NTA increased markedly following DSP treatment (Fig. 2B and C, top panels, lanes 3). Indeed, the amount of MexA bound to and eluted from Ni-NTA increased with increasing DSP concentration (compare lanes 3 [top panel] of Fig. 2C and B) despite there being no significant change in the amount of MexB-His recovered from Ni-NTA at the different DSP concentrations (Fig. 2B and C, bottom panels, lanes 3). As expected, too, the amounts of MexA and MexB-His present in K1119 harboring pDN3 and pDN25 were not influenced by DSP treatment (Fig. 2, lanes 1). Thus, increased recovery of MexA from Ni-NTA in the presence of increasing DSP is best explained by increased stabilization of existing MexA-MexB complexes by DSP in vivo. To confirm that the recovery of MexA from Ni-NTA following DSP treatment was, in fact, due to association of MexA with MexB-His, attempts were made to show that such recovery was MexB-His dependent. To this end, K1119 expressing MexA with and without MexB-His was treated with DSP, cell extracts were incubated with Ni-NTA, and recovery of MexA on the Ni-NTA was assessed. As seen in Fig. 3, while substantial levels of MexA were recovered from the Ni-NTA when extracts from DSP-treated K1119 expressing both MexA and MexB-His were applied to the Ni-NTA (Fig. 3A, lane 2), almost undetectable levels of the protein were bound to and eluted from Ni-NTA when extracts were from K1119 expressing only MexA (Fig. 3A, lane 4). These differences were not attributable to, e.g., more MexA being produced by K1119 harboring pDN3 alone (Fig. 3A, lane 3) versus both pDN3 and pDN25 (Fig. 3A, lane 1), and they clearly confirm that MexA recovery from Ni-NTA requires MexB-His and thus represents a specific in vivo interaction between MexA and MexB.
FIG. 2.

Western immunoblot demonstrating in vivo cross-linking of MexA to MexB-His6. P. aeruginosa strain K2275 carrying plasmids pDN3 (pRK415::mexA) and pDN25 (pMMB206::mexB-6his) was treated with 0 (A), 1 (B), or 4 (C) mM DSP, and Triton X-100-soluble extracts of cell envelope preparations were incubated with Ni-NTA. Triton X-100-soluble cell envelope extracts (1 μl) (lane 1), wash fractions from Ni-NTA (10 μl) (lane 2), and elution fractions from Ni-NTA (10 μl) (lane 3) were immunoblotted and developed with antibodies to MexA (top panels) and MexB (bottom panels). Note that treatment of elution fractions with DTT (to break cross-links) and subsequent heating prior to electrophoresis and immunoblotting permitted visualization of monomeric MexA and MexB-His only.
FIG. 3.
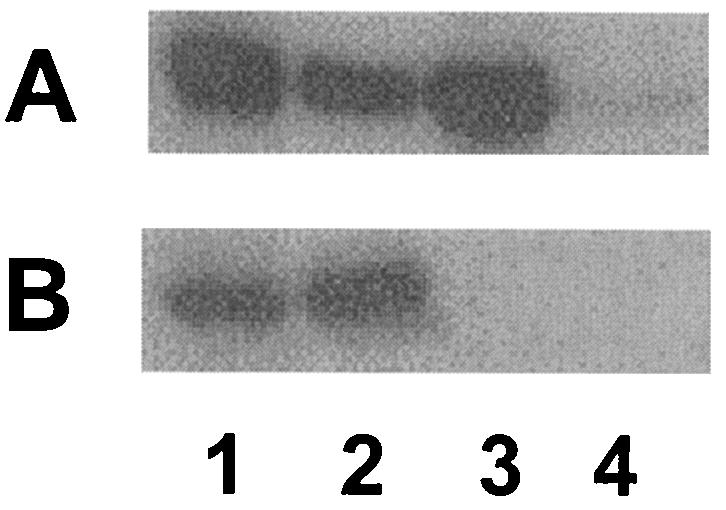
Western immunoblot demonstrating MexB-His-dependent recovery of MexA on Ni-NTA. P. aeruginosa strain K2275 carrying plasmid pDN3 (pRK415::mexA) and either pDN25 (pMMB206:: mexB-6his) (lanes 1 and 2) or pMMB206 (lanes 3 and 4) was treated with 4 mM DSP, and Triton X-100-soluble extracts of a cell envelope preparation were incubated with Ni-NTA. Triton X-100-soluble cell envelope extracts (1 μl) applied to Ni-NTA (lanes 1 and 3) and DTT-treated elution fractions (10 μl) recovered from Ni-NTA (lanes 3 and 4) were immunoblotted and developed with antibodies to MexA (A) and MexB (B).
Mutations in MexA compromising MexA-MexB association.
To assess which if any of the above-described MexA mutations interfered with in vivo MexA association with MexB, DSP-mediated cross-linking of MexB-His with the various mutant MexA proteins expressed in K1119 and their subsequent recovery on Ni-NTA were assessed. Somewhat surprisingly, all mutant MexA proteins examined could be cross-linked to MexB-His and thus could be recovered on Ni-NTA (data not shown). Still, mutations that compromised a productive or functional interaction between MexA and MexB might nonetheless permit a sufficiently close association such that cross-linking could stabilize an otherwise weak interaction with MexB and provide for recovery of the mutant MexA proteins with MexB-His on Ni-NTA. Unfortunately, while demonstration of a MexA-MexB-His interaction without the use of a cross-linking agent would certainly be a better indication of their in vivo association, such an interaction was generally not measurable in strain K1119 in the absence of DSP (Fig. 2A, upper panel, lane 3). (The limited recovery of MexA with MexB-His on Ni-NTA in this instance was comparable to the background level of MexA recovered on Ni-NTA in the absence of MexB-His [Fig. 3A, lane 4] and as such is not indicative of any MexA-MexB association.) When these studies were carried out with an OprM-producing strain (e.g., K2275), however, pDN3-encoded wild-type MexA was readily recovered from Ni-NTA in the absence of prior cross-linking with DSP but only when MexB-His was also expressed (Fig. 4A lane 2, wild type [c.f. lane 4]). This MexB-His-dependent recovery of MexA from Ni-NTA in the absence of prior cross-linking thus provided a cross-linker-free measure of in vivo MexA-MexB association. When extracts from K2275 expressing MexB-His and the various mutant MexA proteins (Fig. 4, lanes 1) were then applied to Ni-NTA, only one mutant MexA protein (G72S) showed evidence of MexB-His association (Fig. 4A, G72S, lane 2). Thus, the bulk of inactivating mutations in MexA compromised, to some extent at least, MexB association in vivo.
FIG. 4.

Western immunoblot assessing in vivo binding of wild-type (WT) and mutant MexA proteins to MexB-His6. Cell envelopes of P. aeruginosa strain K2275 carrying plasmid pDN25 (pMMB206:: mexB-6his) (lanes 1 and 2) or pMMB206 (lanes 3 and 4) and pRK415 derivatives expressing the indicated MexA proteins were extracted with Triton X-100 and applied to Ni-NTA. Triton X-100-soluble cell envelope extracts (1 μl) applied to Ni-NTA (lanes 1 and 3) and DTT-treated elution fractions (10 μl) recovered from Ni-NTA (lanes 2 and 4) were immunoblotted and developed with antibodies to MexA (A) and MexB (B). See Table 1 for designations of the pRK415 derivatives expressing the indicated MexA proteins.
Mutations in MexA compromising MexA-MexA interaction.
Previous studies with MFPs in other organisms indicate that these proteins are oligomeric (7, 77, 85), and, indeed, a preliminary cross-linking study suggested that MexA forms multimers (79). To confirm a MexA-MexA interaction and subsequently assess the impact of the above-described MexA mutations on such self-association, the mexA genes were fused in frame to coding sequences for the DNA-binding domain of LexA on pMS604, and repression of a chromosomal lacZ gene under the control of a LexA operator (i.e., sulA) was assessed in E. coli SU101. LexA binding to sulA and subsequent repression of lacZ in SU101 requires prior dimerization of the LexA-binding domains encoded by pMS604, necessitating interaction of the MexA sequences fused to the LexA DNA-binding domains. Thus, any reduction of β-galactosidase activity produced by SU101 carrying pMS604::mexA relative to a plasmid without an insert provides a measure of MexA-MexA interaction in vivo. The observation, then, that SU101 expressing a fusion of wild-type MexA and LexA (from pMS604 derivative pDN14 [Fig. 1B, lane 1]) produced sevenfold less β-galactosidase activity than the same strain carrying pMS604 (Table 2) clearly confirms a MexA-MexA interaction and indicates that MexA is multimeric (at least dimeric) in vivo. When the mutant mexA genes were subsequently cloned into pMS604, all but the MexAL91P-LexA (Fig. 1B, lane 13) and MexAL110P-LexA (Fig. 1B, lane 14) fusion proteins were detectable in SU101 (Fig. 1B) and could thus be assessed for MexA-MexA interaction. Interestingly, both of the previously unstable (in P. aeruginosa [Fig. 1A, lanes 9 and 12]) MexAN270H and MexAG297Dmutant proteins produced stable fusions with LexA (Fig. 1B, lanes 9 and 12) and so could also be examined for an impact on MexA-MexA interaction. Of the four mutations at the N terminus whose impact on MexA-MexA interaction could be examined in this assay, two had a negative impact (G72S and V129M) (Table 2). Interestingly, four of six C-terminal mutations that compromised MexA function did not affect the MexA-MexA interaction (only the T256I and G297D mutations did). Intriguingly, the pDN18-encoded MexAV129M yielded β-galactosidase activity that was almost threefold higher than that of the negative control pMS604 lacking a mexA insert. Whether this reflects some ability of the pMS604-encoded LexA sequences to self-associate and this is fully disrupted in the mexA (V129M) construct is unclear.
TABLE 2.
Influence of MexA mutations on MexA-MexA interactiona
| Plasmid | MexA mutation | β-Galactosidase activityb (Miller units) |
|---|---|---|
| pMS604 | 386 ± 9 | |
| pDN14 | Wild type | 56 ± 4 |
| pDN15 | P68S | 21 ± 3 |
| pDN16 | G72S | 247 ± 8 |
| pDN17 | A108T | 64 ± 6 |
| pDN18 | V129M | 919 ± 40 |
| pDN19 | T256I | 253 ± 20 |
| pDN20 | V264E | 6 ± 1 |
| pDN21 | N270S | 8 ± 1 |
| pDN22 | H279Y | 64 ± 27 |
| pDN23 | V286A | 7 ± 6 |
| pDN24 | G297D | 206 ± 23 |
E. coli SU101 carrying the indicated pMS604 plasmid derivatives expressing LexA fusions to wild-type or mutant MexA was grown to late log phase and assayed for β-galactosidase activity as described in Materials and Methods. Boldface indicates that MexA-MexA interaction is compromised.
Results are the means ± standard deviations from two or three independent experiments carried out in triplicate.
Conservation in other MFP components of residues whose mutation in MexA compromises function.
To determine if MexA residues shown here to be of functional significance were conserved in other MFPs and thus were of general functional importance for this family of proteins, MexA was aligned with the MFPs of several organisms, including MFPs that function in drug efflux and protein export (Table 3). Intriguingly, the N-terminal residues of MexA whose mutation compromised function were all highly conserved, although P68 could be replaced with other turn-promoting residues (e.g., alanine) and V129 was often replaced with another branched hydrophobic residue (e.g., isoleucine). In contrast, of the C-terminal residues whose mutation compromised MexA function, only T256 was highly conserved, with V264, N270, H279, and V286 not conserved at all and G297 only weakly conserved (although a small residue appeared to predominate at this position in most MFPs). The conservation of functionally significant residues in the N terminus and the lack of this in the C terminus tends to reflect a generally greater degree of amino acid sequence homology within the N terminus of the MFP family than in the C terminus (e.g., in Fig. 5, 26 of 35 absolutely conserved residues in the three MFPs shown occur in the first half of the mature proteins).
TABLE 3.
Conservation in MFP components of multidrug and protein exporters of amino acid residues whose mutation in MexA compromises drug resistancea
| Mutated residue in MexAb | Corresponding residue inc:
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MexC | MexE | MexX | MexJ | MexH | MexV | AcrA | MtrC | AdeA | AmeA | CeoA | TtgA | SmeD | AmrA | CmeA | COG0864 (AcrA) | HlyD family | COG1566 (EmrA) | |
| P68 | A | P | A | F | A | A | P | A | P | P | P | P | P | A | P | A | P | P |
| G72 | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G |
| L91 | L | L | L | L | L | L | L | L | L | L | L | L | L | L | L | L | L | L |
| A108 | G | A | G | A | A | A | G | A | A | A | A | A | A | A | A | A | A | A |
| L110 | L | L | L | V | L | L | L | L | L | V | L | L | A | H | F | L | L | L |
| V129 | V | I | I | V | A | I | I | I | I | A | I | V | V | V | I | A | I | V |
| T256 | T | T | T | S | S | T | T | T | T | T | A | T | T | T | S | T | L | —d |
| V264 | K | V | L | A | L | A | I | A | E | L | R | L | K | L | V | E | V | — |
| N270 | G | G | R | T | G | G | H | N | R | D | R | H | G | R | S | G | A | — |
| H279 | R | R | Q | R | S | N | R | R | R | R | A | H | R | R | T | E | L | — |
| V286 | I | K | V | E | A | G | L | A | S | — | R | L | K | L | V | D | G | — |
| G297 | A | A | L | A | A | A | G | A | A | A | A | E | G | G | A | G | A | — |
Amino acid sequences of the listed MFP components were aligned with MexA by using ClustalW (http://www.ebi.ac.uk/clustalw/) (78), and residues of these MFPs that aligned with MexA residues whose mutation in this study was shown to compromise drug resistance were identified.
Original MexA amino acid residue whose mutation in this study compromised drug resistance. See Table 1 for a description of the mutations.
Corresponding amino acid residue in the MFP components of multidrug transporters from P. aeruginosa (MexC [accession number AAB41956], MexE [T30829], MexX [also called PA2019; see Pseudomonas genome project at http://www.pseudomonas.com], MexJ [also called PA3677] [CAA67864], MexH [also called PA4206] [C83120], MexV [also called PA4374] [D83099]), E. coli (AcrA [P31223]), Neisseria gonorrhoeae (MtrC [AAA80193]), Acinetobacter baumannii (AdeA [AAL14439]), Agrobacterium tumefaciens (AmeA [AAG09745]), Burkholderia cepacia (CeoA [T43023]), Pseudomonas putida (TtgA [AAD39553]), Stenotrophomonas maltophilia (SmeD [CAC14594]), Burkholderia pseudomallei (AmrA [AAC27753]), and Campylobacter jejuni (CmeA [AAL74244]). In identifying corresponding residues in members of COG0845 and COG1566 (http://www.ncbi.nlm.nih.gov/COG/), the consensus sequences of these two groups of AcrA-like (COG0845; http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=COG0845&version=) and EmrA-like (COG1566; http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=COG1566&version=) MFP components were used in the alignment. Similarly, the consensus sequence of the HlyD family (pfam00529; http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=pfam00529&version=v1.63) was aligned with MexA in identifying corresponding residues in this group of proteins. Aligned residues that are conserved in MexA are indicated in boldface.
—, a Blast search identified conserved domains in the N-terminal portion only of the EmrA group (COG1566) and MexA, and thus only this portion was aligned with MexA by the program.
FIG. 5.

Multiple alignment of MFPs. The MexA, AcrA, and HlyD (accession number AAA23977) proteins were aligned on-line by using ClustalW (http://www.ebi.ac.uk/clustalw/) (78). Residues in MexA whose mutation in this study compromised MexA function are indicated in lowercase, and the impact of the mutation on MexA-MexA interaction is indicated above the residue (D−, dimerization deficient; D+, dimerization proficient; D?, dimerization could not be assessed because the mutant protein was not expressed in the E. coli SU101 strain used to assess MexA dimerization). Regions of MexA predicted to form α-helices (overlined) were identified by using programs available on-line from the JPred server (http://www.compbio.dundee.ac.uk/∼www-jpred/) (9), and two of these (shown in italics) were predicted by Johnson and Church (30) to form coiled-coil structures. The N-terminal region of HlyD implicated in promoting an interaction of HlyD with the OMF TolC (69) is indicated in italics. The C-terminal region of AcrA predicted to be involved in the interaction of this MFP with its cognate RND component, AcrB (16), is also indicated in italics.
DISCUSSION
Mutations that completely inactivate MexA in regard to its contribution to MexAB-OprM-mediated multidrug resistance in P. aeruginosa mapped to two very distinct regions of the protein. While this implies, perhaps, the existence of only two major functional domains in this MFP, our selection requires production of wild-type levels of mutant MexA protein, and should mutations in other functionally important domains also compromise protein stability, these would, of course, be excluded from our “screen.” Nonetheless, we have identified two regions of functional importance in MexA, one in a highly conserved region of the protein (N terminus) and one within a poorly conserved region (C terminus). The bulk of the inactivating mutations within the N-terminal half of MexA occur within or immediately preceding a possible interrupted coiled-coil/helix structure (Fig. 5) proposed to form an antiparallel coiled coil or “helical hairpin” (30). Interestingly, two of the more destabilizing mutations (L91P and L110P, which fail to yield a hybrid MexA-LexA fusion protein) introduce a classical helix breaker into or very near the proposed helical hairpin structure and would be expected to have a large impact on its structure (and thus on its function and, possibly, stability).
The possible presence of a coiled-coil/helical hairpin region in MexA (and, indeed, the corresponding regions of all MFPs [30]) could provide for, in this instance, MexA-MexA interaction and thus the expected multimerization of the protein. Indeed, the results presented here confirm the multimeric nature of MexA, and the multimeric nature of many other MFPs (e.g., AcrA [multidrug efflux] [85], HlyD [hemolysin export] [77], and EmrA [multidrug efflux] [7]) has similarly been confirmed, with many of these apparently forming trimers (77, 85). The conserved nature of the N termini of MFPs in general and, more specifically, of residues whose mutation in MexA compromises function is certainly consistent with these playing a role in a shared function such as MFP multimerization. Certainly, some mutations in this region interfere with MexA-MexA interaction, indicating that multimerization is necessary for activity and that this region contributes to multimerization. Still, mutations in the C-terminal half of the protein also compromise MexA-MexA interaction, and so it is not entirely possible to rule out a contribution from this part of the protein as well. Also, while the C terminus is, in general, poorly conserved in the MFP family, as are residues whose mutation inactivates MexA (which is inconsistent with these being involved in MFP multimerization, should MFPs use a shared mechanism for multimerization), the only C-terminal mutations that compromise MexA-MexA interaction occur either at residues that are very well conserved (T256) or at residues whose size is well conserved (G297 [G or A predominates at this position in most MFPs]). Whether these might represent a second domain for MexA-MexA interaction (i.e., separate N- and C-terminal domains associate to form multimeric MexA) or whether these are in close apposition in the three-dimensional structure and thus contribute to a single multimerization domain is unclear. If, however, the N terminus of MexA interacts with OprM and the C terminus interacts with MexB (see below), the latter possibility is unlikely. Attempts to address this directly by assessing the ability of the MexA N-terminal region alone to promote MexA-MexA association (using the LexA fusion system described above) were unsuccessful owing to a failure to obtain a stable MexA-LexA fusion protein.
Intriguingly, the helical hairpin implicated above in MexA/MFP multimerization also corresponds to a region of the HlyD MFP component of hemolysin export that may promote an association with its cognate OMF component, TolC (69). There is, in fact, compelling genetic evidence in favor of MFP-OMF association in vivo (2, 6, 36, 69), and binding of the CvaA (25) and HlyD (77) MFPs to the OMF components of the CvaAB-TolC colicin V and HlyBD-TolC hemolysin exporters, respectively, has been demonstrated. The fact that two N-terminal mutations (P68S and A108T) that inactivate MexA do not affect MexA-MexA interaction might be explained by their having a negative impact on MexA-OprM binding. Consistent with this, one of these mutations (A108T) maps to a region of MexA that corresponds to the proposed OMF (i.e., TolC)-binding domain of HlyD, while the second (P68S) occurs just upstream of this sequence (Fig. 5). Whether the N-terminal regions of MFPs play a dual role in multimerization and OMF binding remains to be seen, although given the conservation of functional residues in this region of the protein, this would suggest a conserved approach to both multimerization and OMF binding. In this vein, it is worth noting that pumps of the RND-MFP-OMF type show considerable flexibility in regard to the OMF component that can be used in assembling a functional tripartite efflux system. Studies of artificially created hybrid multidrug efflux systems in P. aeruginosa have confirmed, for example, that OprM can replace the OprJ (19, 74) and OprN (46) OMF components of the MexCD-OprJ and MexEF-OprN efflux systems, respectively. Similarly, OprJ can replace the OprM component of MexAB-OprM (74), and the E. coli OMF TolC can replace OprJ (73). In addition, at least two OMFs, the aforementioned OprM and TolC, function naturally as the OMF components of several multidrug efflux systems in P. aeruginosa (MexXY-OprM [1, 50], MexJK-OprM [8], and MexVW-OprM [44]) and E. coli (MdtABC-TolC [54], YhiUV-TolC [56], AcrAD-TolC [24], and AcrEF-TolC [33]). This general, though not absolute, lack of specificity in regard to OMF recruitment by RND-MFP pairs would be consistent with the presence of conserved sequences in the OMF-binding regions of MFPs, with these proteins thus employing a common strategy for promoting OMF binding.
Whether or not N-terminal regions of MFPs such as MexA play a role in multimerization and/or OMF recruitment, clearly the bulk of N-terminal mutations that inactivate MexA have a negative impact on MexB association, and this itself would likely be sufficient to compromise activity. Still, in light of information implicating a C-terminal domains(s) in MFP binding to their cognate IM export components and, apparently, determining the specificity of this interaction (16), it seems unlikely that the N terminus of MexA plays a specific role in MexB binding. More likely, proper assembly of the MexA multimer, perhaps aided by OprM, is needed to promote a functional association with MexB, and mutations affecting these processes may indirectly interfere with MexA binding to MexB. Still, in at least one, as yet unexplained, instance, a mutation that compromises MexA-MexA association (G72S) does not impede MexB binding.
Genetic (2, 6, 36, 69) and cross-linking (25, 77, 85; this report) studies confirm an interaction between MFPs and their cognate IM efflux-export components, with studies of HlyD (69) and AcrA (16) indicating that C-terminal regions of these proteins are responsible for this binding. Intriguingly, the bulk of C-terminal mutations that inactivate MexA occur within or very near a region of MexA that corresponds to the AcrB-binding domain of AcrA (Fig. 5). It is tempting to speculate, then, that these residues contribute to a MexB-binding domain of MexA. Certainly, the fact that most of these mutations (e.g., V264, N270, H279, and V286 [Fig. 5]) do not impede MexA multimerization but do block MexA binding to MexB is consistent with this. It is also noteworthy that none of these four residues is conserved in the MFP family, suggesting that they play a specific role in promoting MexA binding to MexB. Indeed, the observation that MFPs tend to show a preference for their cognate IM efflux components (16, 53, 82) (with AcrA being a noted exception [16]) would be consistent with a general lack of sequence similarity within the proposed C-terminal binding domain for the IM counterparts. Interestingly, the only C-terminal MexA-inactivating mutations that are conserved (T256, which is highly conserved, and G297, whose small size is conserved throughout the MFP family) are also unique in their disruption of MexA multimerization. While these may play a role in, e.g., MexA-MexA association at the C terminus (see above), they may also contribute to MexB binding, possibly by maintaining the actual binding/specificity domain in a conformation that is compatible with MexB association. In any case, the observation that mutation of the G297-equivalent residue in the MFP CvaA (G313) also yields an unstable and inactive protein (24) highlights the probable functional significance of this residue in other MFPs.
Acknowledgments
We thank R. Srikumar for construction of plasmid pRSP48 and L. Mutharia for providing the protocol for adsorption of the polyclonal antiserum.
This work was supported by an operating grant from the Canadian Cystic Fibrosis Foundation (CCFF). D.N. holds a CCFF studentship. K.P. is a CCFF scholar.
REFERENCES
- 1.Aires, J. R., T. Köhler, H. Nikaido, and P. Plesiat. 1999. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 43:2624-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akatsuka, H., R. Binet, E. Kawai, C. Wandersman, and K. Omori. 1997. Lipase secretion by bacterial hybrid ATP-binding cassette exporters: molecular recognition of the LipBCD, PrtDEF, and HasDEF exporters. J. Bacteriol. 179:4754-4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1992. Short protocols in molecular biology, 2nd ed. John Wiley & Sons, Inc., New York, N.Y.
- 4.Avila-Sakar, A. J., S. Misaghi, E. M. Wilson-Kubalek, K. H. Downing, H. Zgurskaya, H. Nikaido, and E. Nogales. 2001. Lipid-layer crystallization and preliminary three-dimensional structural analysis of AcrA, the periplasmic component of a bacterial multidrug efflux pump. J Struct. Biol. 136:81-88. [DOI] [PubMed] [Google Scholar]
- 5.Barcak, G. J., M. S. Chandler, R. J. Redfield, and J. F. Tomb. 1991. Genetic systems in Haemophilus influenzae. Methods Enzymol. 204:321-342. [DOI] [PubMed] [Google Scholar]
- 6.Binet, R., and C. Wandersman. 1995. Protein secretion by hybrid bacterial ABC-transporters: specific functions of the membrane ATPase and the membrane fusion protein. EMBO J. 14:2298-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borges-Walmsley, M. I., J. Beauchamp, S. M. Kelly, K. Jumel, D. Candlish, S. E. Harding, N. C. Price, and A. R. Walmsley. 2003. Identification of oligomerization and drug-binding domains of the membrane fusion protein EmrA. J. Biol. Chem. 278:12903-12912. [DOI] [PubMed] [Google Scholar]
- 8.Chuanchuen, R., C. T. Narasaki, and H. P. Schweizer. 2002. The MexJK efflux pump of Pseudomonas aeruginosa requires OprM for antibiotic efflux but not for efflux of triclosan. J. Bacteriol. 184:5036-5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuff, J. A., M. E. Clamp, A. S. Siddiqui, M. Finlay, and G. J. Barton. 1998. JPred: a consensus secondary structure prediction server. Bioinformatics 14:892-893. [DOI] [PubMed] [Google Scholar]
- 10.Dean, C. R., M. A. Visalli, S. J. Projan, P.-E. Sum, and P. A. Bradford. 2003. Efflux-mediated resistance to tigecycline (GAR-936) in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 47:972-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Lorenzo, V., L. Eltis, B. Kessler, and K. Timmis. 1993. Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 123:17-24. [DOI] [PubMed] [Google Scholar]
- 12.Dinh, T., I. T. Paulsen, and M. H. Saier, Jr. 1994. A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. J. Bacteriol. 176:3825-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dmitrova, M., G. Younes-Cauet, P. Oertel-Buchheit, D. Porte, M. Schnarr, and M. Granger-Schnarr. 1998. A new LexA-based genetic system for monitoring and analyzing protein heterodimerization in Escherichia coli. Mol. Gen. Genet. 257:205-212. [DOI] [PubMed] [Google Scholar]
- 14.Eda, S., H. Maseda, and T. Nakae. 2003. An elegant means of self-protection in Gram-negative bacteria by recognizing and extruding xenobiotics from the periplasmic space. J. Biol. Chem. 278:2085-2088. [DOI] [PubMed] [Google Scholar]
- 15.Elkins, C. A., and H. Nikaido. 2002. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominately by two large periplasmic loops. J. Bacteriol. 184:6490-6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elkins, C. A., and H. Nikaido. 2003. Chimeric analysis of AcrA function reveals the importance of its C-terminal domain in its interaction with the AcrB multidrug efflux pump. J. Bacteriol. 185:5349-5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans, K., L. Passador, R. Srikumar, E. Tsang, J. Nezezon, and K. Poole. 1998. Influence of the MexAB-OprM multidrug efflux system on quorum-sensing in Pseudomonas aeruginosa. J. Bacteriol. 180:5443-5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17a.Garinot-Schneider, C., A. J. Pommer, G. R. Moore, C. Kleanthous, and R. James. 1996. Identification of putative active-site residues in the DNase domain of colicin E9 by random mutagenesis. J. Mol. Biol. 260:731-742. [DOI] [PubMed] [Google Scholar]
- 18.Germ, M., E. Yoshihara, H. Yoneyama, and T. Nakae. 1999. Interplay between the efflux pump and the outer membrane permeability barrier in fluorescent dye accumulation in Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 261:452-455. [DOI] [PubMed] [Google Scholar]
- 19.Gotoh, N., H. Tsujimoto, A. Nomura, K. Okamoto, M. Tsuda, and T. Nishino. 1998. Functional replacement of OprJ by OprM in the MexCD-OprJ multidrug efflux system of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 165:21-27. [DOI] [PubMed] [Google Scholar]
- 20.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20a.Hancock, R. E. W., and A. Carey. 1979. Outer membrane of Pseudomonas aeruginosa: heat and 2-mercaptoethanol-modifiable proteins. J. Bacteriol. 140:902-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hancock, R. E. W., and D. P. Speert. 2000. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Res. Update 3:247-255. [DOI] [PubMed] [Google Scholar]
- 22.Hirakata, Y., R. Srikumar, K. Poole, N. Gotoh, T. Suematsu, S. Kohno, S. Kamihira, R. E. Hancock, and D. P. Speert. 2002. Multidrug efflux systems play an important role in the invasiveness of Pseudomonas aeruginosa. J Exp. Med. 196:109-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 24.Hwang, J., and P. C. Tai. 1999. Mutational analysis of CvaA in the highly conserved domain of the membrane fusion protein family. Curr. Microbiol. 39:195-199. [DOI] [PubMed] [Google Scholar]
- 25.Hwang, J., X. Zhong, and P. C. Tai. 1997. Interactions of dedicated export membrane proteins of the colicin V secretion system: CvaA, a member of the membrane fusion protein family, interacts with CvaB and TolC. J. Bacteriol. 179:6264-6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoue, H., H. Nojima, and H. Okayama. 1991. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23-28. [DOI] [PubMed] [Google Scholar]
- 27.Jalal, S., O. Ciofu, N. Hoiby, N. Gotoh, and B. Wretlind. 2000. Molecular mechanisms of fluoroquinolone resistance in Pseudomonas aeruginosa isolates from cystic fibrosis. Antimicrob. Agents Chemother. 44:710-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jalal, S., and B. Wretlind. 1998. Mechanisms of quinolone resistance in clinical strains of Pseudomonas aeruginosa. Microbiol. Drug Resist. 4:257-261. [DOI] [PubMed] [Google Scholar]
- 29.Jo, J. T., F. S. Brinkman, and R. E. Hancock. 2003. Aminoglycoside efflux in Pseudomonas aeruginosa: involvement of novel outer membrane proteins. Antimicrob. Agents Chemother. 47:1101-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson, J. M., and G. M. Church. 1999. Alignment and structure prediction of divergent protein families: periplasmic and outer membrane proteins of bacterial efflux pumps. J. Mol. Biol. 287:695-715. [DOI] [PubMed] [Google Scholar]
- 31.Kawabe, T., E. Fujihira, and A. Yamaguchi. 2000. Molecular construction of a multidrug exporter system, AcrAB: molecular interaction between AcrA and AcrB, and cleavage of the N-terminal signal sequence of AcrA. J. Biochem. (Tokyo) 128:195-200. [DOI] [PubMed] [Google Scholar]
- 32.Keen, N. T., S. Tamaki, D. Kobayashi, and D. Trollinger. 1988. Improved broad-host-range plasmids for DNA cloning in Gram-negative bacteria. Gene 70:191-197. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi, K., N. Tsukagoshi, and R. Aono. 2001. Suppression of hypersensitivity of Escherichia coli acrB mutant to organic solvents by integration activation of the aceEF operon with the IS1 or IS2 element. J. Bacteriol. 183:2646-2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohler, T., M. Kok, M. Michea-Hamzehpour, P. Plesiat, N. Gotoh, T. Nishino, L. Kocjanici Curty, and J.-C. Pechere. 1996. Multidrug efflux in intrinsic resistance to trimethoprim and sulfamethoxazole in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 40:2288-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koronakis, V., A. Sharff, E. Koronakis, B. Luisi, and C. Hughes. 2000. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405:914-919. [DOI] [PubMed] [Google Scholar]
- 36.Letoffe, S., P. Delepelaire, and C. Wandersman. 1996. Protein secretion in gram-negative bacteria: assembly of the three components of ABC protein-mediated exporters is ordered and promoted by substrate binding. EMBO J. 15:5804-5811. [PMC free article] [PubMed] [Google Scholar]
- 37.Li, X.-Z., K. Poole, and H. Nikaido. 2003. Contributions of MexAB-OprM and an EmrE homologue to intrinsic resistance of Pseudomonas aeruginosa to aminoglycosides and dyes. Antimicrob. Agents Chemother. 47:27-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li, X.-Z., H. Nikaido, and K. Poole. 1995. Role of MexA-MexB-OprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39:1948-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li, X.-Z., L. Zhang, and K. Poole. 1998. Role of the multidrug efflux systems of Pseudomonas aeruginosa in organic solvent tolerance. J. Bacteriol. 180:2987-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li, X.-Z., L. Zhang, and K. Poole. 2000. Interplay between the MexAB-OprM multidrug efflux system and the outer membrane barrier in the multiple antibiotic resistance of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 45:433-436. [DOI] [PubMed] [Google Scholar]
- 41.Li, X.-Z., L. Zhang, R. Srikumar, and K. Poole. 1998. β-lactamase inhibitors are substrates of the multidrug efflux pumps of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 42:399-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li, X. Z., and K. Poole. 1999. Organic solvent-tolerant mutants of Pseudomonas aeruginosa display multiple antibiotic resistance. Can J. Microbiol. 45:18-22. [DOI] [PubMed] [Google Scholar]
- 43.Li, X. Z., and K. Poole. 2001. Mutational analysis of the OprM outer membrane component of the MexA-MexB-OprM multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 183:12-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li, Y., T. Mima, Y. Komori, Y. Morita, T. Kuroda, T. Mizushima, and T. Tsuchiya. 2003. A new member of the tripartite multidrug efflux pumps, MexVW-OprM, in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 52:572-575. [DOI] [PubMed] [Google Scholar]
- 45.Mao, W., M. S. Warren, D. S. Black, T. Satou, T. Murata, T. Nishino, N. Gotoh, and O. Lomovskaya. 2002. On the mechanism of substrate specificity by resistance nodulation division (RND)-type multidrug resistance pumps: the large periplasmic loops of MexD from Pseudomonas aeruginosa are involved in substrate recognition. Mol. Microbiol. 46:889-901. [DOI] [PubMed] [Google Scholar]
- 46.Maseda, H., H. Yoneyama, and T. Nakae. 2000. Assignment of the substrate-selective subunits of the MexEF-OprN multidrug efflux pump of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44:658-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masuda, N., and S. Ohya. 1992. Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36:1847-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masuda, N., E. Sakagawa, S. Ohya, N. Gotoh, H. Tsujimoto, and T. Nishino. 2000. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-OprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44:3322-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller, J. H. 1992. A short course in bacterial genetics. A laboratory manual and handbook for Escherichia coli and related bacteria, p. 72-74. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 50.Mine, T., Y. Morita, A. Kataoka, T. Mitzushima, and T. Tsuchiya. 1999. Expression in Escherichia coli of a new multidrug efflux pump, MexXY, from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43:415-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morales, V. M., A. Backman, and M. Bagdasarian. 1991. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97:39-47. [DOI] [PubMed] [Google Scholar]
- 52.Murakami, S., R. Nakashima, E. Yamashita, and A. Yamaguchi. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419:587-593. [DOI] [PubMed] [Google Scholar]
- 53.Murata, T., M. Kuwagaki, T. Shin, N. Gotoh, and T. Nishino. 2002. The substrate specificity of tripartite efflux systems of Pseudomonas aeruginosa is determined by the RND component. Biochem. Biophys. Res. Commun. 299:247-251. [DOI] [PubMed] [Google Scholar]
- 54.Nagakubo, S., K. Nishino, T. Hirata, and A. Yamaguchi. 2002. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J. Bacteriol. 184:4161-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nikaido, H. 1989. Outer membrane barrier as a mechanism of antimicrobial resistance. Antimicrob. Agents Chemother. 33:1831-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishino, K., and A. Yamaguchi. 2002. EvgA of the two-component signal transduction system modulates production of the YhiUV multidrug transporter in Escherichia coli. J. Bacteriol. 184:2319-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pearson, J. P., C. Van Delden, and B. H. Iglewski. 1999. Active efflux and diffusion are involved in transport of Pseudomonas aeruginosa cell-to-cell signals. J. Bacteriol. 181:1203-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poole, K. 2001. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J. Mol. Microbiol. Biotechnol. 3:255-264. [PubMed] [Google Scholar]
- 59.Poole, K. 2001. Multidrug resistance in Gram-negative bacteria. Curr. Opin. Microbiol. 4:500-508. [DOI] [PubMed] [Google Scholar]
- 60.Poole, K. 2002. Outer membranes and efflux: the path to multidrug resistance in gram-negative bacteria. Curr. Pharm. Biotechnol. 3:77-98. [DOI] [PubMed] [Google Scholar]
- 61.Poole, K. 2004. Efflux-mediated multiresistance in Gram-negative bacteria. Clin. Microbiol. Infect. 10:1-14. [DOI] [PubMed] [Google Scholar]
- 62.Poole, K., D. E. Heinrichs, and S. Neshat. 1993. Cloning and sequence analysis of an EnvCD homologue in Pseudomonas aeruginosa: regulation by iron and possible involvement in the secretion of the siderophore pyoverdine. Mol. Microbiol. 10:529-544. [DOI] [PubMed] [Google Scholar]
- 63.Poole, K., K. Krebes, C. McNally, and S. Neshat. 1993. Multiple antibiotic resistance in Pseudomonas aeruginosa: evidence for involvement of an efflux operon. J. Bacteriol. 175:7363-7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poole, K., and R. Srikumar. 2001. Multidrug efflux in Pseudomonas aeruginosa: components, mechanisms and clinical significance. Curr. Top. Med. Chem. 1:59-71. [DOI] [PubMed] [Google Scholar]
- 65.Poole, K., K. Tetro, Q. Zhao, S. Neshat, D. Heinrichs, and N. Bianco. 1996. Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob. Agents Chemother. 40:2021-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Porte, D., P. Oertel-Buchheit, M. Granger-Schnarr, and M. Schnarr. 1995. Fos leucine zipper variants with increased association capacity. J. Biol. Chem. 270:22721-22730. [DOI] [PubMed] [Google Scholar]
- 67.Redly, A., and K. Poole. 2003. Pyoverdine-mediated regulation of FpvA synthesis in Pseudomonas aeruginosa: involvement of a probable ECF sigma factor, FpvI. J. Bacteriol. 185:1261-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 69.Schlor, S., A. Schmidt, E. Maier, R. Benz, W. Goebel, and I. Gentschev. 1997. In vivo and in vitro studies on interactions between the components of the hemolysin (HlyA) secretion machinery of Escherichia coli. Mol. Gen. Genet. 256:306-319. [DOI] [PubMed] [Google Scholar]
- 70.Schweizer, H. P. 1998. Intrinsic resistance to inhibitors of fatty acid biosynthesis in Pseudomonas aeruginosa is due to efflux: application of a novel technique for generation of unmarked chromosomal mutations for the study of efflux systems. Antimicrob. Agents Chemother. 42:394-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simon, R., U. Priefer, and A. Puehler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 72.Sobel, M. L., G. A. McKay, and K. Poole. 2003. Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 47:3202-3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srikumar, R., T. Kon, N. Gotoh, and K. Poole. 1998. Expression of Pseudomonas aeruginosa multidrug efflux pumps MexA-MexB-OprM and MexC-MexD-OprJ in a multidrug-sensitive Escherichia coli strain. Antimicrob. Agents Chemother. 42:65-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Srikumar, R., X.-Z. Li, and K. Poole. 1997. Inner membrane efflux components are responsible for the β-lactam specificity of multidrug efflux pumps in Pseudomonas aeruginosa. J. Bacteriol. 179:7875-7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Srikumar, R., C. J. Paul, and K. Poole. 2000. Influence of mutations in the mexR repressor gene on expression of the MexA-MexB-OprM multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 182:1410-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Srikumar, R., and K. Poole. 1999. Demonstration of ethidium bromide efflux by multiresistant pumps of Pseudomonas aeruginosa. Clin. Microbiol. Infect. 5:5S58-5S59. [Google Scholar]
- 77.Thanabalu, T., E. Koronakis, C. Hughes, and V. Koronakis. 1998. Substrate-induced assembly of a contiguous channel for protein export from E. coli: reversible bridging of an inner-membrane translocase to an outer membrane exit pore. EMBO J. 17:6487-6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tikhonova, E. B., Q. Wang, and H. I. Zgurskaya. 2002. Chimeric analysis of the multicomponent multidrug efflux transporters from gram-negative bacteria. J. Bacteriol. 184:6499-6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wong, K. K. Y., F. S. Brinkman, R. S. Benz, and R. E. W. Hancock. 2001. Evaluation of a structural model of Pseudomonas aeruginosa outer membrane protein OprM, an efflux component involved in intrinsic antibiotic resistance. J. Bacteriol. 183:367-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoneyama, H., H. Maseda, H. Kamiguchi, and T. Nakae. 2000. Function of the membrane fusion protein, MexA, of the MexA, B-OprM efflux pump in Pseudomonas aeruginosa without an anchoring membrane. J. Biol. Chem. 275:4628-4634. [DOI] [PubMed] [Google Scholar]
- 82.Yoneyama, H., A. Ocaktan, N. Gotoh, T. Nishino, and T. Nakae. 1998. Subunit swapping in the Mex-extrusion pumps in Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 244:898-902. [DOI] [PubMed] [Google Scholar]
- 83.Yu, E. W., G. McDermott, H. I. Zgurskaya, H. Nikaido, and D. E. Koshland, Jr. 2003. Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science 300:976-980. [DOI] [PubMed] [Google Scholar]
- 84.Zgurskaya, H. I., and H. Nikaido. 1999. AcrA is a highly asymmetric protein capable of spanning the periplasm. J. Mol. Biol. 285:409-420. [DOI] [PubMed] [Google Scholar]
- 85.Zgurskaya, H. I., and H. Nikaido. 2000. Cross-linked complex between oligomeric periplasmic lipoprotein AcrA and the inner-membrane-associated multidrug efflux pump AcrB from Escherichia coli. J. Bacteriol. 182:4264-4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zgurskaya, H. I., and H. Nikaido. 2000. Multidrug resistance mechanisms: drug efflux across two membranes. Mol. Microbiol. 37:219-225. [DOI] [PubMed] [Google Scholar]
- 87.Zhao, Q., X.-Z. Li, R. Srikumar, and K. Poole. 1998. Contribution of outer membrane efflux protein OprM to antibiotic resistance in Pseudomonas aeruginosa independent of MexAB. Antimicrob. Agents Chemother. 42:1682-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ziha-Zarifi, I., C. Llanes, T. Koehler, J.-C. Pechere, and P. Plesiat. 1999. In vivo emergence of multidrug-resistant mutants of Pseudomonas aeruginosa overexpressing the active efflux system MexA-MexB-OprM. Antimicrob. Agents Chemother. 43:287-291. [DOI] [PMC free article] [PubMed] [Google Scholar]