

Abstract
Alcohol-induced liver fibrosis and eventually cirrhosis is a leading cause of death. Acetaldehyde, the first metabolite of ethanol, up-regulates expression of the human α2(I) collagen gene (COL1A2). Early acetaldehyde-mediated effects involve phosphorylation and nuclear translocation of SMAD3/4–containing complexes that bind to COL1A2 promoter to induce fibrogenesis. We used human and mouse hepatic stellate cells to elucidate the mechanisms whereby acetaldehyde up-regulates COL1A2 by modulating the role of Ski and the expression of SMADs 3, 4, and 7. Acetaldehyde induced up-regulation of COL1A2 by 3.5-fold, with concomitant increases in the mRNA (threefold) and protein (4.2- and 3.5-fold) levels of SMAD3 and SMAD4, respectively. It also caused a 60% decrease in SMAD7 expression. Ski, a member of the Ski/Sno oncogene family, is colocalized in the nucleus with SMAD4. Acetaldehyde induces translocation of Ski and SMAD4 to the cytoplasm, where Ski undergoes proteasomal degradation, as confirmed by the ability of the proteasomal inhibitor lactacystin to blunt up-regulation of acetaldehyde-dependent COL1A2, but not of the nonspecific fibronectin gene (FN1). We conclude that acetaldehyde up-regulates COL1A2 by enhancing expression of the transactivators SMAD3 and SMAD4 while inhibiting the repressor SMAD7, along with promoting Ski translocation from the nucleus to cytoplasm. We speculate that drugs that prevent proteasomal degradation of repressors targeting COL1A2 may have antifibrogenic properties.
Alcohol-induced liver fibrosis is a multifactorial event characterized by increased collagen production as a result of up-regulation of α2(I) collagen (COL1A2) gene.1 This is induced primarily by its immediate oxidation product, acetaldehyde, as well as by other events occurring during alcohol metabolism, such as changes in redox state,2 formation of free radicals and generation of reactive oxygen species,2–4 and depletion of antioxidant defenses and generation of aliphatic aldehydes derived from lipid peroxidation (namely, 4-hydroxy-nonenal and malonyldialdehyde).5 Because ethanol also increases the circulation of bacterial-derived endotoxin,6–8 and this in turn up-regulates expression of inflammatory cytokines, the aforementioned events also play a role in the inflammatory response and therefore in fibrogenesis and complications resulting from chronic alcoholic liver disease.7,9
To unravel key molecular mechanisms involved in acetaldehyde-mediated up-regulation of type I collagen, we have investigated the key roles played by this ethanol metabolite in up-regulation of type I collagen genes in hepatic stellate cells (HSCs).1 We have shown that several different transcription factors are involved in acetaldehyde-dependent up-regulation of the type I collagen genes. Although CAAT/enhancing binding protein p35С (p35C/EBPβ) is required for expression of the α1(I) collagen mRNA,10 Sp1 and SMAD3 are essential for up-regulation of COL1A2 gene.11 We and others have also shown that reactive oxygen species in general,2–4 and H2O2 in particular, play a key role in the acetaldehyde-elicited response and that H2O2 acts as a second messenger in both acetaldehyde-dependent and transforming growth factor β1 (TGF-β1)–dependent up-regulation of type I collagen genes.2,10
Acetaldehyde up-regulates expression of COL1A2 gene via a de novo protein synthesis–independent, PI3K-dependent mechanism.12 In addition, the early acetaldehyde-mediated effects, occurring during the first 6 to 12 hours after acetaldehyde treatment, are independent of TGF-β1.12 However, the mechanisms whereby acetaldehyde modulates expression and activity of members of the SMAD family, including SMAD3, SMAD4, and SMAD7, leading to COL1A2 up-regulation are not well understood. Here, we show that SMAD3 and SMAD4 are the limiting factors in COL1A2 gene up-regulation and that this ethanol metabolite enhances expression of SMAD3 and SMAD4 at the mRNA and protein levels.
c-Ski, a homolog of v-Ski in cells and a versatile transcriptional regulator that is widely distributed in different tissues, has been reported to be a corepressor of TGF-β/SMAD signaling.13 Binding of TGF-β to its receptor serine/threonine kinases results in the regulation of SMAD2 and SMAD3 proteins. The phosphorylated SMADs then form heteromeric complexes with a common mediator SMAD4 (co-SMAD).14 Together, they translocate into the nucleus, where they bind to DNA and activate transcription of the target genes.15 We further show that Ski, a member of the Ski/Sno family of oncogenes, is colocalized in the nucleus with SMAD4 and that acetaldehyde induces their cotranslocation to the cytosol, where Ski is degraded by proteasomes. We also demonstrate that inhibiting proteasomal degradation of Ski by lactacystin blunts the acetaldehyde-dependent up-regulation of COL1A2 gene, but has no effect on expression of the nonspecific protein fibronectin.
Materials and Methods
Plasmids and Reagents
TGF-β1 was purchased from Roche Diagnostics (Indianapolis, IN). Lactacystin was purchased from Calbiochem–Novabiochem (Millipore, Billerica, MA). Acetaldehyde was purchased from Thermo Fisher Scientific (Waltham, MA). The construction of the −378COL1A2LUC chimeric plasmid containing the −378 to +54 region of COL1A2 linked to the firefly luciferase gene has been described previously.12 SMAD3, -4, and -7 expression plasmids cloned into the pcDNA3 cytomegalovirus expression vector (Life Technologies, Carlsbad, CA) have been described previously.11,16 The cDNAs of COL1A2, fibronectin, and TGF-β1 have been described previously.17–19 S14 ribosomal protein cDNA was obtained from ATCC (Manassas, VA). cDNA fragments of human SMAD3 (nucleotides 501 to 878) and SMAD4 (nucleotides 601 to 1050) were used to determine steady-state levels of SMAD3 and SMAD4 mRNAs. Nonspecific IgG rabbit, mouse, and goat polyclonal antibodies against Ski (sc-9140), SMAD3 (sc-101154), and SMAD4 (sc-1909), respectively, were obtained from Santa Cruz Biotechnology (Dallas, TX). Neutralizing antibody to TGF-β1 was obtained from Promega (Madison, WI).
HHSC Isolation and Culture
Human HSCs (HHSCs) were isolated from a consenting study subject with clinically proven normal healthy liver during gastric bypass surgery for morbid obesity as described previously.1 Informed consent in writing was obtained from the study patient, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a prior approval by the Institutional Review Committee. For some experiments, mouse HSCs were isolated as described previously.20 Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (HyClone; Thermo Fisher Scientific, Waltham, MA) and antibiotics. Experiments were performed in triplicate, using cells obtained from at least three different patients cultured for 2 to 8 passages.
Northern Blot Hybridization and Run-On Transcription Assay
Confluent HHSCs were placed in a serum-free medium containing glutamine, nonessential amino acids, and antibiotics. Approximately 14 hours later, acetaldehyde was added at a final concentration of 200 μmol/L. Viability of cells was estimated using the trypan blue exclusion test. In all cases, cellular viability was greater than 90%. Total RNA was extracted at 30 minutes and 1, 3, 24, and 48 hours and was processed for Northern blot hybridizations according to standard protocols.12 Likewise, a standard protocol was used to determine rates of COL1A2, fibronectin, and TGF-β1 transcription in control and acetaldehyde-treated cells, using S14 and pBR322 DNA as controls.11
In some run-on transcription experiments, cells were preincubated with 30 μmol/L lactacystin for 2 hours before acetaldehyde administration; in others, cells were preincubated for 2 hours with a neutralizing antibody to TGF-β1 (Promega) or an unrelated IgG (Santa Cruz Biotechnology) at a final concentration of 5 μg/mL. To determine the effectiveness of the anti–TGF-β1 antibody, some HHSCs were also incubated with 8 ng/mL of recombinant TGF-β1 in the presence or absence of the corresponding neutralizing antibody, or an unrelated IgG, according to a protocol described previously.21 Nuclei were isolated at 15 and 30 minutes and were used in run-on transcription assays as described previously.12 Relative intensity of the signals was determined by laser densitometric analysis of the radiographic films. Data were corrected for loading differences, using S14 as control.
RT-PCR Analysis
Transcript levels of SMADs 3, 4, and 7 in HHSCs were measured using a quantitative RT-PCR technique (RT-qPCR). Experiments were performed as described previously.22 Primer sequences for qPCR amplification were as follows: SMAD3 mRNA , forward 5′-GAGGGCAGGCTTGGGGAAAATG-3′ and reverse 5′-GGGAGGGTGCCGGTGGTGTAATAC-3′; SMAD4 mRNA, forward 5′-AAAGGTGAAGGTGATGTTTGGGTC-3′ and reverse 5′-CTGGAGCTATTCCACCTACTGATCC-3′; SMAD7 mRNA, forward 5′-CGAGACCCTTCTCACTCCTG-3′ and reverse 5′-GCATCCTTGGTTAGGGTCAA-3′; and GAPDH mRNA, forward 5′-GGCCTCCAAGGAGTAAGACC-3′ and reverse 5′-CTGTGAGGAGGGGAGATTTCA-3′. All reagents were purchased from Life Technologies. Relative gene expression was calculated as
| (1) |
Western Blot Analysis
Total, nuclear, and cytosolic extracts were prepared from control and acetaldehyde-treated HHSCs as described previously.1,12 In some experiments, HHSCs were treated with 8 ng/mL recombinant TGF-β1; in other experiments, cells were preincubated for 60 minutes with 30 μmol/L lactacystin before administration of acetaldehyde or TGF-β1. Total extracts (20 μg) were separated by SDS-PAGE on 10% gels, transferred onto a nitrocellulose membrane, and probed with SMAD3 or SMAD4 goat antibodies (1:1000; Santa Cruz Biotechnology), followed by incubation with horseradish peroxidase–conjugated rabbit anti-goat IgG (1:5000; Santa Cruz Biotechnology). Nuclear and cytosolic extracts were separated by SDS-PAGE on 4% to 12% gels, transferred onto polyvinylidene difluoride membranes, and probed with Ski rabbit antibody (1:1000; Santa Cruz Biotechnology), followed by incubation with horseradish peroxidase–conjugated chicken anti-rabbit secondary antibody (1:2000; Santa Cruz Biotechnology). Proteins were detected with a NEN Life Science Products Renaissance enhanced chemiluminescence system (PerkinElmer, Waltham, MA), according to the manufacturer’s recommendations.
Coimmunoprecipitation of Ski with SMAD4 and 20S Proteasome
The coimmunoprecipitation experiment was performed as described previously.23 In brief, SMAD4 was immunoprecipitated with a SMAD4 antibody (Santa Cruz Biotechnology) bound to protein l-agarose beads (Santa Cruz Biotechnology), and the amount of Ski coimmunoprecipitated with SMAD4 was quantified by Western analysis using Ski antibody (1:1000; Santa Cruz Biotechnology).
Cell Transfections
Conditions for the preparation and transfection of plasmids into HHSCs by the calcium phosphate procedure have been described previously.12,16 HHSCs were treated with 10% glycerol for 90 seconds, at 6 hours after transfection, and then were placed in medium containing 0.1% fetal bovine serum. Twelve hours later, acetaldehyde was added at the final concentration of 200 μmol/L, unless otherwise indicated. For some experiments, 8 ng/mL TGF-β1 (unless otherwise indicated) was added, alone or in combination with acetaldehyde. Cells were procured at 36 hours after the addition of acetaldehyde and were used to determine luciferase activity as described previously.
In some experiments, cells were preincubated for 60 minutes with 30 μmol/L lactacystin before administration of acetaldehyde. Transcriptional activity of acetaldehyde of the chimeric constructs was normalized against cotransfected pSV2CAT. Transfections were performed multiple times and in duplicate. For some experiments, HHSCs were cotransfected with 7.0 μg of the −378COL1A2LUC reporter construct and 2.5 μg of a SMAD4 vector, followed by treatment with acetaldehyde using the conditions described above. For other experiments, HHSCs were cotransfected with 7.0 μg of the −378COL1A2LUC reporter construct and 2.5 μg of vectors overexpressing SMAD3, SMAD4, both SMAD3 and SMAD4, or SMAD7, followed by treatment with acetaldehyde using the conditions described above.
Immunostaining
Mouse HSCs plated on coverslips were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with −20°C cold acetone for 30 minutes, rinsed with PBS, and blocked with 3% bovine serum albumin–PBS for 1 hour. After blocking, cells were incubated overnight with a goat polyclonal antibody against Ski (1:10) and rabbit polyclonal antibody against SMAD4 (1:10), followed by the appropriate fluorophore-conjugated secondary antibody for 1 hour (1:300); between acetaldehyde procedures, coverslips were washed with 0.1% Tween 20–PBS and mounted on glass slides with Gel-Mount medium (BioMeda, Foster City, CA). Images were captured using an AX70 light microscope (Olympus, Tokyo, Japan) and were processed with Photoshop software version 5.0.2 (Adobe Systems, San Jose, CA).
Statistical Analysis
Statistical differences between experimental groups were analyzed by Student’s t-test (normally distributed data with equal variances) or U test (normally distributed data with different variances). All P values of ≤0.05 were considered significant. Data represent three to six independent experiments using HHSCs obtained from a single patient. Data are expressed as means ± SEM.
Results
Acetaldehyde and TGF-β1 Have an Additive Effect on Expression of a Reporter Vector Driven by the Acetaldehyde-Responsive Element −378COL1A2LUC
Because TGF-β1 also induced expression of SMADs 3 and 4, and given that the acetaldehyde and TGF-β1 responsive elements are localized in the same region of the COL1A2 promoter, it was important to determine whether acetaldehyde and TGF-β1 up-regulation of COL1A2 gene expression is additive or synergistic. To this end, we transfected HSCs with the −378COL1A2LUC reporter vector and treated these cells with 100 μmol/L acetaldehyde, 4 ng/mL TGF-β1, or both. It is important to note that the doses used were half of those normally used in our previous experiments,12 and we therefore expected a weaker individual response. Neither acetaldehyde nor TGF-β1 alone was sufficient to up-regulate expression of the COL1A2 reporter vector to previously observed levels (Figure 1).12 However, cells treated with the combination of acetaldehyde and TGF-β1 responded with a 4.2-fold increase in reporter activity (P < 0.05), indicating that acetaldehyde and TGF-β1 have an additive stimulatory effect on the activity of COL1A2 reporter vector.
Figure 1.

Effect of acetaldehyde, TGF-β1, or the two in combination on the expression of a reporter vector driven by the −378COL1A2 promoter in HSCs. First, HHSCs were transfected with the reporter vector. Next, 12 hours later, cells were incubated with either 100 μmol/L acetaldehyde, 4 ng/mL TGF-β1, or both. Then, 36 hours later, cells were harvested and luciferase activity was determined. Cells treated with the combination of acetaldehyde and TGF-β1 showed a 4.2-fold increase in reporter activity. Controls were transfected cells without treatment. Data are expressed as means ± SEM. ∗P < 0.05 versus control.
The Late Acetaldehyde-Dependent Up-Regulation of COL1A2 Transcription Is Mediated by TGF-β1
We have shown that acetaldehyde is directly responsible for the early expression of α2(I) collagen mRNA and for changes in cell signaling occurring early (before 6 to 12 hours). However, the late events appear to be associated with acetaldehyde-induced expression of TGF-β1. To test this possibility, we measured the late acetaldehyde-dependent transcription of COL1A2 gene in the presence of a TGF-β1 neutralizing antibody. We used a nonspecific immunoglobulin as a control for these experiments. Transcription of COL1A2 was up-regulated 3.5-fold by acetaldehyde (P < 0.05) (Figure 2). Whereas the neutralizing antibody to TGF-β1 strongly inhibited COL1A2 gene transcription of cells treated with acetaldehyde by 60% (P < 0.05), the nonspecific immunoglobulin had no effect.
Figure 2.

TGF-β1 is involved in late acetaldehyde-dependent up-regulation of COL1A2. Run-on transcription assays of control (white bars; untreated cells in serum-free medium) and acetaldehyde-treated (black bars) HHSCs for 24 hours in the presence or absence of a nonspecific IgG or a neutralizing antibody to TGF-β1 (gray bars; anti–TGF-β1). As a control for these experiments, some HSCs were treated with 2 ng/mL of TGF-β1 in the absence or presence of the neutralizing antibody to the cytokine. COL1A2 gene transcription was up-regulated 3.5-fold by acetaldehyde, whereas the neutralizing antibody to TGF-β1 strongly inhibited COL1A2 gene transcription in cells treated with acetaldehyde by 60%. The nonspecific immunoglobulin had no effect. Data are expressed as means ± SEM. ∗P < 0.05 versus control. †P < 0.05 versus acetaldehyde-treated cells.
Acetaldehyde Induces the Late Expression of SMAD3 and SMAD4 mRNAs in HSCs
We have already shown that acetaldehyde induces formation of SMAD3/4 complexes that bind to the COL1A2 promoter and phosphorylates SMAD3.12 Thus, we considered it important to investigate whether acetaldehyde induces expression of SMADs 3 and 4 and whether this is an early event directly induced by acetaldehyde per se or a late event resulting from up-regulation of TGF-β1. We determined the time course of expression of SMAD3 and SMAD4 mRNA and protein in HHSCs incubated with acetaldehyde. Acetaldehyde up-regulated the mRNA expression of SMADs 3 and 4 approximately threefold (P < 0.05 for both) and up-regulated SMAD3 protein expression 4.2-fold (P < 0.05) and SMAD4 protein expression 3.5-fold (P < 0.05) (Figure 3). This effect was observed by approximately 24 hours after incubation with acetaldehyde and lasted up to 48 hours, a time period that coincided with acetaldehyde-induced up-regulation of TGF-β1 mRNA expression.
Figure 3.

Increase in SMAD3 and SMAD4 mRNA and protein is delayed after acetaldehyde treatment. A: Time-course analysis of SMAD3 and SMAD4 mRNA expression levels after acetaldehyde treatment. Total RNA obtained from HHSCs cultured in the absence or presence of 200 μmol/L acetaldehyde for times ranging from 30 minutes to 48 hours was used to determine steady-state levels of SMAD3 and SMAD4 mRNAs by using RT-PCR analysis. B: Replica dishes were used to obtain cell extracts and to determine levels of SMAD3 and SMAD4 protein by Western analysis. Data are expressed as means ± SEM. ∗P < 0.05 versus control (A); †P < 0.05 versus 30-minute time point (B).
Both SMAD3 and SMAD4 Are Limiting Factors in Acetaldehyde-Elicited Up-Regulation of COL1A2
To further test the requirement of SMAD3 and SMAD4 in acetaldehyde-elicited up-regulation of COL1A2, we cotransfected HHSCs with the −378COL1A2LUC reporter vector and a vector that overexpressed either SMAD3 or SMAD4 and determined the reporter activity in cells treated with or without 200 μmol/L acetaldehyde. Overexpression of either SMAD3 or SMAD4 alone did not significantly enhance reporter activity induced by acetaldehyde. However, when HHSCs were cotransfected with the reporter vector and also the two vectors expressing SMAD3 and SMAD4, there was a twofold increase in reporter activity (P < 0.05), compared with that induced by acetaldehyde alone or cells expressing either SMAD3 or SMAD4 (Figure 4).
Figure 4.
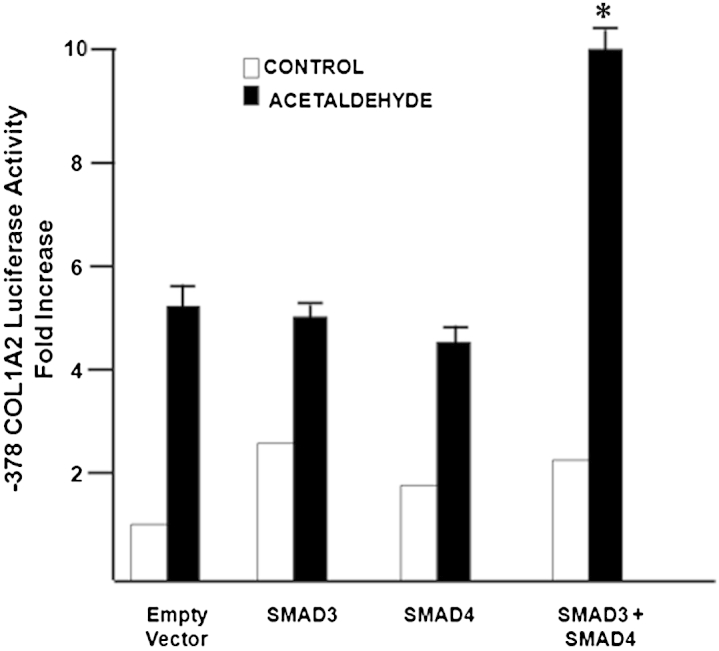
SMAD3 and SMAD4 are limiting factors for acetaldehyde-mediated COL1A2 up-regulation in HSCs. Effect of acetaldehyde on luciferase activity of HSCs transiently cotransfected with 7 μg −378COL1A2LUC reporter vector and either a control empty cytomegalovirus vector or 2.5 μg expression vectors for SMAD3 or SMAD4. At 12 hours after transfection, cells were treated with 200 μmol/L acetaldehyde for 36 hours. Cells were harvested and used to measure luciferase activity. Up-regulation of either SMAD3 or SMAD4 alone does not significantly enhance reporter activity induced by acetaldehyde; however, in HHSCs cotransfected with both the reporter vector and the two vectors expressing SMAD3 and SMAD4, reporter activity increased twofold, compared with that induced by acetaldehyde alone or cells expressing either SMAD3 or SMAD4. Data are expressed as means ± SEM. ∗P < 0.05.
Acetaldehyde Down-Regulates Expression of SMAD7 and Overexpression of SMAD7 Blunts Up-Regulation of COL1A2
We and others have shown that SMAD7 is a repressor of collagen gene expression.24,25 In the present study, therefore, we investigated whether acetaldehyde has any effect on expression of SMAD7. Acetaldehyde down-regulated expression of SMAD7 mRNA in a time-dependent manner, reaching the maximum down-regulation of 60% (P < 0.05) after 3 hours, relative to the value at 30 minutes, after which time it increased steadily and reached values threefold (P < 0.05) above basal levels by 48 hours (Figure 5A). Interestingly, when HHSCs were cotransfected with both a vector overexpressing SMAD7 and the −378COL1A2LUC reporter vector, acetaldehyde failed to up-regulate expression of COL1A2 reporter vector (Figure 5B). Overall, these findings suggest that acetaldehyde stimulates COL1A2 gene expression at least in part by down-regulating expression of SMAD7 mRNA.
Figure 5.
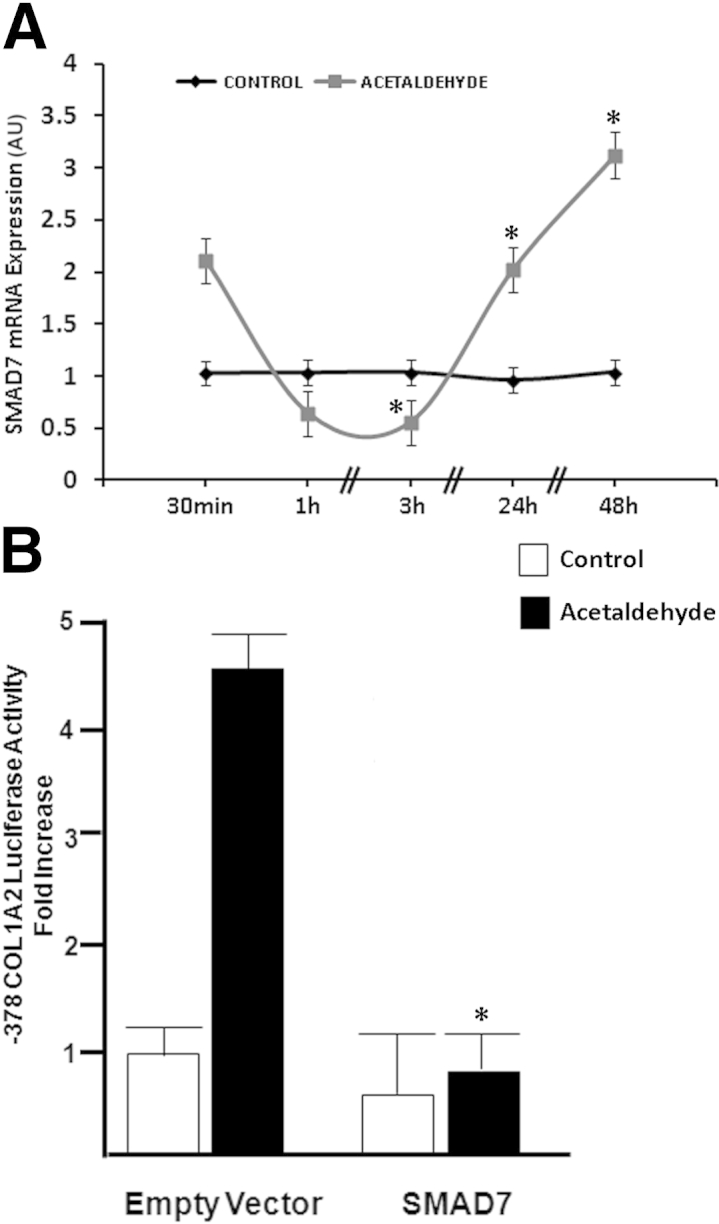
Acetaldehyde modulates SMAD7 expression. A: Total RNA was obtained from HHSCs cultured in the absence or presence of 200 μmol/L acetaldehyde for times ranging from 30 minutes to 48 hours. Acetaldehyde down-regulates the expression of SMAD7 mRNA in a time-dependent manner, reaching the maximum down-regulation of 60% after 3 hours, relative to the value at 30 minutes, after which time expression increases steadily and reaches values threefold above basal levels by 48 hours. SMAD7 mRNA expression levels were corrected for differences in loading using GAPDH as a housekeeping gene. ∗P < 0.05 versus control. B: HHSCs were transiently cotransfected with the −378COL1A2 reporter plasmid and a cytomegalovirus-driven SMAD7 expression vector. At 12 hours after transfection, cells were treated with 200 μmol/L acetaldehyde for 36 hours. Cells were harvested and used to measure luciferase activity. Acetaldehyde failed to up-regulate the expression of COL1A2 reporter vector in HHSCs cotransfected with a vector overexpressing SMAD7 and the −378COL1A2LUC reporter vector. Data are expressed as means ± SEM. ∗P < 0.05 versus acetaldehyde-treated empty vector. AU, arbitrary units.
Ski, a Known Repressor of TGF-β1–Responsive Genes, Is Colocalized with SMAD4 in the Nucleus
Ski/SnoN is a member of an oncogene family of proteins that are involved in TGF-β1–mediated transcription and repression of some cytokine target genes.26 In mouse myoblasts, the activity of Ski is mediated primarily through its binding with SMAD4 (but not with SMAD2).14 Having found that acetaldehyde-mediated up-regulation of COL1A2 requires SMAD4, we next investigated whether mouse HSCs also express Ski and whether acetaldehyde has any effect on its expression and subcellular localization. Immunocytochemical analysis of control and acetaldehyde-treated mouse HSCs revealed that Ski is localized mainly in the nucleus. With acetaldehyde treatment, however, Ski is translocated to the cytosol, where it has a granular appearance and extends from the perinuclear area to the plasma membrane (Figure 6).
Figure 6.
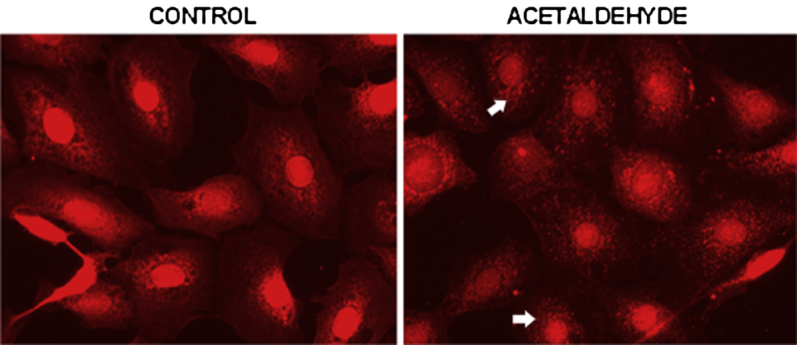
Acetaldehyde modifies cellular distribution of Ski. Immunofluorescence microscopy of mouse HSCs cultured in the absence or presence of 200 μmol/L acetaldehyde for 30 minutes. Cells were fixed and stained with a rabbit anti-Ski antibody followed by a Cy3 (red) labeled anti-rabbit immunoglobulin. Although fluorescence in control untreated cells was concentrated predominantly in the nuclei, in acetaldehyde-treated cells the intensity of the nuclear signal decreased and the cytosol signal increased. In addition, acetaldehyde-treated cells appear more granular and diffused (arrows). Original magnification, ×20.
To determine whether Ski cytosolic translocation could be clinically relevant, we analyzed Ski status in response to acetaldehyde and TGF-β1 in HHSCs. Western blot analysis of nuclear Ski in control, acetaldehyde-treated, and TGF-β1–treated HHSCs revealed that nuclear Ski decreased by 40% (P < 0.05) within 15 minutes and by 80% (P < 0.05) after 30 minutes of TGF-β1 treatment, compared with control. By contrast, acetaldehyde treatment decreased nuclear Ski by 40% (P < 0.05) of the control by 15 minutes, and had not decreased further at 30 minutes (Figure 7, A and B). As a consequence of Ski nuclear translocation, cytosolic Ski increased threefold (P < 0.001) after 15 minutes of acetaldehyde exposure and fivefold (P < 0.001) after 15 minutes of TGF-β1 treatment. Significantly, cytosolic Ski increased by ninefold with acetaldehyde and TGF-β1 treatment within 30 minutes (P < 0.05 and P < 0.05, respectively) (Figure 7, C and D).
Figure 7.
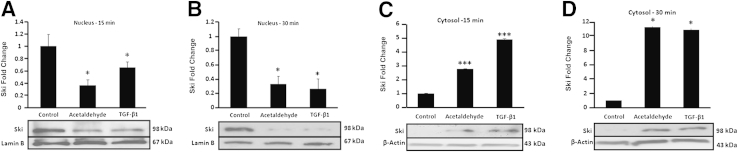
Acetaldehyde and TGF-β1 down-regulate the expression of Ski in HSCs. A–D: Western analysis of Ski in nuclear proteins extracted from HHSCs at 15 (A) and 30 minutes (B) and in cytosolic extracts at 15 (C) and 30 minutes (D) after treatment with either 200 μmol/L acetaldehyde or 8 ng/mL of TGF-β1. Data are expressed as means ± SEM of triplicate experiments. ∗P < 0.05, ∗∗∗P < 0.001 versus control.
Additional fluorescence microscopy data using antibodies to simultaneously detect both Ski and SMAD4 revealed that SMAD4 is also localized in the nucleus of untreated HSCs. Moreover, SMAD4 appears to colocalize with Ski, and both appear to be translocated together to the cytosol after acetaldehyde treatment. However, a significant amount of SMAD4 remains in the nucleus, as demonstrated by positive staining with anti-SMAD4 antibodies (Figure 8A). To further test whether SMAD4 and Ski interact with each other, we performed coimmunoprecipitation assays using an antibody against SMAD4 and then verified whether Ski coprecipitated with SMAD4 by Western blot analysis using Ski antibody. SMAD4 and Ski formed complexes in both untreated control and acetaldehyde-treated cells (Figure 8B).
Figure 8.

Ski and SMAD4 colocalize in the nuclei of HSCs and translocate to the cytosol after acetaldehyde administration. A: Immunofluorescence microscopy of mouse HSCs cultured in the absence or presence of 200 μmol/L acetaldehyde for 30 minutes. Cells were stained with antibodies to SMAD4 and Ski, followed by the corresponding secondary antibodies labeled with fluorescein isothiocyanate (green) for Ski and Cy3 (red) for SMAD4. Both Ski and SMAD4 localized in the nuclei of HSCs; yellow fluorescence indicates colocalization. After administration of acetaldehyde, both proteins are exported to the cytosol (arrow). However, a fair amount of SMAD4 is present in the nucleus (red). B: Immunoprecipitation (IP) of SMAD4, followed by Western analysis for the detection of Ski in HHSCs cultured in the absence or presence of 200 μmol/L acetaldehyde (B). Results are representative of three independent experiments. Original magnification, ×40.
Lactacystin, an Inhibitor of Proteasomal Degradation, Inhibits Acetaldehyde-Mediated COL1A2 Promoter-Driven Reporter Activity and Gene Transcription
Because Ski is degraded by proteasomes,27 we investigated whether lactacystin, an inhibitor of proteasomal degradation, has any effect on the levels of Ski and on the level of response of the −378COL1A2LUC reporter vector to acetaldehyde. In HHSCs treated with lactacystin alone, Ski levels were higher than in control cells (P < 0.05) (Figure 9A). Furthermore, lactacystin prevented acetaldehyde-mediated decrease in nuclear Ski by approximately 50% (P < 0.05). Likewise, lactacystin also prevented acetaldehyde-mediated up-regulation of the −378COL1A2LUC reporter vector activity by 50% (P < 0.05) (Figure 9B).
Figure 9.

Lactacystin, an inhibitor of proteasomal degradation, inhibits acetaldehyde-mediated expression of COL1A2 promoter-driven reporter activity. A: Western analysis of Ski in nuclear proteins extracted from HHSCs treated with 200 μmol/L acetaldehyde, 30 μmol/L lactacystin, or both. In HHSCs treated with lactacystin alone, Ski levels were higher than in control cells. B: HHSCs were transfected with the −378COL1A2LUC reporter vector; 12 hours later, cells were incubated with 30 μmol/L lactacystin. After 30 minutes, acetaldehyde (200 μmol/L final concentration) was added; 36 hours later, cells were harvested and luciferase activity was determined. Controls for these experiments were transfected cells without treatment, cells treated with lactacystin alone, and cells treated with acetaldehyde alone. Lactacystin prevented acetaldehyde-mediated decrease in nuclear Ski by approximately 50% and up-regulation of the −378COL1A2LUC reporter vector activity by 50%. Data are expressed as means ± SEM. ∗P < 0.05 versus control. †P < 0.05 versus lactacystin treated cells. ‡P < 0.05 versus acetaldehyde-treated cells.
To determine the specificity of this event, we tested the effect of lactacystin on acetaldehyde-induced transcription of COL1A2 (Figure 10A) and of FN1, the nonspecific fibronectin gene (Figure 10B). Lactacystin prevented acetaldehyde-mediated up-regulation of COL1A2 by 60% (P < 0.05), but had no effect on up-regulation of fibronectin transcription (Figure 10). This is in agreement with our previous studies suggesting that acetaldehyde-dependent regulation of collagen and of fibronectin follows different pathways,9 and that TGF-β1–dependent up-regulation of fibronectin is SMAD independent.28,29
Figure 10.

Lactacystin blocks acetaldehyde-mediated COL1A2 gene transcription, but not acetaldehyde-mediated fibronectin gene transcription. A and B: Lactacystin prevented acetaldehyde-mediated up-regulation of COL1A2 by 60% (P < 0.05) (A), but had no effect on the up-regulation of fibronectin transcription (B). Controls are assigned a value of 100%. All values were corrected for loading differences using an S14 ribosomal protein cDNA. Data are expressed as means ± SEM, representative of three independent experiments. ∗P < 0.05 versus control. †P < 0.05 versus acetaldehyde-treated cells.
Discussion
Acetaldehyde, the first metabolite of ethanol, is fibrogenic and induces expression of both COL1A1 and COL1A2 genes by a mechanism dependent on the generation of H2O2. These events are partially reproduced by adding this reactive oxygen species to cultured HSCs and can be prevented by the addition of catalase.2,12 Although the mechanisms whereby acetaldehyde induces generation of H2O2 remain to be established, our findings suggest that it may derive from mitochondrial dysfunction and not from the activation of the NADP(H) oxidase.12 This suggestion is based on the fact that phenyliodonium, an inhibitor of NADP(H) oxidase, had no effect on acetaldehyde-dependent up-regulation of α1(I) collagen mRNA.30 By contrast, ethanol is well known to cause mitochondrial alterations and redox changes.9,31 However, these results do not rule out the possibility that other cell types, such as inflammatory and Kupffer cells, known to express NADP(H) oxidase could contribute to the overall pool of H2O2 in vivo.8
Previous work from our laboratory has shown that some fibrogenic actions of acetaldehyde are mediated and/or enhanced by TGF-β1.1,12 Acetaldehyde induces expression and activation of TGF-β1 and of its type II receptor.1,12,16 However, the early events triggered by acetaldehyde occurring within the first 6 to 12 hours after acetaldehyde administration are not dependent on TGF-β1 and protein synthesis. Furthermore, both acetaldehyde and TGF-β1 stimulate phosphorylation and binding of SMAD3/4–containing transcriptional complexes to the COL1A2 promoter region.12 However, in contrast to the ability of TGF-β1 to up-regulate expression of SMADs 3 and 4 at the mRNA and protein levels and to enhance the phosphorylation of SMAD2,32 acetaldehyde has no effect on SMAD2 phosphorylation12 and does not significantly up-regulate expression of SMAD2 protein. In the present study, although neither acetaldehyde nor TGF-β1 alone had significant stimulatory effect, the combination elicited the maximum additive stimulatory activity of COL1A2 reporter vector (Figure 1).
Overall, these findings strongly suggest that the fibrogenic signaling pathways triggered by acetaldehyde and TGF-β1 are distinctly different. The fact that the stimulatory effect of acetaldehyde after 24 hours on COL1A2 gene expression was significantly blunted by TGF-β1 neutralizing antibody, but not by a nonspecific antibody (Figure 2), confirms that acetaldehyde-induced expression of TGF-β1 is responsible for the up-regulation of COL1A2. That the concomitant expression of both SMAD3 and SMAD4 (Figure 3) is obligatory for this effect of acetaldehyde on TGF-β1 expression is shown by the fact that neither SMAD3 nor SMAD4 vector overexpression alone caused acetaldehyde-induced stimulation of COL1A2 reporter activity, whereas in combination the two SMADs caused marked stimulation (Figure 4). On the other hand, the ability of acetaldehyde to up-regulate COL1A2 reporter activity in normal HSCs, but not in SMAD7-overexpressing HSCs (Figure 5), indicates that the action of acetaldehyde involves the suppression of SMAD7, a potent repressor of COL1A2 gene expression.
Our immunocytochemical studies demonstrated that Ski is preferentially localized in the nuclei of activated HSCs (Figures 6 and 7) and that acetaldehyde induces translocation of the repressor protein Ski from the nucleus to the cytoplasm by forming a complex with SMAD4 and thereby leading to the activation of nuclear SMAD4 (Figure 8). The key repressor role of Ski in the regulation of COL1A2 gene expression was clearly established (Figures 9 and 10). Lactacystin, a potent proteasomal inhibitor, blunted acetaldehyde-dependent up-regulation of COL1A2 gene transcription and reporter activity of a luciferase vector driven by the acetaldehyde-responsive element of COL1A2, but had no effect on the induction of housekeeping fibronectin gene transcription.
Thus, we have shown that, in addition to up-regulating COL1A2 gene expression by enhancing expression and activity of positive transactivators, acetaldehyde also down-regulates expression of the gene transcription inhibitor SMAD7. More importantly, acetaldehyde effectively promotes the translocation of the other transcription repressor, Ski, from the nucleus to the cytoplasmic compartment to undergo proteasomal degradation via the ubiquitin pathway. Our findings suggest that transcriptional up-regulation of COL1A2 by acetaldehyde occurs via two distinct mechanisms. The first occurs very rapidly, is transient, and involves the elimination of repressors of COL1A2 (such as Ski and SMAD7) and the phosphorylation of SMAD3. The second mechanism is more sustained and corresponds to the expression of TGF-β1 and consequent up-regulation of SMAD3 and SMAD4, a process that starts after 6 to 12 hours of exposure to acetaldehyde. Based on these new findings, as well as on earlier work,16 we speculate that the cytokine TGF-β1 augments acetaldehyde-dependent expression of COL1A2. This suggestion is also supported by the fact that the effects of acetaldehyde and TGF-β1 are additive.
We have shown here that acetaldehyde induces a rapid down-regulation of SMAD7. This SMAD is a negative regulator of the COL1A2 gene,24,25 so its decrease occurs at the time when collagen mRNA is being up-regulated. Interestingly, the expression of SMAD7 starts to increase at a time when SMAD3 and SMAD4 are being up-regulated and reaches its maximal level when the expression of COL1A2 has leveled off or even started to decrease. These findings may explain the up-regulation and down-regulation of collagen gene expression after a single acetaldehyde dose of 200 μmol/L. Thus, although collagen mRNA is up-regulated when repressors are down-regulated, collagen gene expression is shut down when the repressors return to normal levels.
The SMAD7 promoter has binding motifs for SMAD3 and SMAD4.33,34 Accordingly, it is conceivable that the up-regulation of SMAD7 mRNA by acetaldehyde results from the late up-regulation of SMAD3 and SMAD4 and subsequent binding to the SMAD7 promoter region. Taken together, our findings suggest that the regulation of the COL1A2 by acetaldehyde involves the concerted actions of several transactivators and repressors, as observed in the present study. In the basal state, several repressors limit COL1A2 expression and thus control the amount of type I collagen present in the normal extracellular matrix. However, acetaldehyde induces degradation of these repressors, thus allowing the binding of transcriptional activators involved in COL1A2 up-regulation. As shown here and previously,1,11,12 this process occurs via rapid phosphorylation of SMAD3 and formation of SMAD3/4 complexes that interact with Sp1.
In conclusion, based on the present findings we speculate that Ski plays a major role in the fibrogenic action of acetaldehyde by sequestering the SMAD4–Ski complex from the nucleus to the cytoplasm, leading to the proteasomal degradation of Ski via the ubiquitin pathway and consequent activation of SMAD4. Activated SMAD4 and phosphorylated SMAD3, the two key transcription factors, in turn up-regulate COL1A2 in the nucleus by interacting with its promoter region. Thus, we suggest that the proteasomal degradation of Ski is an important event in acetaldehyde-mediated up-regulation of COL1A2 in HSCs. The present findings unveil additional critical steps in the acetaldehyde-mediated fibrogenic process and thus suggest possible new targets for antifibrogenic therapy.
Acknowledgments
We thank Marcos Amorim, Cindy Else, Gianluca Svegliati-Baroni, and Francesco Ramirez for their assistance.
Footnotes
Supported in part by NIH grants AA-010541 and AA-009231 (M.R.L.), a grant-in aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (Y.I.), and CONACYT–Mexico National Council of Science and Technology fellowships 137122 (K.R.-G.) and 128405 and 151478 (J.A.-R.).
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Department of the Army or the Department of Defense.
K.R.-G. and R.S. contributed equally to this work.
M.R. is deceased.
References
- 1.Svegliati-Baroni G., Ridolfi F., Di Sario A., Saccomanno S., Bendia E., Benedetti A., Greenwel P. Intracellular signaling pathways involved in acetaldehyde-induced collagen and fibronectin gene expression in human hepatic stellate cells. Hepatology. 2001;33:1130–1140. doi: 10.1053/jhep.2001.23788. [DOI] [PubMed] [Google Scholar]
- 2.Greenwel P., Domínguez-Rosales J.A., Mavi G., Rivas-Estilla A.M., Rojkind M. Hydrogen peroxide: a link between acetaldehyde-elicited alpha1(I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology. 2000;31:109–116. doi: 10.1002/hep.510310118. [DOI] [PubMed] [Google Scholar]
- 3.Lin W., Wu G., Li S., Weinberg E.M., Kumthip K., Peng L.F., Méndez-Navarro J., Chen W.C., Jilg N., Zhao H., Goto K., Zhang L., Brockman M.A., Schuppan D., Chung R.T. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFkappaB. J Biol Chem. 2011;286:2665–2674. doi: 10.1074/jbc.M110.168286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novitskiy G., Traore K., Wang L., Trush M.A., Mezey E. Effects of ethanol and acetaldehyde on reactive oxygen species production in rat hepatic stellate cells. Alcohol Clin Exp Res. 2006;30:1429–1435. doi: 10.1111/j.1530-0277.2006.00171.x. [DOI] [PubMed] [Google Scholar]
- 5.Parola M., Marra F. Adipokines and redox signaling: impact on fatty liver disease. Antioxid Redox Signal. 2011;15:461–483. doi: 10.1089/ars.2010.3848. [DOI] [PubMed] [Google Scholar]
- 6.Enomoto N., Schemmer P., Ikejima K., Takei Y., Sato N., Brenner D.A., Thurman R.G. Long-term alcohol exposure changes sensitivity of rat Kupffer cells to lipopolysaccharide. Alcohol Clin Exp Res. 2001;25:1360–1367. [PubMed] [Google Scholar]
- 7.Paik Y.H., Schwabe R.F., Bataller R., Russo M.P., Jobin C., Brenner D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- 8.Nagata K., Suzuki H., Sakaguchi S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J Toxicol Sci. 2007;32:453–468. doi: 10.2131/jts.32.453. [DOI] [PubMed] [Google Scholar]
- 9.Lieber C.S. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 10.García-Trevijano E.R., Iraburu M.J., Fontana L., Domínguez-Rosales J.A., Auster A., Covarrubias-Pinedo A., Rojkind M. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960–970. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W., Ou J., Inagaki Y., Greenwel P., Ramirez F. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor beta1 stimulation of alpha 2(I)-collagen (COL1A2) transcription. J Biol Chem. 2000;275:39237–39245. doi: 10.1074/jbc.M003339200. [DOI] [PubMed] [Google Scholar]
- 12.Svegliati-Baroni G., Inagaki Y., Rincon-Sanchez A.R., Else C., Saccomanno S., Benedetti A., Ramirez F., Rojkind M. Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology. 2005;42:343–352. doi: 10.1002/hep.20798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piek E., Roberts A.B. Suppressor and oncogenic roles of transforming growth factor-beta and its signaling pathways in tumorigenesis. Adv Cancer Res. 2001;83:1–54. doi: 10.1016/s0065-230x(01)83001-3. [DOI] [PubMed] [Google Scholar]
- 14.Takeda M., Mizuide M., Oka M., Watabe T., Inoue H., Suzuki H., Fujita T., Imamura T., Miyazono K., Miyazawa K. Interaction with Smad4 is indispensable for suppression of BMP signaling by c-Ski. Mol Biol Cell. 2004;15:963–972. doi: 10.1091/mbc.E03-07-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J., Li P., Zhang Y., Li G.B., Zhou Y.G., Yang K., Dai S.S. c-Ski inhibits the proliferation of vascular smooth muscle cells via suppressing Smad3 signaling but stimulating p38 pathway. Cell Signal. 2013;25:159–167. doi: 10.1016/j.cellsig.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Inagaki Y., Truter S., Ramirez F. Transforming growth factor-beta stimulates alpha 2(I) collagen gene expression through a cis-acting element that contains an Sp1-binding site. J Biol Chem. 1994;269:14828–14834. [PubMed] [Google Scholar]
- 17.Inagaki Y., Mamura M., Kanamaru Y., Greenwel P., Nemoto T., Takehara K., Ten Dijke P., Nakao A. Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J Cell Physiol. 2001;187:117–123. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1059>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 18.Myers J.C., Dickson L.A., de Wet W.J., Bernard M.P., Chu M.L., Di Liberto M., Pepe G., Sangiorgi F.O., Ramirez F. Analysis of the 3′ end of the human pro-alpha 2(I) collagen gene. Utilization of multiple polyadenylation sites in cultured fibroblasts. J Biol Chem. 1983;258:10128–10135. [PubMed] [Google Scholar]
- 19.Schwarzbauer J.E., Tamkun J.W., Lemischka I.R., Hynes R.O. Three different fibronectin mRNAs arise by alternative splicing within the coding region. Cell. 1983;35:421–431. doi: 10.1016/0092-8674(83)90175-7. [DOI] [PubMed] [Google Scholar]
- 20.Inagaki Y., Truter S., Bou-Gharios G., Garrett L.A., de Crombrugghe B., Nemoto T., Greenwel P. Activation of proalpha2(I) collagen promoter during hepatic fibrogenesis in transgenic mice. Biochem Biophys Res Commun. 1998;250:606–611. doi: 10.1006/bbrc.1998.9345. [DOI] [PubMed] [Google Scholar]
- 21.Lieber C.S. Metabolism of alcohol. Clin Liver Dis. 2005;9:1–35. doi: 10.1016/j.cld.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Choi S.S., Omenetti A., Witek R.P., Moylan C.A., Syn W.K., Jung Y., Yang L., Sudan D.L., Sicklick J.K., Michelotti G.A., Rojkind M., Diehl A.M. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1093–G1106. doi: 10.1152/ajpgi.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cenni V., Sirri A., Riccio M., Lattanzi G., Santi S., de Pol A., Maraldi N.M., Marmiroli S. Targeting of the Akt/PKB kinase to the actin skeleton. Cell Mol Life Sci. 2003;60:2710–2720. doi: 10.1007/s00018-003-3349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bitzer M., von Gersdorff G., Liang D., Dominguez-Rosales A., Beg A.A., Rojkind M., Böttinger E.P. A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 2000;14:187–197. [PMC free article] [PubMed] [Google Scholar]
- 25.Dooley S., Hamzavi J., Breitkopf K., Wiercinska E., Said H.M., Lorenzen J., Ten Dijke P., Gressner A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178–191. doi: 10.1016/s0016-5085(03)00666-8. [DOI] [PubMed] [Google Scholar]
- 26.Liu X., Sun Y., Weinberg R.A., Lodish H.F. Ski/Sno and TGF-beta signaling. Cytokine Growth Factor Rev. 2001;12:1–8. doi: 10.1016/s1359-6101(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko T., Murata S. Using siRNA techniques to dissect proteasome assembly pathways in mammalian cells. Methods Mol Biol. 2012;832:433–442. doi: 10.1007/978-1-61779-474-2_30. [DOI] [PubMed] [Google Scholar]
- 28.Hocevar B.A., Brown T.L., Howe P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuchida K., Zhu Y., Siva S., Dunn S.R., Sharma K. Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int. 2003;63:2000–2009. doi: 10.1046/j.1523-1755.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- 30.Ooyabu J., Ohtsuka M., Kashino Y., Koike H., Satoh K. The expression pattern of NAD(P)H oxidases and the cyclic electron transport pathway around photosystem I of Synechocystis sp. PCC6803 depend on growth conditions. Biosci Biotechnol Biochem. 2008;72:3180–3188. doi: 10.1271/bbb.80370. [DOI] [PubMed] [Google Scholar]
- 31.Chen A. Acetaldehyde stimulates the activation of latent transforming growth factor-beta1 and induces expression of the type II receptor of the cytokine in rat cultured hepatic stellate cells. Biochem J. 2002;368:683–693. doi: 10.1042/BJ20020949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Gersdorff G., Susztak K., Rezvani F., Bitzer M., Liang D., Bottinger E.P. Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor beta. J Biol Chem. 2000;275:11320–11326. doi: 10.1074/jbc.275.15.11320. [DOI] [PubMed] [Google Scholar]
- 33.Cutroneo K.R., Phan S.H. TGF-beta1-induced Smad 3 binding to the Smad 7 gene: knockout of Smad 7 gene transcription by sense phosphorothioate oligos, autoregulation, and effect on TGF-beta1 secretion: bleomycin acts through TGF-beta1. J Cell Biochem. 2003;89:474–483. doi: 10.1002/jcb.10528. [DOI] [PubMed] [Google Scholar]
- 34.Poncelet A.C., Schnaper H.W. Sp1 and Smad proteins cooperate to mediate transforming growth factor-beta 1-induced alpha 2(I) collagen expression in human glomerular mesangial cells [Erratum appeared in J Biol Chem 2001, 276:47746] J Biol Chem. 2001;276:6983–6992. doi: 10.1074/jbc.M006442200. [DOI] [PubMed] [Google Scholar]