

Abstract
The chemotherapeutic drug cisplatin is actively transported into proximal tubules, leading to acute renal injury. Previous studies suggest that the multidrug resistance–associated protein 2 (Mrp2) transporter may efflux cisplatin conjugates from cells. We sought to determine whether the absence of Mrp2 alters the accumulation and toxicity of platinum in the kidneys of mice and whether transgenic expression of the human MRP2 gene could protect against cisplatin injury in vivo. Plasma, kidneys, and livers from vehicle- and cisplatin-treated wild-type and Mrp2-null mice were collected for quantification of platinum and toxicity. By 24 hours, twofold higher concentrations of platinum were detected in the kidneys and livers of Mrp2-null mice compared with wild types. Enhanced platinum concentrations in Mrp2-null mice were observed in DNA and cytosolic fractions of the kidneys. Four days after cisplatin treatment, more extensive proximal tubule injury was observed in Mrp2-null mice compared with wild-type mice. Kidneys from naive Mrp2-null mice had elevated glutathione S-transferase mRNA levels, which could increase the formation of cisplatin-glutathione conjugates that may be metabolized to toxic thiol intermediates. Transgenic expression of the human MRP2 gene in Mrp2-null mice reduced the accumulation and nephrotoxicity of cisplatin to levels observed in wild-type mice. These data suggest that deficiency in Mrp2 lowers platinum excretion and increases susceptibility to kidney injury, which can be rescued by the human MRP2 ortholog.
Cisplatin is commonly used in chemotherapy regimens for the treatment of solid cancers. The success of cisplatin therapy is limited, in part, by kidney injury. Up to 37% of patients develop signs of nephrotoxicity after receiving a single dose of cisplatin despite strategies such as hydration to limit renal exposure.1 This is problematic for patients because kidney injury can delay further treatment and limit the total number of chemotherapy cycles received, thereby reducing the overall efficacy of cisplatin-containing regimens.
Previous studies have identified the contribution of uptake and efflux transporters to the renal secretion of cisplatin. The organic cation transporter 2 (Oct2; gene Slc22a2) is highly expressed in the kidneys and mediates the renal uptake of cisplatin.2–5 Transgenic mice lacking Oct1 and Oct2 transporters exhibit decreased platinum (Pt) excretion and reduced nephrotoxicity.6 In addition, patients with cancer with a loss-of-function polymorphism in OCT2 (808G>T) are protected against cisplatin renal injury.6,7 Additional data suggest that the copper transporter 1 (Ctr1; gene Slc31a1) participates in the renal uptake of cisplatin,8–12 although recent work has found conflicting results.13 After uptake of cisplatin by Oct2 or Ctr1, the subsequent efflux of cisplatin into the renal lumen is mediated by the multidrug and toxin extrusion protein 1 (Mate1; gene Slc47a1).14 As expected, Mate1 knockout mice have increased renal concentrations of cisplatin and enhanced nephrotoxicity.14
Once inside the renal cell, cisplatin is biotransformed by spontaneous hydrolysis to monoaquated and diaquated species. Formation of cisplatin aquated intermediates leads to electrophilic attack of cellular macromolecules, such as DNA and protein. The tripeptide glutathione (GSH) can also bind cisplatin and has been suggested to form a variety of nontoxic cisplatin conjugates, including monoplatinum-monoglutathione, diplatinum-monoglutathione, and bis-(glutathionato)-platinum.15,16 It has been postulated that these conjugates have the potential for metabolism to reactive thiol intermediates. Previous studies suggest that the multidrug resistance–associated transporter 2 (Mrp2; gene Abcc2) can efflux GSH conjugates of cisplatin from cancer cells and confer resistance to cytotoxicity.15,17,18 Overexpression of human (h)MRP2 increases cisplatin resistance by 10-fold in human embryonic kidney cells17 and reduces cisplatin accumulation by 30% in porcine kidney LLC-PK1 cells.18 Moreover, primary hepatocytes isolated from transport-deficient rats (Mrp2-deficient strain) exhibit increased binding of Pt to DNA and enhanced susceptibility to cisplatin cytotoxicity.19 However, the ability of Mrp2 to reduce the accumulation of cisplatin and/or its conjugates and limit renal injury in vivo is not well understood. Therefore, the purpose of this study was to determine whether loss of Mrp2 function enhances renal Pt accumulation and nephrotoxicity in mice and to evaluate the ability of the human MRP2 gene to rescue Mrp2-null mice from heightened cisplatin-induced nephrotoxicity in vivo.
Materials and Methods
Chemicals
Unless otherwise specified, all chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Vesicle Transport Assays
Inside-out Sf9 insect cell plasma membrane vesicles expressing mouse (m)Mrp2 or hMRP2 were purchased from Life Technologies (Carlsbad, CA). To assess the ability of cisplatin to inhibit Mrp2/MRP2-mediated transport of 5,6-carboxy-2′,7′-dichlorofluorescein (CDCF), 20-μg vesicles were incubated with 10 μmol/L CDCF, 4 mmol/L adenosine triphosphate, 2 mmol/L GSH, and increasing concentrations of cisplatin (50 to 200 μmol/L) in reaction buffer at 37°C for 10 minutes according to the manufacturer's protocol. Vesicles were washed, vacuum filtered, and solubilized with 50% methanol. Fluorescence was read at excitation wavelength 504 nm; emission wavelength 529 nm.
Animal Treatment
Wild-type (WT) C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). Mrp2-null mice were obtained from Taconic Laboratories (Hudson, NY) and were backcrossed until they were >99% congenic with the C57BL/6 strain. Congenic analysis was performed by the Bionomics Research and Technology Core at Rutgers University (New Brunswick, NJ). hMRP2 mice on a C57BL/6 background were purchased from Taconic. Cisplatin was dissolved in saline after heating to 50°C. Groups of 9- to 14-week-old adult male mice were injected i.p. with 5 mL/kg of saline vehicle or 20 mg/kg of cisplatin after overnight fasting. The doses of cisplatin used in this study were similar to those used clinically.1 Feed was returned 4 hours after cisplatin injection. Kidney, liver, and plasma samples were collected 1 hour to 4 days after cisplatin treatment. Groups of mice were placed into metabolism cages for quantification of urine Pt concentrations (24-hour periods between 0 and 3 days) and urine output (24-hour period from 3 to 4 days). Tissues were fixed in 10% formalin or were snap frozen.
Analytes and Pathology
Blood urea nitrogen (BUN) and serum creatinine levels were quantified as indicators of renal injury (Thermotrace, Melbourne, Australia; and Pointe Scientific, Canton, MI). Total and direct bilirubin concentrations as well as alanine aminotransferase activity were also quantified (Pointe Scientific). GSH was quantified using the GSH/GSSG-Glo assay (Promega, Madison, WI). Five-micron paraffin-embedded kidney sections were stained with H&E and were examined for histopathological changes by a board-certified anatomic veterinary pathologist (M.J.G.) using a published grading scale.20
RNA Isolation and mRNA Quantification
Total RNA was isolated using RNA-Bee reagent (Tel-Test Inc., Friendswood, TX). The mRNA expression of mouse kidney injury molecule-1, heme oxygenase-1, metallothionein-1, glutathione S-transferase (Gst) isoforms, glutamate cysteine ligase catalytic subunit, Oct2, Mrp4, breast cancer resistance protein (Bcrp), and multidrug resistance protein 1a was quantified using the branched DNA 1.0 signal amplification assay (Affymetrix Inc., Santa Clara, CA).21 Oct1, Mate1, and Ctr1 mRNAs were quantified by quantitative PCR. cDNA was generated using the SuperScript first-strand cDNA synthesis kit (Life Technologies). Specific forward and reverse primers (Integrated DNA Technologies, Coralville, IA) were added to 1 μg of cDNA from each sample. The following primers (forward and reverse, respectively) were used for mouse Mate1 (5′-GTTGGCCTTACGGAGAGGAC-3′ and 5′-AATCCCACCCACCAAGACTAA-3′), mouse Ctr1 (5′-ACACACAAAACTGTTGGGCAG-3′ and 5′-AGAGAAAGTATCCCGTCCCAG-3′), mouse Oct1 (5′-TGTCGGCTCTGGCTACAGGAGA-3′ and 5′-GGGGGATTCTGGGACAAACCAGTAA-3′), and mouse ribosomal protein l 13a (Rpl13a) (5′-CAAGAAAAAGCGGATGGTGG-3′ and 5′-TCCGTAACCTCAAGATCTGC-3′). SYBR Green (Applied Biosystems, Carlsbad, CA) was used for the detection of amplified products. Quantitative PCR was performed in a 384-well plate format using the ABI 7900HT PCR system (Applied Biosystems). CT values were converted to ΔΔCT values by comparing with a reference gene, Rpl13a.
Inductively Coupled Plasma Mass Spectrometry
Total Pt Analysis
Concentrated EMD OmniTace Ultra high-purity nitric acid (VWR, Radnor, PA) was added to all the samples to a final concentration of 5%, followed by digestion using a CEM Mars X microwave system (CEM Corp, Matthews, NC) (Table 1). Large samples were pretreated in an ultrasonic bath, whereas samples not completely digested were reacted, postdigestion, with 30% hydrogen peroxide (TraceSelect, Fluka; Sigma-Aldrich). Digested samples were analyzed by inductively coupled plasma mass spectrometry using a Thermo-Elemental X5 instrument (Thermo Fisher). Multiple Pt ions were monitored for quality control. Reported concentrations were determined using m/z = 195. This method has been used previously for metals analysis in blood samples.22
Table 1.
Methods for Inductively Coupled Plasma Mass Spectrometry Analysis
| Matrix | Approximate weight (g) or volume (μL) | Nitric acid (mL) | Microwave program, 300 W |
|---|---|---|---|
| Urine∗ | 20–100 μL | 0.25–0.5 | 50% power, 5 minutes, once, then 75% power, 5 minutes, 5× |
| Liver†‡ | 0.1–0.25 g | 1–1.5 | 75% power, 5 minutes, once, then 100% power, 5 minutes, 5×, then 100% power, 10 minutes, 1–2× |
| Kidney† | 0.1–0.25 g | 1–1.5 | Same as liver |
| Plasma∗†§ | 50 μL | 0.25 | Same as urine |
| Vesicles | 100 μL | 0.15 | Same as urine |
The approximate rule for a largely aqueous matrix is at least two parts acid to one part matrix by volume.
Microwaving was discontinued when the samples were clear and pale yellow.
Hydrogen peroxide (0.5 mL, 30% H2O2) was occasionally added to complete the digestion of liver. If so, an additional microwave treatment (100% power, 10 minutes) was used to decompose the residual hydrogen peroxide.
Samples in the 0.25-mL range can be microwaved in 15-mL centrifuge tubes provided that less time and lower percentage power are used.
Fractionation Pt Studies
Kidneys were homogenized in 250 mmol/L sucrose–10 mmol/L Tris buffer and were centrifuged at 100,000 × g for the collection of cytosol and crude membrane fractions. DNA was isolated using the DNeasy blood and tissue kit (Qiagen, Valencia, CA).
Western Blot Analysis
Kidneys were homogenized in sucrose-Tris buffer containing protease inhibitors. Western blot staining was performed as described elsewhere.23 The following antibodies were used: Mrp2 (M2III-5), Mrp4 (M4I-10), and Bcrp (BXP-53) from Enzo Life Sciences (Farmingdale, NY); Oct2 (sc292622) and Mate1 (sc138983) from Santa Cruz Biotechnology (Santa Cruz, CA); and Ctr1 (ab129067), multidrug resistance protein 1a/b (C219), and β-actin (ab8227) from Abcam Inc. (Cambridge, MA).
Immunofluorescence
Immunofluorescence analysis was performed for hMRP2 (M2III-6), mMrp2 (EAG15), and mouse Bcrp (Bxp-53). Sections were washed and incubated with goat anti-mouse or anti-rabbit IgG Alexa 488 IgG and anti-rat 563 IgG antibodies (Invitrogen, Carlsbad, CA). Images were acquired using a Zeiss Observer D1 microscope with an X-Cite series 120Q fluorescent illuminator (Zeiss Inc., Thornwood, NY) and a ProgRes camera with CapturePro version 2.8 software (Jenoptik, Easthampton, MA). Negative controls without primary antibody were included to ensure minimal nonspecific staining (data not shown).
Statistical Analysis
The software program GraphPad Prism version 5 (GraphPad Software Inc., La Jolla, CA) was used for statistical analysis. Pharmacokinetic curves were generated using area-under-the-curve analysis with one-phase nonlinear decay. Differences among groups were evaluated by Student's unpaired t-test (two groups) or one-way analysis of variance followed by the Newman-Keuls multiple comparison tests (three or more groups). Histopathological data were rank ordered before analysis. Statistical significance was set at P < 0.05.
Results
Interaction of mMrp2 and hMRP2 Transporters with Cisplatin in Vitro
An indirect approach (competition for transport) was used to investigate cisplatin as a potential Mrp2/MRP2 substrate in the presence of GSH using inverted plasma membrane vesicles. Commercial vesicles expressed the Mrp2 and MRP2 proteins, with notably lower expression of mMrp2 compared with hMRP2 protein (Figure 1A). The probe Mrp2/MRP2 substrate, CDCF, was used for inhibition assays as previously described.24,25 Cisplatin inhibited the adenosine triphosphate–dependent transport of CDCF in plasma membrane vesicles (Figure 1B). Transport of CDCF by both orthologs was inhibited up to 70% to 80% at 100 to 200 μmol/L cisplatin. mMrp2-mediated transport was also markedly inhibited at 50 μmol/L cisplatin. Note that serum concentrations as high as 20 μmol/L have been observed clinically,26 although it is unclear how this value translates to intracellular tubule levels.
Figure 1.

Interaction of mMrp2 and hMRP2 transporters with cisplatin in vitro. A: Western blot analysis of commercially available mMrp2 and hMRP2 vesicles. β-Actin was used as a loading control. B: Twenty micrograms of inverted Mrp2- and MRP2-expressing vesicles were incubated with 10 μmol/L CDCF for 10 minutes in the presence and absence of adenosine triphosphate (ATP), GSH, and increasing concentrations of cisplatin. The total mean transport rates of CDCF in mMrp2- and hMRP2-expressing vesicles were 1051 and 1933 pmol/min/mg of protein, respectively (data not shown). The ATP-independent transport rates of CDCF in mMrp2- and hMRP2-expressing vesicles were 12 and 222 pmol/min/mg protein, respectively (data not shown). Data are presented as means ± SEM (n = 4 to 5) normalized to ATP-dependent MRP2 transport of CDCF (no cisplatin). ∗P < 0.05 compared with 0 μmol/L cisplatin.
Renal and Hepatic Injury
Cisplatin caused time-dependent renal injury in WT and Mrp2-null mice (Figure 2A). BUN levels increased similarly in both genotypes on day 3. By 4 days, BUN levels in cisplatin-treated Mrp2-null mice were twofold higher than those in WT mice. Likewise, elevations in serum creatinine levels were observed only on day 4, with a greater increase in Mrp2-null mice. Mild increases in alanine aminotransferase activity were also seen in cisplatin-treated Mrp2-null mice, with no change detected in WT mice.
Figure 2.

Renal and hepatic injury markers in wild-type (black bars) and Mrp2-null (gray bars) mice treated with cisplatin. A: BUN, serum creatinine (SCr), and alanine aminotransferase (ALT) levels in plasma from WT and Mrp2-null mice 2 through 4 days after 20 mg/kg of i.p. cisplatin treatment (n = 5 to 15). B: mRNA expression of kidney injury molecule-1 (Kim-1), metallothionein-1 (Mt-1), and heme oxygenase-1 (Ho-1) was quantified using total kidney RNA from control and 20-mg/kg cisplatin-treated WT and Mrp2-null mice on day 4 (n = 4 to 11). C: Urine flow rate of control and 20-mg/kg cisplatin-treated WT and Mrp2-null mice from day 3 to 4. Urine volume was quantified from mice in metabolic cages for 24 hours and was normalized to body weight (n = 3 to 8). Data are presented as means ± SEM. mRNA data were normalized to WT control mice. ∗P < 0.05, compared with genotype control mice; †P < 0.05, compared with cisplatin-treated WT mice.
Cisplatin-treated Mrp2-null mice exhibited significant elevations in the renal mRNA expression of kidney injury molecule-1 (fourfold), metallothionein-1 (twofold), and heme oxygenase-1 (2.5-fold) on day 4 compared with WT mice (Figure 2B). In addition, Mrp2-null mice had a 44% reduction in urine output between 3 and 4 days, although this decrease was not statistically significant (Figure 2C).
Histopathologic evaluation of kidneys from cisplatin-treated mice revealed S1/S2 proximal tubule degeneration and necrosis as well as sloughing of tubule epithelium that was more severe in Mrp2-null mice at 4 days (Figure 3 and Table 2). Necrotic tubules contained renal casts with eosinophilic material and were more numerous in Mrp2-null mice. Livers from WT and Mrp2-null mice showed no evidence of hepatocellular degeneration and/or necrosis (data not shown).
Figure 3.

Histopathologic injury in WT and Mrp2-null mice treated with cisplatin. WT and Mrp2-null mice were treated with vehicle or 20 mg/kg of i.p. cisplatin, and kidneys were collected on day 4. Samples were fixed in zinc formalin before routine processing and paraffin embedding. Five-micron sections of kidneys were stained with H&E and were examined by light microscopy for the presence and severity of renal cast formation (∗), proximal tubule degeneration (+), apoptosis, and necrosis (ˆ) as well as neutrophil infiltration. Original magnification, ×40.
Table 2.
Histopathologic Analysis of Kidneys from WT and Mrp2-Null Mice after Cisplatin Injection
| Mice and time after injection | Histopathologic grade |
Mice with grade ≥2 (%) | |||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | ||
| WT | |||||||
| Control | 13 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 Days | 1 | 6 | 0 | 0 | 0 | 0 | 0∗ |
| 3 Days | 1 | 2 | 2 | 0 | 0 | 0 | 40∗ |
| 4 Days | 4 | 3 | 6 | 1 | 0 | 0 | 50∗ |
| Mrp2-null | |||||||
| Control | 13 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 Days | 1 | 4 | 2 | 0 | 0 | 0 | 29∗ |
| 3 Days | 1 | 1 | 3 | 0 | 0 | 0 | 60∗ |
| 4 Days | 0 | 1 | 3 | 6 | 2 | 0 | 91∗† |
Kidneys were removed 2 to 4 days after 20 mg/kg of i.p. cisplatin or vehicle injection and were fixed in zinc formalin before paraffin embedding and staining with H&E. Kidney slices were evaluated for the severity of degeneration and necrosis in proximal tubule segments. Histopathologic scoring of renal proximal tubule degeneration and necrosis by a veterinary pathologist was as follows: grade 0, no injury; grade 1, minimal injury involving <10% of cells with degeneration or necrosis; grade 2, mild injury involving 10% to 25% of cells; grade 3, moderate injury involving >25% to 40% of cells; grade 4, marked injury involving >40% to 50% of cells; grade 5, severe injury involving >50% of cells. The number of mice with a particular histopathologic grade is shown in each column. Mice with grades ≥2 are considered to have significant kidney injury. The ratios of mice with grades ≥2 compared with the total number of mice are presented as percentages. Histopathologic grades were rank ordered before statistical analysis.
P < 0.05 compared with genotype control mice.
P < 0.05 compared with cisplatin-treated WT mice.
Pt Concentrations
Kidney concentrations of Pt in Mrp2-null mice were elevated above WT levels at 24 and 48 hours (30% increase in the area under the curve) (Figure 4A). Similar to the kidneys, Pt concentrations were higher in the livers of Mrp2-null mice through 72 hours. The clearance of Pt from the plasma was similar between WT and Mrp2-null mice, suggesting similar uptake between genotypes. Using metabolic cages, total Pt excretion into urine was high and variable in both genotypes at 24 hours and decreased by 48 and 72 hours, with no differences observed (Figure 4A). Fractionation of kidneys from WT and Mrp2-null mice 24 hours after cisplatin treatment revealed 50% increases in Pt levels in cytosol and DNA but not in the crude membrane fraction, suggesting specificity in the subcellular localization of cisplatin intermediates and/or adducts (Figure 4B).
Figure 4.

Pt concentrations in WT and Mrp2-null mice treated with cisplatin. Quantification of Pt concentrations by inductively coupled plasma mass spectrometry in the kidneys, livers, plasma, and urine (A) and cytosol, crude membrane, and DNA fractions from kidneys (B) of WT and Mrp2-null mice treated with 20 mg/kg of i.p. cisplatin. There were no differences in urine output between WT and Mrp2-null mice up to 72 hours. Data are presented as means ± SEM. n = 4 to 10 (A); n = 5 (B). ∗P < 0.05 compared with cisplatin-treated WT mice.
Kidney Uptake and Efflux Transporters
Because additional proteins can transport cisplatin, we quantified the basal expression of Oct2, Ctr1, and Mate1 mRNAs and proteins as well as additional apical efflux transporters, Mrp4, Bcrp, and multidrug resistance protein 1 in both genotypes. Expression of Oct2, Ctr1, Mate1, Bcrp, and multidrug resistance protein 1 mRNA and protein was similar in kidneys from naive WT and Mrp2-null mice (Figure 5, A and B). Because of the high expression of Oct1 mRNA in mice,27 mRNA levels of this isoform were also assessed and unchanged in Mrp2-null mice (Figure 5A). Oct3 mRNA levels were very low in both genotypes (data not shown). Mrp4 mRNA and protein levels were elevated in Mrp2-null mice, similar to previous reports.28,29 It is unclear whether Mrp4 can transport cisplatin; however, this basal increase does not seem to compensate for reduced Pt excretion and enhanced toxicity in Mrp2-null mice.
Figure 5.

Basal expression of transporters and GSH-related enzymes in kidneys of naive WT and Mrp2-null mice. A: mRNA levels of uptake and efflux transporters in WT and Mrp2-null mice. B: Western blot analysis of transporter proteins in WT and Mrp2-null mice. β-Actin was used as a loading control. mRNA expression of glutathione S-transferase (Gst) and glutamate cysteine ligase catalytic subunit (Gclc) (C) and GSH concentrations (D) in WT and Mrp2-null mice. Data are presented as means ± SEM. n = 4 to 5 (A); n = 4 to 6 (C and D). mRNA data were normalized to WT mice. ∗P < 0.05 compared with naive WT mice.
Kidney GSH Pathways
Basal differences in the mRNA expression of enzymes involved in GSH production and conjugation were also quantified. Compared with WT mice, expression of Gsta3, p1/2, and m1 was elevated 50% to 100% in kidneys from naive Mrp2-null mice (Figure 5C). There were no notable differences in Gstt1 and glutamate cysteine ligase catalytic subunit mRNA levels between genotypes. Renal GSH concentrations were not significantly different between WT and Mrp2-null mice (Figure 5D).
hMRP2 Mice
hMRP2 mice have been recently generated as a transgenic knock-in in Mrp2-null mice.30 To confirm proper trafficking of hMRP2 protein to the brush border membrane of tubules, we used species-specific antibodies against Mrp2/MRP2 for co-localization with the mouse Bcrp transporter (Figure 6A). As expected, the anti-rodent Mrp2 antibody detected protein in WT mice only, which co-localized with mouse Bcrp on the apical membrane of tubules. The anti-human MRP2 antibody detected MRP2 protein in tubules that co-localized with mouse Bcrp in the humanized mice. Neither mMrp2 nor hMRP2 protein was detected in the Mrp2-null mice. Similar insertion of hMRP2 protein into the canalicular membrane of hepatocytes was seen in humanized mice (Supplemental Figure S1). Bilirubin is a known Mrp2/MRP2 substrate. Similar to patients with Dubin-Johnson syndrome, mice lacking Mrp2 have increased direct and total bilirubin levels.28,29 We similarly observed hyperbilirubinemia in Mrp2-null mice that was reduced in humanized mice (Figure 6B), confirming that the hMRP2 protein is functional in the transgenic animals.
Figure 6.

Characterization of hMRP2 mice. Indirect immunofluorescence against mMrp2 (EAG15 antibody) or hMrp2 (M2III-6 antibody) (green) was conducted on 6-μm kidney cryosections from control WT, Mrp2-null, and hMRP2 mice. Brush border localization of mMrp2 and hMRP2 proteins was confirmed by co-localization with mouse Bcrp protein (mBcrp; Bxp-53 antibody) (red). A: Representative cortex regions are shown. B: Direct and total bilirubin concentrations were quantified in plasma from control WT, Mrp2-null, and hMRP2 mice. Data are presented as means ± SEM. n = 3 to 5. ∗P < 0.05 compared with naive WT mice. Original magnification, ×64 (A).
Similar to Figure 2, administration of cisplatin to Mrp2-null mice increased BUN, serum creatinine, and alanine aminotransferase levels as well as kidney injury molecule-1 and heme oxygenase-1 mRNA levels to a greater extent than in WT mice (Figure 7). Expression of MRP2 in humanized mice restored these end points to levels observed in WT mice. Likewise, Pt levels were 41% higher in the kidneys of Mrp2-null mice compared with WT and hMRP2 mice (Figure 8A). No differences in plasma Pt levels were detected between genotypes (Figure 8B).
Figure 7.

Renal and hepatic injury markers in WT, Mrp2-null, and hMRP2 mice treated with cisplatin. A: BUN, serum creatinine (SCr), and alanine aminotransferase (ALT) levels in plasma from WT, Mrp2-null, and hMRP2 mice 4 days after 20-mg/kg i.p. cisplatin treatment. B: mRNA expression of kidney injury molecule-1 (Kim-1), metallothionein-1 (Mt-1), and heme oxygenase-1 (Ho-1) was quantified using total kidney RNA from control and 20 mg/kg cisplatin–treated WT and Mrp2-null mice on day 4. Data are presented as means ± SEM. n = 3 to 9 (A and B) ∗P < 0.05 compared with genotype control mice; †P < 0.05 compared with cisplatin-treated WT mice.
Figure 8.
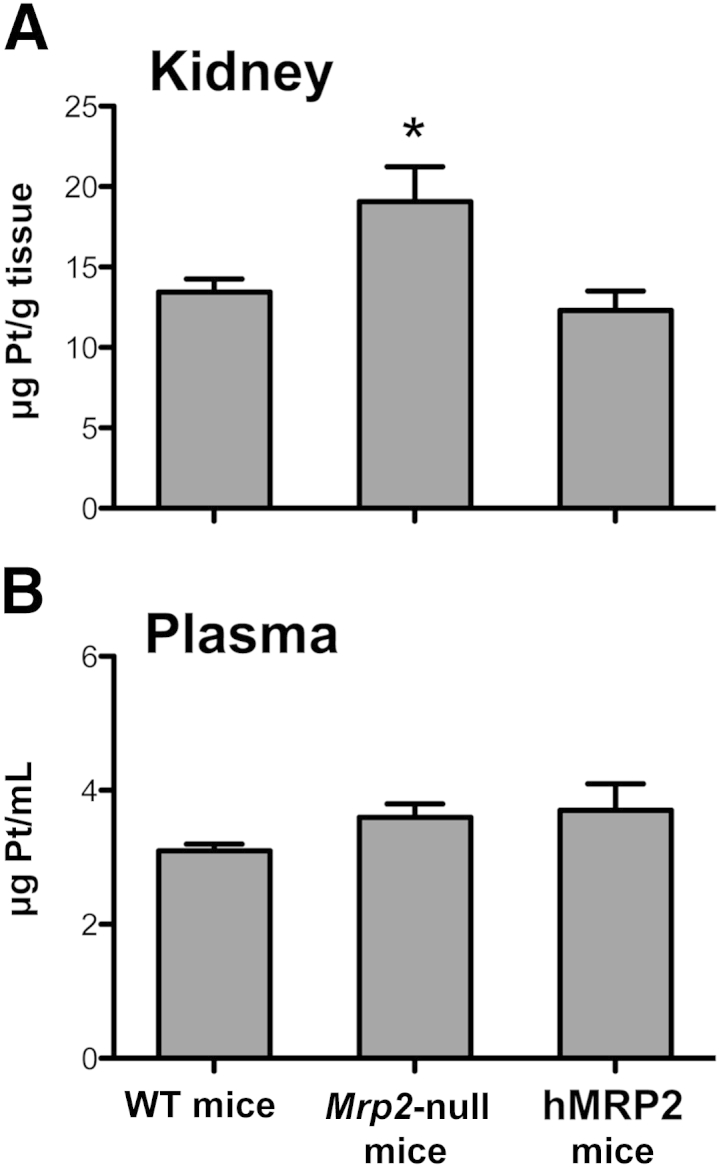
Pt concentrations in WT, Mrp2-null, and hMRP2 mice treated with cisplatin. Pt concentrations were quantified by inductively coupled plasma mass spectrometry in the kidneys (A) and plasma (B) of WT, Mrp2-null, and hMRP2 mice 24 hours after treatment with 20 mg/kg of i.p. cisplatin. Data are presented as means ± SEM. n = 6 to 9 (A and B). ∗P < 0.05 from cisplatin-treated WT mice.
Discussion
We investigated the role of Mrp2 in the disposition and toxicity of cisplatin in mice. We have shown enhanced susceptibility of Mrp2-null mice to renal injury by multiple end points (histopathology, serum analytes, urine output, and biomarker mRNAs). Differences in toxicity are likely due to higher concentrations of Pt in the kidneys of Mrp2-null mice. It can be suggested that elevated tissue Pt concentrations are due to impaired efflux rather than enhanced uptake because levels of Oct2 and Ctr1 mRNA and protein as well as Pt clearance from plasma were similar between genotypes. The present findings are in contrast to a recent study that concluded that Mrp2 does not participate in the renal excretion and toxicity of cisplatin in mice.31 This conclusion was based on histopathological evaluation and urinary Pt concentrations in cisplatin-treated WT and Mrp2-null mice at 3 days. Note that these data are consistent with the present findings because we did not observe differences in renal injury between genotypes until 4 days. Likewise, quantification of Pt concentrations in the kidneys and liver may be a more sensitive measure of pharmacokinetics in mice. These findings strongly support the use of time course studies for comparison of toxicity end points. Furthermore, these data are consistent with previous in vitro studies demonstrating a role for MRP2 in cytoprotection against cisplatin in isolated hepatocytes,19 esophageal squamous carcinoma cells,32 and hepatocellular carcinoma cells33 but extends this function to normal kidneys.
Cisplatin-GSH is proposed to be metabolized by γ-glutamyltranspeptidase to a cysteinyl-glycine conjugate and aminopeptidase to a cysteine conjugate on the extracellular surface of the brush border membrane (Figure 9).34–36 The cysteine conjugates are then transported into proximal tubules and are metabolized by β-lyase to highly reactive thiols. The mechanism(s) for cisplatin conjugate reabsorption is not well-known. Neutral amino acid transporter complexes, such as system L, may mediate influx, similar to observations in carcinoma cells.37 However, additional experiments are needed to identify the exact carriers involved in intact proximal tubules.
Figure 9.

Proposed metabolism and transport of cisplatin renal proximal tubules. Cisplatin is removed from the blood by OCT2 and CTR1. Cisplatin can then be excreted by MATE1. Cisplatin also hydrolyzes spontaneously to aquated species that can bind DNA and cause toxicity or that can be conjugated with GSH by glutathione S-transferases (GSTs). We propose that the MRP2 effluxes cisplatin-GSH to the tubule lumen, where it can be cleaved by γ-glutamyltranspeptidase (GGT) and aminodipeptidase (AP) to cisplatin-cysteine (CYS). The cisplatin-CYS conjugate is then reabsorbed by the proximal tubule through an unknown mechanism. Cisplatin-CYS can then be metabolized by cysteine S-conjugate β-lyase (BL) to form a reactive thiol that contributes to toxicity.
Although conjugation with GSH is generally a detoxification reaction, cisplatin conjugated to GSH, cysteinyl-glycine, and N-acetylcysteine is more toxic to cultured LLC-PK1 kidney cells than cisplatin itself.34 This is further supported by studies demonstrating that pharmacological inhibition of γ-glutamyltranspeptidase and β-lyase protects mice from nephrotoxicity35,36 and that γ-glutamyltranspeptidase–null mice are resistant to cisplatin injury.35 Because of the important role of GSH conjugation in the formation of reactive Pt metabolites, the higher expression of Gsts, despite similar GSH levels in Mrp2-null mice, may contribute to the higher susceptibility of these mice to cisplatin toxicity. Therefore, as more sensitive technologies for detecting cisplatin conjugates in vivo are developed, it will be important to identify the cisplatin species that are retained in the cytosol of Mrp2-null mice.
The hMRP2 protein traffics to the brush border and canalicular membranes and restores normal circulating bilirubin concentrations in Mrp2-null mice.30 We have demonstrated that hMRP2 mice have a similar susceptibility to cisplatin nephrotoxicity as WT mice. In fact, BUN and serum creatinine levels were slightly lower in hMRP2 mice. This may be due to the higher MRP2 mRNA level in humanized mice compared with WT mice.30 The ability of hMRP2 to protect against the toxicity of various pharmaceuticals has been demonstrated in multiple tissues based on single nucleotide polymorphism analyses. For example, hMRP2 polymorphisms alter the sensitivities of individuals to tenofovir nephrotoxicity,38 irinotecan-induced diarrhea,39 doxorubicin-induced cardiotoxicity,40 and carbamezepine-induced neurotoxicity.41 A preliminary investigation revealed that MRP2 polymorphisms do not predict cisplatin pharmacokinetics and toxicity; however, this study was underpowered, and few variants were detected in the study population.31 Although nephrotoxicity was not evaluated, a polymorphism in the MRP2 gene has been shown to predict greater cisplatin efficacy in patients with non–small cell lung cancer.42 Based on these findings, further work is needed to investigate MRP2 polymorphisms in predicting cisplatin efficacy and toxicity.
We observed elevated Pt concentrations in the kidneys and livers of Mrp2-null mice compared with WT mice (Figure 4A). In fact, the higher hepatic Pt concentrations persisted through 72 hours. The ability of the kidneys to ultimately excrete the Pt in Mrp2-null mice at a faster rate than the liver may reflect the higher expression of Mate1 mRNA in the kidneys.43 The greater hepatic burden of Pt in the livers of Mrp2-null mice did not result in toxicity. Although mild increases in serum alanine aminotransferase activity were observed between 3 and 4 days in the null mice, no histopathologic changes consistent with hepatocyte degeneration or necrosis were observed. This difference in organ sensitivity to cisplatin may reflect the unique ability of the kidneys to generate reactive thiols via β-lyase–mediated metabolism or possibly enhanced detoxification mechanisms in the liver.
The present data add to the existing literature regarding the transporters that participate in the renal disposition and toxicity of cisplatin (Figure 9). In conclusion, the absence of Mrp2 exacerbates cisplatin-induced nephrotoxicity in mice due to elevated concentrations of Pt in the DNA and cytosol, and possibly due to enhanced Gst mRNA expression. Moreover, the MRP2 gene protects Mrp2-null mice from cisplatin toxicity, demonstrating a renoprotective role for the human ortholog in vivo.
Acknowledgments
We thank Dr. Bruno Steiger (University Hospital, Zurich, Switzerland) for the Mrp2 antibody, Myrna Trumbauer (Rutgers University, Piscataway, NJ) for performing tissue collections, and Drs. Curtis Klaassen (University of Kansas Medical Center, Kansas City, KS) and Thomas Raub (Eli Lilly & Co, Indianapolis, IN) for assistance with conducting preliminary studies.
Footnotes
Supported by National Institute of Diabetes and Digestive and Kidney Diseases grants DK080774 and DK093903 (L.M.A.), National Institute of Environmental Health Sciences grants ES020522 and ES005022, components of the National Institutes of Health, and by the Pharmacokinetics and Pharmacodynamics Shared Resource of The Cancer Institute of New Jersey (grant CA072720). A portion of this work was supported by startup funding from the Environmental and Occupational Health Sciences Institute.
Disclosures: None declared.
Supplemental Data
mMrp2 and hMRP2 immunostaining (green) in livers of WT, Mrp2-null, and hMRP2 mice. Canalicular localization was confirmed by co-localization with mouse Bcrp protein (red). Original magnification, ×64.
References
- 1.Shord S.S., Thompson D.M., Krempl G.A., Hanigan M.H. Effect of concurrent medications on cisplatin-induced nephrotoxicity in patients with head and neck cancer. Anticancer Drugs. 2006;17:207–215. doi: 10.1097/00001813-200602000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Ciarimboli G., Ludwig T., Lang D., Pavenstadt H., Koepsell H., Piechota H.J., Haier J., Jaehde U., Zisowsky J., Schlatter E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol. 2005;167:1477–1484. doi: 10.1016/S0002-9440(10)61234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciarimboli G., Deuster D., Knief A., Sperling M., Holtkamp M., Edemir B., Pavenstadt H., Lanvers-Kaminsky C., am Zehnhoff-Dinnesen A., Schinkel A.H., Koepsell H., Jurgens H., Schlatter E. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am J Pathol. 2010;176:1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burger H., Zoumaro-Djayoon A., Boersma A.W., Helleman J., Berns E.M., Mathijssen R.H., Loos W.J., Wiemer E.A. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2) Br J Pharmacol. 2010;159:898–908. doi: 10.1111/j.1476-5381.2009.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filipski K.K., Loos W.J., Verweij J., Sparreboom A. Interaction of cisplatin with the human organic cation transporter 2. Clin Cancer Res. 2008;14:3875–3880. doi: 10.1158/1078-0432.CCR-07-4793. [DOI] [PubMed] [Google Scholar]
- 6.Filipski K.K., Mathijssen R.H., Mikkelsen T.S., Schinkel A.H., Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86:396–402. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwata K., Aizawa K., Kamitsu S., Jingami S., Fukunaga E., Yoshida M., Yoshimura M., Hamada A., Saito H. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol. 2012;16:843–851. doi: 10.1007/s10157-012-0638-y. [DOI] [PubMed] [Google Scholar]
- 8.Pabla N., Murphy R.F., Liu K., Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2009;296:F505–F511. doi: 10.1152/ajprenal.90545.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson C.A., Blair B.G., Safaei R., Howell S.B. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol Pharmacol. 2009;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H.H., Kuo M.T. Role of glutathione in the regulation of cisplatin resistance in cancer chemotherapy. Met Based Drugs. 2010;2010:1–7. doi: 10.1155/2010/430939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin X., Okuda T., Holzer A., Howell S.B. The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae. Mol Pharmacol. 2002;62:1154–1159. doi: 10.1124/mol.62.5.1154. [DOI] [PubMed] [Google Scholar]
- 12.Ishida S., Lee J., Thiele D.J., Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A. 2002;99:14298–14302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivy K.D., Kaplan J.H. A re-evaluation of the role of hCTR1, the human high-affinity copper transporter, in platinum-drug entry into human cells. Mol Pharmacol. 2013;83:1237–1246. doi: 10.1124/mol.113.085068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura T., Yonezawa A., Hashimoto S., Katsura T., Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80:1762–1767. doi: 10.1016/j.bcp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Ishikawa T., Ali-Osman F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells: molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem. 1993;268:20116–20125. [PubMed] [Google Scholar]
- 16.Townsend D.M., Marto J.A., Deng M., Macdonald T.J., Hanigan M.H. High pressure liquid chromatography and mass spectrometry characterization of the nephrotoxic biotransformation products of Cisplatin. Drug Metab Dispos. 2003;31:705–713. doi: 10.1124/dmd.31.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Y., Konig J., Buchholz J.K., Spring H., Leier I., Keppler D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol Pharmacol. 1999;55:929–937. [PubMed] [Google Scholar]
- 18.Kawabe T., Chen Z.S., Wada M., Uchiumi T., Ono M., Akiyama S., Kuwano M. Enhanced transport of anticancer agents and leukotriene C4 by the human canalicular multispecific organic anion transporter (cMOAT/MRP2) FEBS Lett. 1999;456:327–331. doi: 10.1016/s0014-5793(99)00979-5. [DOI] [PubMed] [Google Scholar]
- 19.Guminski A.D., Balleine R.L., Chiew Y.E., Webster L.R., Tapner M., Farrell G.C., Harnett P.R., Defazio A. MRP2 (ABCC2) and cisplatin sensitivity in hepatocytes and human ovarian carcinoma. Gynecol Oncol. 2006;100:239–246. doi: 10.1016/j.ygyno.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 20.Manautou J.E., Silva V.M., Hennig G.E., Whiteley H.E. Repeated dosing with the peroxisome proliferator clofibrate decreases the toxicity of model hepatotoxic agents in male mice. Toxicology. 1998;127:1–10. doi: 10.1016/s0300-483x(98)00013-4. [DOI] [PubMed] [Google Scholar]
- 21.Hartley D.P., Klaassen C.D. Detection of chemical-induced differential expression of rat hepatic cytochrome P450 mRNA transcripts using branched DNA signal amplification technology. Drug Metab Dispos. 2000;28:608–616. [PubMed] [Google Scholar]
- 22.Xie R., Johnson W., Rodriguez L., Gounder M., Hall G.S., Buckley B. A study of the interactions between carboplatin and blood plasma proteins using size exclusion chromatography coupled to inductively coupled plasma mass spectrometry. Anal Bioanal Chem. 2007;387:2815–2822. doi: 10.1007/s00216-007-1147-9. [DOI] [PubMed] [Google Scholar]
- 23.Aleksunes L.M., Slitt A.L., Maher J.M., Augustine L.M., Goedken M.J., Chan J.Y., Cherrington N.J., Klaassen C.D., Manautou J.E. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol Appl Pharmacol. 2008;226:74–83. doi: 10.1016/j.taap.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kidron H., Wissel G., Manevski N., Hakli M., Ketola R.A., Finel M., Yliperttula M., Xhaard H., Urtti A. Impact of probe compound in MRP2 vesicular transport assays. Eur J Pharm Sci. 2012;46:100–105. doi: 10.1016/j.ejps.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Lechner C., Reichel V., Moenning U., Reichel A., Fricker G. Development of a fluorescence-based assay for drug interactions with human Multidrug Resistance Related Protein (MRP2; ABCC2) in MDCKII-MRP2 membrane vesicles. Eur J Pharm Biopharm. 2010;75:284–290. doi: 10.1016/j.ejpb.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Schelman W.R., Mohammed T.A., Traynor A.M., Kolesar J.M., Marnocha R.M., Eickhoff J., Keppen M., Alberti D.B., Wilding G., Takebe N., Liu G. A phase I study of AT-101 with cisplatin and etoposide in patients with advanced solid tumors with an expanded cohort in extensive-stage small cell lung cancer. Invest New Drugs. 2014;32:295–302. doi: 10.1007/s10637-013-9999-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alnouti Y., Petrick J.S., Klaassen C.D. Tissue distribution and ontogeny of organic cation transporters in mice. Drug Metab Dispos. 2006;34:477–482. doi: 10.1124/dmd.105.006932. [DOI] [PubMed] [Google Scholar]
- 28.Chu X.Y., Strauss J.R., Mariano M.A., Li J., Newton D.J., Cai X., Wang R.W., Yabut J., Hartley D.P., Evans D.C., Evers R. Characterization of mice lacking the multidrug resistance protein MRP2 (ABCC2) J Pharmacol Exp Ther. 2006;317:579–589. doi: 10.1124/jpet.105.098665. [DOI] [PubMed] [Google Scholar]
- 29.Vlaming M.L., Mohrmann K., Wagenaar E., de Waart D.R., Elferink R.P., Lagas J.S., van Tellingen O., Vainchtein L.D., Rosing H., Beijnen J.H., Schellens J.H., Schinkel A.H. Carcinogen and anticancer drug transport by Mrp2 in vivo: studies using Mrp2 (Abcc2) knockout mice. J Pharmacol Exp Ther. 2006;318:319–327. doi: 10.1124/jpet.106.101774. [DOI] [PubMed] [Google Scholar]
- 30.Scheer N., Balimane P., Hayward M.D., Buechel S., Kauselmann G., Wolf C.R. Generation and characterization of a novel MRP2 humanized mouse line. Drug Metab Dispos. 2012;40:2212–2218. doi: 10.1124/dmd.112.047605. [DOI] [PubMed] [Google Scholar]
- 31.Sprowl J.A., Gregorc V., Lazzari C., Mathijssen R.H., Loos W.J., Sparreboom A. Associations between ABCC2 polymorphisms and cisplatin disposition and efficacy. Clin Pharmacol Ther. 2012;91:1022–1026. doi: 10.1038/clpt.2011.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamasaki M., Makino T., Masuzawa T., Kurokawa Y., Miyata H., Takiguchi S., Nakajima K., Fujiwara Y., Matsuura N., Mori M., Doki Y. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br J Cancer. 2011;104:707–713. doi: 10.1038/sj.bjc.6606071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korita P.V., Wakai T., Shirai Y., Matsuda Y., Sakata J., Takamura M., Yano M., Sanpei A., Aoyagi Y., Hatakeyama K., Ajioka Y. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol Rep. 2010;23:965–972. doi: 10.3892/or_00000721. [DOI] [PubMed] [Google Scholar]
- 34.Townsend D.M., Deng M., Zhang L., Lapus M.G., Hanigan M.H. Metabolism of Cisplatin to a nephrotoxin in proximal tubule cells. J Am Soc Nephrol. 2003;14:1–10. doi: 10.1097/01.asn.0000042803.28024.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Townsend D.M., Hanigan M.H. Inhibition of γ-glutamyl transpeptidase or cysteine S-conjugate β-lyase activity blocks the nephrotoxicity of cisplatin in mice. J Pharmacol Exp Ther. 2002;300:142–148. doi: 10.1124/jpet.300.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenderny S., Lin H., Garrett T., Tew K.D., Townsend D.M. Protective effects of a glutathione disulfide mimetic (NOV-002) against cisplatin induced kidney toxicity. Biomed Pharmacother. 2010;64:73–76. doi: 10.1016/j.biopha.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamauchi K., Sakurai H., Kimura T., Wiriyasermkul P., Nagamori S., Kanai Y., Kohno N. System L amino acid transporter inhibitor enhances anti-tumor activity of cisplatin in a head and neck squamous cell carcinoma cell line. Cancer Lett. 2009;276:95–101. doi: 10.1016/j.canlet.2008.10.035. [DOI] [PubMed] [Google Scholar]
- 38.Izzedine H., Hulot J.S., Villard E., Goyenvalle C., Dominguez S., Ghosn J., Valantin M.A., Lechat P., Deray A.G. Association between ABCC2 gene haplotypes and tenofovir-induced proximal tubulopathy. J Infect Dis. 2006;194:1481–1491. doi: 10.1086/508546. [DOI] [PubMed] [Google Scholar]
- 39.de Jong F.A., Scott-Horton T.J., Kroetz D.L., McLeod H.L., Friberg L.E., Mathijssen R.H., Verweij J., Marsh S., Sparreboom A. Irinotecan-induced diarrhea: functional significance of the polymorphic ABCC2 transporter protein. Clin Pharmacol Ther. 2007;81:42–49. doi: 10.1038/sj.clpt.6100019. [DOI] [PubMed] [Google Scholar]
- 40.Wojnowski L., Kulle B., Schirmer M., Schluter G., Schmidt A., Rosenberger A., Vonhof S., Bickeboller H., Toliat M.R., Suk E.K., Tzvetkov M., Kruger A., Seifert S., Kloess M., Hahn H., Loeffler M., Nurnberg P., Pfreundschuh M., Trumper L., Brockmoller J., Hasenfuss G. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112:3754–3762. doi: 10.1161/CIRCULATIONAHA.105.576850. [DOI] [PubMed] [Google Scholar]
- 41.Kim W.J., Lee J.H., Yi J., Cho Y.J., Heo K., Lee S.H., Kim S.W., Kim M.K., Kim K.H., In Lee B., Lee M.G. A nonsynonymous variation in MRP2/ABCC2 is associated with neurological adverse drug reactions of carbamazepine in patients with epilepsy. Pharmacogenet Genomics. 2010;20:249–256. doi: 10.1097/FPC.0b013e328338073a. [DOI] [PubMed] [Google Scholar]
- 42.Sun N., Sun X., Chen B., Cheng H., Feng J., Cheng L., Lu Z. MRP2 and GSTP1 polymorphisms and chemotherapy response in advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2010;65:437–446. doi: 10.1007/s00280-009-1046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lickteig A.J., Cheng X., Augustine L.M., Klaassen C.D., Cherrington N.J. Tissue distribution, ontogeny and induction of the transporters Multidrug and toxin extrusion (MATE) 1 and MATE2 mRNA expression levels in mice. Life Sci. 2008;83:59–64. doi: 10.1016/j.lfs.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
mMrp2 and hMRP2 immunostaining (green) in livers of WT, Mrp2-null, and hMRP2 mice. Canalicular localization was confirmed by co-localization with mouse Bcrp protein (red). Original magnification, ×64.