

Abstract
Protein tyrosine phosphatases (PTPs) play an important role in the regulation of mammalian signal transduction. During some cell signaling processes, the generation of endogenous hydrogen peroxide inactivates selected PTPs via oxidation of the enzyme's catalytic cysteine thiolate group. Importantly, low molecular weight and protein thiols in the cell have the potential to regenerate the catalytically active PTPs. Here we examined the recovery of catalytic activity from two oxidatively-inactivated PTPs (PTP1B and SHP-2) by various low molecular weight thiols and the enzyme thioredoxin. All thiols examined regenerated the catalytic activity of oxidized PTP1B, with apparent rates constants that varied by a factor of approximately eight. In general, molecules bearing low pKa thiol groups were particularly effective. The biological thiol, glutathione repaired oxidized PTP1B with an apparent second-order rate constant of 0.023 ± 0.004 M−1 s−1, while the dithiol, DTT, displayed an apparent second-order rate constant of 0.325 ± 0.007 M−1 s−1. The enzyme thioredoxin regenerated the catalytic activity of oxidized PTP1B at a substantially faster rate than DTT. Thioredoxin (2 μM) converted oxidized PTP1B to the active form with an observed rate constant of 1.4 × 10−3 s−1. The rates at which these agents regenerated oxidized PTP1B followed the trend Trx > DTT > GSH, with comparable values observed at 2 μM Trx, 4 mM DTT and 60 mM GSH. Various disulfides that are byproducts of the reactivation process did not inactivate native PTP1B at concentrations of 1-20 mM. The common biochemical reducing agent tris(2-carboxyethyl)phosphine (TCEP) regenerates enzymatic activity from oxidized PTP1B somewhat faster than the thiol-based reagents, with a rate constant of 1.5 ± 0.5 M−1 s−1. We observed profound kinetic differences between the thiol-dependent regeneration of activity from oxidized PTP1B and SHP-2, highlighting the potential for structural differences in various oxidized PTPs to play a significant role in the rates at which low molecular weight thiols and thiol-containing enzymes such as thioredoxin and glutaredoxin return catalytic activity to these enzymes during cell signaling events.
Many important mammalian signaling pathways are regulated by phosphorylation of specific tyrosine residues on target proteins.1-4 The phosphorylation status of these proteins is controlled by the coordinated action of protein tyrosine kinases that catalyze the addition of phosphoryl groups and protein tyrosine phosphatases (PTPs) that catalyze their hydrolytic removal.2-6 The catalytic activity of selected PTPs is down-regulated as part of some signal transduction events.3,7 This involves the activation of NADPH oxidases that generate a burst of hydrogen peroxide (H2O2) that oxidizes the catalytic cysteine thiolate group at the active site of selected PTPs.8-14 The oxidatively-inactivated forms of various PTPs may exist with the catalytic cysteine residue either as a sulfenic acid, a disulfide, or a sulfenyl amide (Scheme 1).15 Reaction of biological thiols with oxidized PTPs can regenerate the catalytically active enzyme, with the active site cysteine in the thiolate form (Scheme 2).15
The oxidative inactivation and subsequent thiol mediated reactivation of PTPs during signaling events constitutes an important biochemical “timing device” that helps control the duration and intensity of cellular responses to various stimuli.3,7,15 A number of studies have investigated the mechanisms by which hydrogen peroxide inactivates PTPs;9-12,16-20 however, the mechanisms by which cellular thiols regenerate the catalytic activity of these proteins has received less attention. Low molecular weight thiols, including the biological thiol glutathione (GSH), can mediate the recovery of activity from oxidized PTPs.12,15,18,21-24 In addition, enzymes such as thioredoxin, glutaredoxin, and sulfiredoxin can repair oxidized PTPs, employing both single cysteine thiol and vicinal dithiol mechanisms in the reduction of oxidized proteins.15,18,21,25-27 In general, the rates, mechanisms, and exact identity of the thiols that regenerate catalytic activity from oxidized PTPs remains an important, yet poorly understood, aspect of many receptor protein tyrosine kinase-mediated cell signaling pathways. In the work described here, we employed various low molecular weight thiols and the enzyme thioredoxin as probes to explore fundamental chemical and biochemical features surrounding the regeneration of catalytic activity from two structurally distinct oxidized PTPs.
EXPERIMENTAL PROCEDURES
Materials
All thiols used in this study were from Sigma-Aldrich, and were of no less than purum reagent grade. Buffer components Tris, Bis-Tris, sodium acetate, and diethylenetriaminepentaacetic acid (DTPA) were also from Sigma. Sodium chloride was from Fisher Scientific, and the non-ionic detergent Surfact Amps® 80 (Tween 80) was from Thermo Scientific. Catalase from Corynebacterium glutamicum (844,000 U/mL) and 30% (wt/wt) aqueous hydrogen peroxide were from Sigma. The chromogenic substrate 4-nitrophenyl phosphate disodium salt hexahydrate (pNPP), and sodium hydroxide were also from Sigma. Recombinant thioredoxin from E. coli (product T0910), thioredoxin reductase (product T7915), and NADPH-tetra(cyclohexylammonium) salt (product N5130) were from Sigma-Aldrich and were used as received. Absorption spectra were recorded on an Agilent 8453 Hewlett-Packard G1103A spectrophotometer. Zeba mini buffer exchange/desalting columns used in the preparation of thiol-free PTP1B or SHP-2 were from Pierce (catalog no. 89882), and were used according to the manufacturer's protocol. The catalytic domains of PTP1B and SHP-2 were expressed and purified as previously described.10 The previously characterized active site directed PTP1B inhibitor 1 was a gift from Dr. Ernest Asante-Appiah (Merck). The previously characterized active-site directed PTP1B inhibitor 2 was prepared as described previously.28
Oxidative Inactivation of Native PTP1B and SHP-2
Prior to use in kinetics assays, both PTPs were removed from stock storage solutions and exchanged into Buffer A (Tris (50 mM), Bis-Tris (50 mM), DTPA (10 mM), and sodium acetate (100 mM), pH 7.0) containing 0.5% (v/v) Tween 80. Subsequently, the PTPs were diluted in the same buffer and completely inactivated by treatment with hydrogen peroxide (1 mM) for 5 min at 25 °C. Following the inactivation incubation period, catalase (100-300 units, final) was added to quench excess H2O2. The reaction tube was then allowed to stand open to air for 2 min to ensure complete evolution of O2 gas, and the oxidized PTPs stored on ice until used. Generally 0.7 μM oxidized PTP1B and 0.35 μM oxidized SHP-2 were the final concentrations for discontinuous assays. The aqueous solubility of PTP1B is greatly decreased upon oxidation, and protein precipitation was observed at concentrations of oxidized PTP1B as low as 4 μM.
Determination of Approximate Rate Constants Via Continuous Spectrophotometric assay
Stock solutions of 25.5 mM pNPP, 250 mM monothiol, and 125 mM dithiol were prepared in Buffer A. For thiols bearing acidic functionalities (thioglycolic acid (TGA), GSH, and N-acetyl-l-cysteine (NAC)), one mole equivalent of acid was neutralized via addition of the appropriate amount of 10 M NaOH in ddH2O to prevent acidification of the assay buffer, and the pH checked against buffer alone using a four-color pH test strip. In a manner similar to that described above, oxidized PTP1B (1.5 μM) and oxidized SHP-2 (0.75 μM) were prepared. To a 1 mL quartz cuvette were added 628 μL of 25.5 mM pNPP and 160 μL of 250 mM monothiol, or 125 mM dithiol. Following zeroing of the spectrophotometer against this solution, 12 μL of oxidized PTP1B or SHP-2 were added, the mixture immediately mixed by gentle vortexing, and the release of 4-nitrophenol followed at 410 nm (5 sec cycle times). Because the concentration of substrate in the assay (20 mM) was saturating (as determined in a separate experiment) and effectively constant, we may express the approximation:
where [Eact] is the concentration of native (active) PTP, and [E•S] is the concentration of the PTP-substrate complex. Under conditions of saturating substrate, we may write:
where [P] is the concentration of product 4-nitrophenol and kcat has its usual meaning in the context of Michaelis-Menten kinetics. Thus, if kcat and [Eact] are constant during the experiment, [E•S] would also be constant, and the term dP/dt would be constant and described by a line with the slope, kcat[E•S] (this being the basis for determination of “traditional” kinetic constants Km and Vmax). Here, however, since the concentration of active PTP changed (increased) with time during thiol-mediated reactivation, rising curves are observed in the plot of [P] versus time. Under the pseudo first-order conditions employed, and for a given kcat, these rising curves can be described by the expression:
where kψ is the pseudo first-order rate constant of reactivation in the presence of excess thiol, and Do, Dt, and D∞ are the instantaneous rates of change in [P] versus time initially, at time t during the reactivation process, and at the completion of the reaction. Thus, computing instantaneous rates of change in Abs410 (differentials, D) at time t and replotting Dt versus the median of the time interval considered affords kinetic data that describes the thiol-mediated recovery of PTP activity versus time under pseudo-first-order conditions. It is worth noting that this method for extracting kinetic data from a continuous spectrophotometric assay is reminiscent of that described by Hart and O'Brien for inactivation of acetylcholinesterase by paraoxon in the presence of 4-nitrophenyl acetate.29
Determination of Rate Constants of Reactivation of Oxidatively-Inactivated PTPs Via Discontinuous Assay Methods
Stocks of oxidatively inactivated PTPs were prepared as described (0.7 μM PTP1B and 0.35 μM SHP 2, vide supra). In the same buffer were prepared 2×-concentrated stocks of thiol. Prior to initiation of the reactivation reaction, the stocks were incubated separately at 25 °C for 5 min to allow thermal equilibration. Immediately upon 1:1 (v/v) mixture of the two stocks, a timer was started, and at 1, 2, 4, 7, and 10 min intervals, 10 μL aliquots were removed from the reaction mixture and placed in 2 mL microcentrifuge tubes containing 490 μL of 20 mM pNPP in activity assay buffer (50 mM Bis-Tris, 10 mM DTPA, 150 mM NaCl, pH 6, 30 °C). The activity assays were allowed to proceed for 10 min at 30 °C prior to quenching with 500 μL of 2 M NaOH.14 The absorbance at 410 nm was recorded and the data analyzed as described below.
To account for any potential residual activity in the oxidized PTP stocks, the appropriate concentration of oxidized PTP was added to the blank series, subjected to identical conditions in the activity assay, and the instrument zeroed against this sample. For thiols of limited solubility and/or sluggish reaction times, DTT or 2-mercaptoethanol (2-ME) were used to determine the endpoint of the reaction. In a separate experiment, it was found that these thiol agents all recovered similar amounts of enzyme activity for PTP1B.
Data from these assays were analyzed in Microsoft Excel by fitting to the general form for a first-order reaction:
where kψ is the pseudo-first-order rate constant and [A]o, [A]t, and [A]∞ are the absorbances at 410 nm initially, at time t, and at the end of the reactivation process, respectively. From this data, apparent bimolecular rate constants and rate constants for thiolate attack on oxidized PTP1B were calculated according to the method of Szajewski and Whitesides.30
Similar methodologies to those described above were utilized in determination of the rate of recovery of catalytic activity of oxidized PTP1B by the trialkylphosphine reagent TCEP. Because the material was received as the hydrochloride salt and additionally bears three acidic carboxylic acid groups, 3.5 equiv of base were added to the stock solution via addition of the appropriate volume of NaOH (10 M) to prevent acidification of the assay buffer. The resulting mixture was confirmed to have the same pH as buffer alone by four color pH strip. The amount of total recoverable activity by treatment of oxidized PTP1B with TCEP (20 mM) or DTT (40 mM) for 30 min (pH 7, 25 °C) was found to be effectively the same (within ~7% of one another).
Reactivation of oxidatively-inactivated PTP1B by the thioredoxin-thioredoxin reductase system
A concentrated (40 μM) stock solution of thioredoxin (a lyophilized powder) was prepared via dissolution in Buffer A lacking Tween, and was stored at - 20°C until used. Solutions of thioredoxin reductase (a 16 μM suspension in ammonium sulfate) were prepared fresh via dilution from the stock into Buffer A. Solutions of NADPH in Buffer A were prepared fresh and stored on ice until used. Solutions of the reducing system (thioredoxin/thioredoxin reductase/NADPH) were prepared fresh from stock solutions and used immediately. In general, the ratio of thioredoxin:thioredoxin reductase was held at 20:1, and it was found that these conditions were more than adequate to ensure that reduction of thioredoxin by thioredoxin reductase in the presence of excess NADPH (100 – 200 μM) was not rate-limiting. For kinetics runs with oxidized PTP1B, the thioredoxin system was prepared (thioredoxin/thioredoxin reductase/NADPH : 4 μM/200 nM/200 μM) and allowed to equilibrate at 25 °C several minutes prior to the start of the assay. Recovery of PTP1B activity was measured as described above.
RESULTS AND DISCUSSION
Oxidative Inactivation of PTP1B
We first examined thiol-mediated regeneration of activity from oxidized PTP1B. PTP1B is the archetypal member of the PTP superfamily and is redox regulated as part of the insulin signaling cascade.9,13 For these studies, we employed the catalytic domain of recombinant human PTP1B (aa 1-322). The oxidatively-inactivated enzyme was prepared by treatment of native PTP1B with H2O2 (1 mM) in Buffer A containing Tween-80 (0.5% v/v) for 5 min at 25 °C, followed by addition of catalase (100-300 units) to quench remaining H2O2. Enzyme prepared in this manner was completely inactive, but approximately 75% of the original activity was consistently recovered by treatment with 1,4-dithio-d-threitol (DTT, 40 mM, 20 min, 25 °C).
Evidence suggests that oxidative inactivation of PTP1B proceeds via conversion of the active site cysteine to the cyclic sulfenyl amide both in the crystal and in solution forms of the enzyme (Scheme 1).31,32 Nonetheless, it remains possible that oxidized PTP1B exists in some other form such as the sulfenic acid (Scheme 1). The inability to completely recover the catalytic activity of oxidized PTP1B presumably reflects the formation of some overoxidized forms of the enzyme. These may include cyclic sulfinyl and sulfonyl amide forms of PTP1B (Structures 3 and 4, respectively).22,31,32 Results of previous chemical model studies indicate that the sulfinyl amide form of PTP1B may be thiol recoverable, while the sulfonyl amide likely is not.22
Thiol-Dependent Recovery of Activity from Oxidized PTP1B
We treated oxidized PTP1B with a panel of structurally diverse thiol-containing compounds in the presence of the chromogenic PTP substrate p-nitrophenylphosphate and used a continuous spectrophotometric assay to monitor the recovery of enzyme activity engendered by each agent. All of the thiols examined caused time-dependent recovery of activity from the oxidized enzyme (Figure 1). From these time courses, carried out under pseudo-first-order conditions, an apparent second-order rate constant for the recovery of enzyme activity was estimated for each thiol (Figure 1, legend).
Figure 1.

Thiol-mediated reactivation of oxidatively inactivated PTP1B. (A) PTP1Box (22 nM) was incubated with various thiols in Buffer A containing the chromogenic PTP substrate pNPP (20 mM), pH 7 at room temperature. The time courses monitored the increase in absorbance at 410 nm resulting from the PTP1B-catalyzed release of p-nitrophenol from the substrate. Monothiols were 50 mM and dithiols 25 mM: TGA-OMe (*), DTT ( ), BAL (+), TGA (
), BAL (+), TGA ( ), 2 ME (
), 2 ME ( ), GSH (
), GSH ( ), NAC (-), no thiol (
), NAC (-), no thiol ( ). (B) Instantaneous rates of reactivation (slopes) versus time. Data were fit with pseudo-first order kinetic treatment to give the following estimates of the apparent bimolecular rate constants calculated in units of M−1 s−1: TGA-OMe = 0.45, DTT = 0.33, BAL = 0.33, TGA = 0.12, 2-ME = 0.08, GSH = 0.04, NAC = 0.04.
). (B) Instantaneous rates of reactivation (slopes) versus time. Data were fit with pseudo-first order kinetic treatment to give the following estimates of the apparent bimolecular rate constants calculated in units of M−1 s−1: TGA-OMe = 0.45, DTT = 0.33, BAL = 0.33, TGA = 0.12, 2-ME = 0.08, GSH = 0.04, NAC = 0.04.
We next undertook a more detailed kinetic examination of a subset of these thiols. For example, treatment of oxidized PTP1B with the biological thiol GSH at concentrations ranging from 1-60 mM resulted in time- and concentration-dependent recovery of the enzyme's catalytic activity (Figure 2A and B). The reaction displayed second-order kinetics, with an apparent rate constant of 0.023 ± 0.004 M−1 s−1. We examined several other structurally varied monothiols including 2-mercaptoethanol (2-ME), cysteine, and thioglycolic acid (TGA) (Table 1). Similar to GSH, the recovery of PTP1B activity caused by these thiols followed second-order kinetics, with the observed rate constants varying by a factor of about eight.
Figure 2.

Reactivation of oxidatively-inactivated PTP1B by glutathione (GSH) and DTT. (A) GSH-mediated recovery of activity from oxidized PTP1B. Concentrations of GSH were 10, 20, 30, 40, and 50 mM (bottom to top). (B) Pseudo-first order rate constants (s−1 × 103) were plotted against corresponding concentrations of GSH (mM), affording a straight line of slope kobs (M−1 s−1) which passes through the origin. (C) Time-course for DTT-mediated recovery of activity from oxidized PTP1B. Concentrations of DTT were 2.5, 5, 10, 15, and 20 mM (bottom to top). (D) Pseudo-first order rate constants (s−1 × 103) were plotted against corresponding concentrations of DTT (mM), affording a straight line of slope kobs (M−1 s−1) which passes through the origin. A higher concentration regime of DTT (40-60 mM) was also explored in separate experiments conducted under identical conditions. No saturation behavior was observed under any of these concentration regimes.
Table 1.
Structures, thiol group pKa, and observed second-order rate constants for reactivation of oxidatively-inactivated PTP1B.
| Thiol | Structure | kobs (M−1 s−1) | pKa |
|---|---|---|---|
| 2,3-dimercaptopropanol |
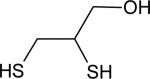
|
0.59 ± 0.02 | 8.6 |
| Dithiothreitol |
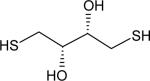
|
0.325 ± 0.007 | 9.2 |
| Cysteamine |
|
0.14 ± 0.01 | 8.2 |
| Cysteine |
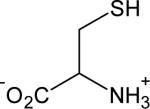
|
0.10 ± 0.02 | 8.2 |
| Thioglycolate |
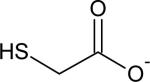
|
0.090 ± 0.006 | 10.1 |
| 2-mercaptoethane sulfonate |
|
0.07 ± 0.03 | 9.5 |
| 2-mercaptoethanol |
|
0.05 ± 0.03 | 9.4 |
| Glutathione |
|
0.023 ± 0.004 | 9.1 |
| 3-mercaptopropionate |
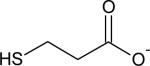
|
0.017 ± 0.002 | 10.6 |
Literature precedents indicate that thiolate (RS–) is the relevant nucleophile in the reactions of thiols with divalent sulfur electrophiles.33,34 Consistent with these precedents, the data presented in Table 1 shows that, with one exception (TGA) that is discussed further below, monothiols with lower pKa values display larger rate constants for their reactions with oxidized PTP1B. Molecules bearing thiol groups with lower pKa values exist with a significantly larger fraction in the reactive thiolate form near neutral pH values. The observed rate constants measured here, along with the literature pKa values for the respective thiol groups,30,35,36 allowed us to estimate the true rate constants for regeneration of activity from oxidized PTP1B by each thiolate (Table 2). This analysis accounts for differences in pKa of the various thiol groups and highlights how structural differences influence reactivity. The results indicate that more basic thiolates were more reactive toward oxidized PTP1B. This is in accord with findings from the Whitesides group regarding the attack of thiolates on low molecular weight disulfides.30,37
Table 2.
Observed second-order rate constants, corrected for thiolate concentration under assay conditions (pH 7, 25 °C).
| Thiol | kRS- (M−1 s−1) | PKa |
|---|---|---|
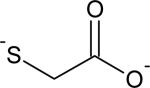
|
113 | 10.1 |
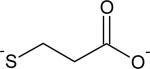
|
69 | 10.6 |
|
|
23 | 9.5 |
|
|
12 | 9.4 |
|
3 | 9.1 |
|
|
2 | 8.2 |
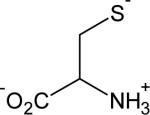
|
2 | 8.2 |
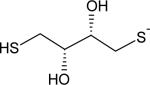
|
52 | 9.2 |
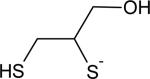
|
24 | 8.6 |
Interestingly, TGA was more reactive than pKa considerations alone would predict. This could reflect favorable electrostatic interactions between the ionized carboxylate group of TGA with positively-charged amino acid side chains such as Arg45, Lys116, and Lys210 located near the catalytic Cys215 in the three-dimensional structure of oxidized PTP1B (see for example: pbd 3SME).10 Alternatively, equilibrium amounts of the neutral carboxylic acid form of TGA could serve as a general acid catalyst to increase the rate k2 as illustrated in Scheme 3.
We also examined reaction of the dithiol DTT with oxidized PTP1B. DTT has been suggested as a reasonable low molecular weight mimic of dithiol-containing enzymes involved in the repair of oxidized proteins.15,18 Treatment of oxidized PTP1B with DTT (1-60 mM, pH 7) produced time- and concentration-dependent recovery of the enzyme's catalytic activity (Figure 2C and D). The data were consistent with a second-order process with a rate constant of 0.325 ± 0.007 M−1 s−1. Importantly, the observed rate of enzyme recovery in the case of DTT was approximately 6.5-fold greater than that measured for the structurally analogous monothiol, 2-ME. Furthermore, the rate constant estimated for the regeneration of active enzyme by the thiolate form of DTT was more than double that of 2-ME thiolate (Table 2). Contrary to the trend identified above, in this case, the less basic thiolate of DTT displays a larger rate constant. The apparent discrepancy in the kinetic behavior of DTT and 2-ME likely reflects a difference in the rate determining step for the regeneration of PTP1B activity by these two thiols. Specifically, the rate constant measured for the recovery of PTP1B activity in the case of DTT may report on the initial attack of thiolate on the oxidized enzyme (k1 in Scheme 2C). This is a reasonable hypothesis given that the second step of the reaction, involving intramolecular attack of the pendent thiol group on the mixed disulfide intermediate, is expected to be fast (Scheme 2D).30,38 In contrast, the observed rates at which monothiols reactivate oxidized PTP1B may reflect the bimolecular attack of a second equivalent of thiolate on the mixed disulfide intermediate (k2 in Scheme 2C). The rate measured here for DTT (0.325 M−1 s−1) can be compared to the value of 0.23 M−1 s−1 reported for the attack of DTT on GSH disulfide under similar conditions (pH 7, 30 °C).30
Unlike DTT, the dithiol 2,3-dimercapto-1-propanol (BAL) exhibited biphasic behavior in the plot of pseudo first-order rate constants versus thiol concentration (Supporting Information). At low concentrations of BAL (up to 5 mM), a line passing through the origin with slope (second-order rate constant) equal to 0.6 M−1 s−1 was observed. At higher concentrations of thiol, a “downward bend” in the plot occurs leading to a second linear region of the plot whose slope corresponds to a different second-order rate constant of approximately 0.17 M−1 s−1 (y-intercept of ~ 0.006 s−1). The different kinetic behaviors of BAL and DTT may arise due to the relatively slow intramolecular ring closure in the case of the PTP-BAL mixed disulfide (k2 in Scheme 2D). At low concentrations of BAL, the intramolecular ring closure of the second, pendent thiol group in the PTP-BAL mixed disulfide (k2, Scheme 2D) may be faster than both the rate of initial attack of BAL on oxidized enzyme (k1[RSH], Scheme 2C) and bimolecular attack of a second equivalent of BAL on the PTP-BAL mixed disulfide (k2[RSH], Scheme 2C). Thus, in the low concentration regime, like DTT, the rate-determining step for recovery of enzyme activity by BAL may be the initial attack of thiol on oxidized PTP1B. However, at higher concentrations of BAL, the rate of bimolecular attack of a second equivalent of BAL on the PTP-BAL mixed disulfide (k2[RSH], Scheme 2C) may surpass that of the intramolecular ring closure (k2, Scheme 2D). If the two forward bimolecular rate constants in Scheme 2C have the relationship k1 > k2 (which is consistent with the interpretation of the data for monothiols discussed above) and the pseudo-first-order rate constant k2[BAL] is greater than the first-order rate constant k2 (Scheme 2D) then the rate-determining step for formation of the active enzyme will be the bimolecular reaction k2 in Scheme 2C. Thus, at low concentrations BAL behaves like the dithiol DTT while at high concentrations it behaves, for all practical purposes, like a monothiol. Along these lines, it is worth noting that BAL typically forms cyclic dimers upon reduction of low molecular weight disulfides;30 however, steric occlusion of the PTP-BAL mixed disulfide at the protein surface may hinder this process, rendering formation of the cyclic dimers relatively slow.
We investigated whether several known, active-site-directed reversible inhibitors of native PTP1B were able to inhibit thiol-mediated reactivation of the oxidized enzyme. We examined the known inhibitors phosphate (Kd = 21 mM),39 and arsenate (Ki = 80 μM).40 In addition, we examined the effects of two previously characterized, active-site-directed, reversible inhibitors of PTP1B, the Merck inhibitor, 1 (IC50 = 47 nM)41 and 2-(oxalylamino)benzoic acid 2 (Ki = 23 μM).28 None of these agents inhibited the ability of thiols to restore oxidized PTP1B to its catalytically active form (data not shown). On the contrary, in the case of sodium phosphate (50 mM), a slight acceleration in the recovery of PTP1B activity by 2-ME and DTT was observed, likely because the significantly greater ionic strength of the phosphate-containing solutions depressed the pKa of the thiol groups in these assays, leading to increased concentration of the reactive thiolate species. The failure of competitive PTP1B inhibitors to slow thiol-mediated recovery of activity from oxidized PTP1B likely reflects the simple fact that the structures of the oxidized and native enzymes are substantially different, and that traditional PTP inhibitors do not bind to the oxidized enzyme with high affinity (for example, compare pdb structures 3SME and 3HNP).10,32
We examined the ability of the thioredoxin/thioredoxin reductase enzyme system to regenerate catalytic activity from oxidized PTP1B. The thioredoxin/thioredoxin reductase system did regenerate active PTP1B, with an observed rate constant of 1.4 × 10−3 s−1 at a thioredoxin concentration of 2 μM (Figure 3). For comparision, a concentration of 5 mM DTT gave approximately the same pseudo-first-order rate constant as 2 μM thioredoxin (Figure 3). It is noteworthy that the yield of active PTP1B generated by the thioredoxin/thioredoxin reductase system was somewhat less than that produced by DTT (Figure 3). The difference in the amount of PTP1B activity regenerated by thioredoxin and the low molecular weight thiols, may be explained by the presence of multiple oxoforms of PTP1B in the assay. As noted above, oxidative inactivation of PTP1B likely generates multiple oxoforms of the enzyme including the sulfenyl amide and higher oxidation states products such as the cyclic sulfinyl amide 3 and the sulfonyl amide 4.22,31,32 Results of previous chemical model studies suggest that low molecular weight thiols can return the sulfinyl amide 3 form of PTP1B to the active state, while the sulfonyl amide likely cannot be recovered.22 As described in this work, it is possible that thioredoxin may selectively process the sulfenyl amide shown in Scheme 1, but not the sulfinyl amide 3. In contrast, the chemical studies reported previously indicated that DTT and other low molecular weight thiols appear indiscriminant with regard to their reactions with small molecules that model these oxoforms of PTP1B.22 While these results suggests the possibility that a mixture of PTP1B oxoforms may be generated during the oxidation of PTP1B with H2O2, the simple bimolecular kinetics observed for the thiol-dependent recovery of activity from oxidized PTP1B further suggest that the oxoforms are converted to active enzyme at similar rates. If multiple processes with significantly different rate constants were operative, biphasic kinetics would be observed.
Figure 3.

Reactivation of oxidatively-inactivated PTP1B by DTT or the thioredoxin-thioredoxin reductase system. To solutions of 2×-concentrated reducing system in Buffer R were added 1:1 (v/v) oxidatively-inactivated PTP1B to final concentrations: PTP1Box (350 nM), DTT (5 mM, blue circles) or thioredoxin/thioredoxin reductase/NADPH (2 μM, 100 nM, 200 μM, respectively, red triangles). Immediately upon mixing, a timer was started, and the amount of recovered enzyme activity monitored at 2, 4, 6, 8, 10, 26, and 45 min intervals.
Disulfide Byproducts Generated in the Reaction of Thiols with Oxidized PTP1B Do Not Inactivate the Catalytically-Active Form of the Enzyme
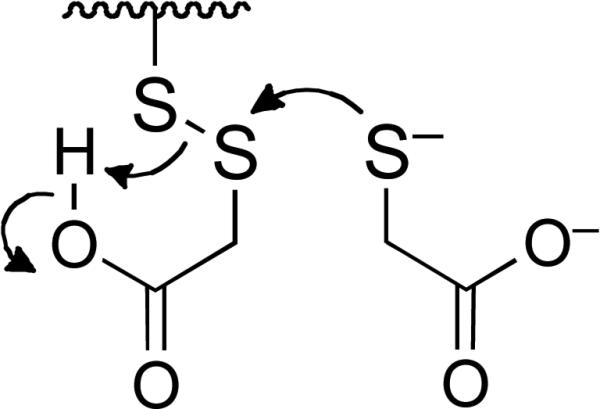
Thiol-mediated reactivation of PTP1B generates a disulfide byproduct (Scheme 2). Previous reports indicate that disulfides such as GSH disulfide, Ellman's reagent, and the natural product gymnorrhizol can inactivate PTP1B.27,42,43 Therefore, we investigated whether 2-ME disulfide, GSH disulfide, and oxidized DTT might inactivate PTP1B under our assay conditions. We found that none of these disulfides inactivated the native enzyme at concentrations of 1-20 mM in Buffer A containing Tween-80 (0.5% v/v) at 25 °C. The results suggest that the process defined by k−2[RSSR] in Scheme 2 was not significant under the conditions of our experiments.
Trialkylphosphine-Mediated Reactivation of Oxidatively-Inactivated PTP1B
Because trialkylphosphine-based reagents are commonly utilized for reduction of protein disulfides to free thiols,44-46 we were curious how the efficiency of the prototypical reagent in this class, tris(2-carboxyethyl)phosphine (TCEP),44 compares to thiols in its ability to regenerate catalytic activity from oxidized PTP1B. As shown in Figure S5, time and concentration-dependent recovery of PTP activity was observed upon treatment of oxidized PTP1B with TCEP under assay conditions identical to those employed in our thiol experiments. The amount of recovered PTP activity was comparable to that recovered by DTT. A replot of the pseudo first-order rate constants versus concentration of TCEP afforded a straight line passing through the origin, suggesting a rate-determining step first-order in TCEP. Reduction of disulfides to two equivalents of thiol by trialkylphosphine reagents involves initial attack of the neutral phosphorus atom on the disulfide, liberating the first equivalent of thiol (Scheme 4).44-46 Subsequent hydrolysis of the thio-phosphonium intermediate liberates the second equivalent of thiol, affording the corresponding trialkylphosphine oxide. The simple second-order kinetics observed here suggest that the initial attack of phosphorus on the oxidized PTP1B sulfenyl sulfur is rate-limiting, and that the hydrolysis step leading to active enzyme is relatively fast (Scheme 4). Additionally, the phosphorus-oxygen bond is sufficiently strong to render k−2 in Scheme 4 negligible, and the reaction irreversible.44-46 Under the conditions employed here (pH 7, 25 °C), the rate constant for reactivation of oxidized PTP1B by TCEP was 1.5 ± 0.5 M−1 s−1. This value is appreciably larger than the rate constants observed for the thiol-containing agents examined here likely due, in large part, to the fact that a substantial fraction of TCEP exists in the unprotonated, reactive form at neutral pH. The pKa of phosphorus-protonated TCEP (R3PH+) has been estimated at 7.66,44 whereas the most acidic thiol examined in our study has a pKa of 8.2 (cysteamine). Adjusting the observed rate constant for concentration of active nucleophile, in analogous fashion to that described above for the thiol/thiolate couples, we calculated a rate constant of 8.0 M−1 s−1 for attack of the TCEP neutral phosphorus nucleophile on oxidized PTP1B. It has been noted previously that, while TCEP reduces low molecular weight disulfides more rapidly than does DTT, the opposite is true in the case of some protein disulfides.45 Here, this not the case. In fact, if initial attack of the phosphorus reagent on oxidized PTP1B is rate-limiting, and the observed rate constant reports on k1 in similar fashion to the dithiol agents, the “pKa-corrected” rate constant is perhaps higher than one might expect for a thiol of pKa = 7.66.
Thiol-Dependent Recovery of Activity from Oxidized SHP-2
Finally, we examined thiol-dependent recovery of activity from oxidatively-inactivated SHP-2, a different member of the human PTP family. The cellular activity of SHP-2 is redox regulated in response to platelet-derived growth factor, endothelin-1, and T-cell receptor stimulation.3,47 Our experiments employed the catalytic domain (aa 246-527) of the recombinant human enzyme and the oxidized enzyme was generated in a manner similar to that described above for PTP1B. Treatment of the oxidized enzyme with DTT (40 mM, 20 min, 25 °C) consistently returned approximately 95% of the original catalytic activity of the enzyme. The amount of activity recovered from oxidatively-inactivated SHP-2 is greater than that observed for PTP1B. This suggests that SHP-2 resists the generation of “overoxidized” oxoforms that cannot returned to active enzyme upon treatment with low molecular weight thiols.
As expected, treatment of oxidized SHP-2 with a panel of thiol-containing molecules gave time-dependent recovery of the enzyme's catalytic activity and, as described above in the context of PTP1B, apparent second-order rate constants for the recovery were estimated from the continuous assay data (Figure 3). A more complete kinetic analysis showed that treatment of oxidized SHP-2 with DTT gave time- and concentration-dependent recovery of PTP activity consistent with a simple, second-order process with rate constant 0.8 ± 0.1 M−1 s−1 (Figure 4). This value matches that measured previously for a slightly different construct (aa 268-525) of the enzyme's catalytic subunit.21 The rate at which DTT regenerates SHP-2 activity, a value expected to reflect the initial attack of DTT thiolate on the oxidized enzyme (k1 in Scheme 2) as discussed above, is approximately 2.5-fold faster than the analogous reaction on PTP1B. This result is particularly interesting in light of previous work suggesting that oxidation of the active site cysteine residue in SHP-2 initiates a disulfide relay that transmits the initial oxidation of the active site cysteine to a distal pair of cysteine residues (Scheme 5).21 While no structural information is available regarding oxidized SHP-2, our result suggests that the proposed disulfide relay in SHP-2 generates a surface-exposed disulfide that is readily accessible to low molecular weight thiols. In contrast, oxidized PTP1B is thought to exist with the catalytic cysteine residue in the cyclic sulfenyl amide form.10,23,32
Figure 4.

Thiol-mediated reactivation of oxidatively-inactivated SHP-2. (A) Oxidized SHP-2 (11 nM) was incubated with various thiols in Buffer A containing the chromogenic PTP substrate pNPP (20 mM) pNPP, pH 7 at room temperature. The time courses monitored the increase in absorbance at 410 nm resulting from the SHP-2-catalyzed release of p-nitrophenol from the substrate. Monothiols were 50 mM and dithiols 25 mM: DTT ( ), TGA OMe (
), TGA OMe ( ), TGA (X), 2 ME (
), TGA (X), 2 ME ( ), GSH (*), NAC (+), no thiol (
), GSH (*), NAC (+), no thiol ( ). (B) Instantaneous rates of reactivation (slopes) versus time. Data were fit with pseudo-first order kinetic treatment to give the following estimates of the apparent bimolecular rate constants calculated in units of M−1 s−1: DTT = 0.89, TGA-OMe = 0.44, TGA = 0.10, 2-ME = 0.02, GSH = 0.02, NAC = 0.01.
). (B) Instantaneous rates of reactivation (slopes) versus time. Data were fit with pseudo-first order kinetic treatment to give the following estimates of the apparent bimolecular rate constants calculated in units of M−1 s−1: DTT = 0.89, TGA-OMe = 0.44, TGA = 0.10, 2-ME = 0.02, GSH = 0.02, NAC = 0.01.
Interestingly, in our survey of thiols using the continuous assay, it was clear that monothiols such as the biological thiol GSH were rather inept at regeneration of activity from oxidized SHP-2, when compared to their action on oxidized PTP1B (Figures 1 and 3). Intrigued, we examined the ability of various concentrations of GSH to regenerate SHP-2 activity. A plot of the resulting observed rate constants versus GSH concentration gave a rising curve that was not consistent with simple second-order kinetics. Indeed, a plot of the observed rate constants obtained under pseudo-first-order conditions versus the square of GSH concentration gave a straight line consistent with the rate law k[RSH]2 and corresponding rate constant of 0.29 ± 0.02 M−2 s−1. Such a rate law could reasonably result from a mechanism involving reversible attack of thiol on a protein disulfide followed by irreversible, rate limiting generation of the active enzyme via attack of a second equivalent of thiol (Scheme 5B). We assume that the thiolated protein intermediate generated by the intial attack of thiol on the oxidized enzyme is catalytically inactive (Scheme 5B), although this assumption need not be true if the species exists at a low steady state concentration. This notion is broadly consistent with the original proposal suggesting that oxidized SHP-2 is inactive despite possessing a native (unoxidized) cysteine thiol group at its active site (Scheme 5A).15,21 This scheme is kinetically analogous to the reduction of flavin by monothiols and dithiols reported previously.38,48-50 Notably, the rate at which physiologically-relevant concentrations of GSH regenerate of active enzyme from oxidized SHP-2 was negligible compared to the analogous reaction with oxidized PTP1B. This behavior is not limited to GSH, as the monothiol 2-ME also displayed kinetics second-order in thiol, with the overall third-order rate constant of 0.25 ± 0.05 M−2 s−1 (Supporting Information).
CONCLUSIONS
All of the thiol-containing molecules examined in this study were able to regenerate active enzyme from oxidized PTP1B. Agents that contain lower pKa thiol groups are superior in their ability to regenerate active enzyme, likely because these compounds present larger amounts of thiolate (RS–) for reaction with the oxidized PTP. Along these lines, our studies suggest that the relatively low pKa of cysteamine (8.3) renders this an effective monothiol reagent for maintaining PTP1B in the active form during biochemical assays. In fact, for this particular application, cysteamine may be superior to DTT from an economical perspective. Approximately two-fold higher concentrations of cysteamine are required to match the rate of DTT under our conditions, but cysteamine is less than one-eighth the cost of DTT on a mole-for-mole basis. Methylthioglycolate (TGA-OMe) also could be effective and economical for such applications but is practically undesirable due to its foul odor.
We found that that the biological thiol GSH regenerates catalytic activity from oxidized PTP1B with an apparent second-order rate constant of 0.023 ± 0.004 M−1 s−1. To provide a general reference point for comparison, this value is approximately 20-times less than the rate at which GSH attacks oxidized GSSG (GSH/GSSG disulfide exchange, 0.41 M−1 s−1, 25 °C, pH 7).51 Based on the rate constant reported here, we can estimate that a physiologically-relevant concentration GSH (5 mM) will convert oxidized PTP1B to the active form with a half-life of 1.7 h (t1/2 = ln 2/(0.023 M−1 s−1 × 0.005 M). Furthermore, the results suggested that, in the case of monothiols, the initial attack on oxidized PTP1B (k1[RSH]) was faster than the second step leading to recovery of the native enzyme (k2[RSH]). This kinetic scenario allows for accumulation of the intermediate enzyme-thiol mixed disulfide (PTP1B-SSR) and may explain why the glutathionylated form of PTP1B has been detected in cell lysates following stimulation of alveolar macrophage respiratory burst.24 Accordingly, enzymes such as the glutaredoxins or sulfiredoxin that repair glutathionylated proteins may be involved in the intracellular conversion of glutathionylated PTP1B to its catalytically active form.25-27 The thioredoxin enzyme system repairs oxidized PTP1B more effectively than the low molecular weight thiols, with comparable rates for the reactivation of oxidized PTP1B obtained at 2 μM Trx, 4 mM DTT and 60 mM GSH. We observed a greater yield of recovered enzyme activity when oxidized PTP1B was treated with low molecular weight thiols than when treated with thioredoxin. This suggests that enzymatic reduction of oxidized PTP1B may occur in an oxoform-selective manner. For example, thioredoxin may rapidly regenerate PTP1B catalytic activity from the sulfenyl amide oxoform, while the higher oxidation states of PTP1B such as the sulfinyl and sulfonyl amide oxoforms are not processed by the enzyme. The rate of reactivation of PTP1B by Trx reported here is substantially faster than the published rate of Trx-dependent reactivation rate measured for a different PTP family member, SHP-2, under somewhat different conditions.21 The observed pseudo-first-order rate constant measured here for the reactivation of oxidized PTP1B by 2 uM Trx, allows us to estimate an apparent second-order rate constant of 700 M−1 s−1 for this process.14 This suggests that the thioredoxin-mediated recovery of oxidized PTP1B proceeds roughly 20-fold faster than the recovery of SHP-2.21
We observed profound kinetic differences between the thiol-dependent regeneration of activity from oxidized PTP1B and SHP-2. This may reflect structural differences between the oxidized enzymes. Along these lines, oxidation of the active site cysteine residue in SHP-2 was proposed to initiate a disulfide relay that transmits the initial oxidation of the active site cysteine to a distal pair of cysteine residues (Scheme 5),21 while oxidized PTP1B is thought to exist with the catalytic cysteine residue in the cyclic sulfenyl amide form.10,23,32 Regeneration of activity from oxidized SHP-2 by monothiols appears to be second-order in thiol concentration and, at physiological concentrations of GSH, recovery of SHP-2 is expected to be quite slow, relative to oxidized PTP1B. An earlier study that examined the regeneration of oxidized SHP-2 by excess glutathione did not note a non-linear dependence on glutathione concentration, although the data was not shown.21 Using the rate constant measured in this work we estimate that, at a steady-state GSH concentration of 5 mM, the half-life for recovery of oxidized SHP-2 would be approximately 27 h (compared to 1.7 h for oxidized PTP1B). The observed kinetics lead us to speculate that the recovery of SHP-2 activity by GSH proceeds via an unfavorable equilibrium addition of the thiol to an enzyme disulfide, followed by irreversible, rate-limiting generation of the active enzyme via attack of a second equivalent of thiol. This kinetic scenario suggests that glutathionylated SHP-2 will not accumulate in cells. Consistent with this analysis, glutathionylated SHP-2 was not observed in cell lysates under conditions where glutathionylated PTP1B was detected.24 From a practical point of view, our results suggest that monothiols will be remarkably poor reagents for protecting SHP-2 against oxidative inactivation, when compared with their activity in the context of PTP1B. On the other hand, the ability of DTT to maintain SHP-2 in its catalytically active form is superior to its activity against oxidized PTP1B. The results presented here, alongside previous work,21 indicate that for both PTP1B and SHP-2, the rates at which the oxidized enzyme is recovered follows the trend Trx > DTT > GSH.
There is increasing recognition that cellular activity of various PTP enzymes is redox regulated as part of both normal and pathogenic processes.3,7,52 The catalytic subunits of the classical PTPs are highly homologous, yet subtle differences among the various family members lead to significant differences in the structures of the oxidized enzymes, with the catalytic cysteine residue existing as either a sulfenic acid, a disulfide, or a sulfenyl amide (Scheme 1).15 Using low molecular weight thiols as probes, we observed here very different kinetic behavior in the regeneration of catalytic activity of two different oxidized PTPs. This highlights the potential for structural differences in oxidized PTPs to play a significant role the rates at which low molecular weight thiols and enzymes such as thioredoxin and glutaredoxin return catalytic activity to these enzymes during cell signaling events.
Supplementary Material
Figure 5.

Reactivation of oxidatively inactivated SHP-2 by DTT and GSH. Assays were conducted as described in the Experimental Section. A. DTT-mediated recovery of activity from oxidized PTP1B. B. Pseudo first order rate constants (s−1 × 103) were plotted against DTT concentration (mM), affording a straight line of slope kobs (M−1 s−1) which passes through the origin. This data is consistent with a bimolecular process in the rate determining step. C. GSH-mediated recovery of activity from oxidized SHP-2. D. Plotting the observed pseud first-order rate constants versus concentration of GSH affords a curvilinear (parabolic) graphical form, indicating a non-second order kinetic process. E. Plotting of the observed pseudo first-order rate constants versus the square of the GSH concentration affords a linear form, which passes through the origin. This suggests a process second-order in thiol concentration in the rate-determining step, with the relevant rate constant being equal to 0.29 ± 0.02 M−2 s−1.
Scheme 1.

Oxidative inactivation of protein tyrosine phosphatases.
Scheme 2.

Thiol-mediated recovery of catalytic activity from oxidized protein tyrosine phosphatases.
Scheme 3.
Scheme 4.
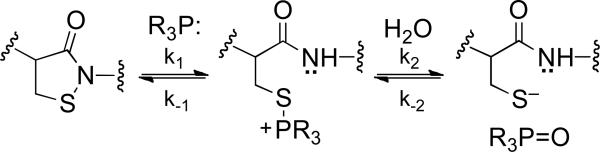

Scheme 5.

Proposed disulfide relay initiated by oxidative inactivation of SHP-2 (Panel A) and proposed mechanism for thiol-dependent recovery of activity from oxidized SHP-2 (Panel B).
ACKNOWLEDGMENTS
We thank Dr. Ernest Asante Appiah (Merck) for providing inhibitor 1. We thank our colleagues Dr. Harkewal Singh Professor Jack Tanner and Professor Thomas Reilly for assistance with enzyme expression and isolation.
Funding
We thank the National Institutes of Health for partial support of this work (CA 100757). In addition, this work was made possible in part by Grant Number P50AT006273 from the National Center for Complementary and Alternative Medicines (NCCAM), the Office of Dietary Supplements (ODS), and the National Cancer Institute (NCI). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NCCAM, ODS, NCI, or the National Institutes of Health.
ABBREVIATIONS
- PTP
protein tyrosine phosphatase
- DTT
1,4-dithio-d-threitol
- pNPP
p-nitrophenylphosphate
- DTPA
diethylenetriamine pentaacetic acid
- BAL
dithiol 2,3-dimercapto-1-propanol
- TGA
thioglycolic acid
- TGA-OMe
methylthioglycolate TCEP, tris(2-carboxyethyl)phosphine
- GSH
glutathione
- 2-ME
2-mercaptoethanol
- NAC
N-acetylcysteine
- Buffer A
Tris (50 mM), Bis-Tris (50 mM), DTPA (10 mM), Tween-80 (0.5% v/v) and sodium acetate (100 mM), pH 7.0
Footnotes
Supporting Information
Plots of kinetic data for the thiol-mediated recovery of catalytic activity from PTP1B and SHP-2, of TCEP-mediated recovery of PTP1B, and of reaction between native PTPs and various disulfide reagents. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Nat. Acad. Sci. USA. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunter T. Signaling - 2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 3.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nature Rev. Cell. Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 4.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tonks NK, Diltz CD, Fischer EH. Purification of the major protein-tyrosine phosphatases of human placenta. J. Biol. Chem. 1988;263:6722–6730. [PubMed] [Google Scholar]
- 6.Tarrant MK, Cole PA. The chemical biology of protein phosphorylation. Ann. Rev. Biochem. 2009;78:797–825. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Östman A, Frijhoff J, Sandin A, Böhmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. Biochem. J. 2011;150:345–356. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 8.Jiang F, Zhang Y, Dusting GJ. NADPH Oxidase-Mediated Redox Signaling: Roles in Cellular Stress Response, Stress Tolerance, and Tissue Repair. Pharmacol. Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 9.Mahedev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou H, Singh H, Parsons ZD, Lewis SM, Bhattacharya S, Seiner DR, LaButti JN, Reilly TJ, Tanner JJ, Gates KS. The biological buffer, bicarbonate/CO2, potentiates H2O2 mediated inactivation of protein tyrosine phosphatases. J. Am. Chem. Soc. 2011;132:15803–15805. doi: 10.1021/ja2077137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LaButti JN, Chowdhury G, Reilly TJ, Gates KS. Redox regulation of protein tyrosine phosphatase 1B by peroxymonophosphate. J. Am. Chem. Soc. 2007;129:5320–5321. doi: 10.1021/ja070194j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 13.Meng T-C, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 14.Parsons ZD, Gates KS. Redox Regulation of Protein Tyrosine Phosphatases: Methods for Kinetic Analysis of Covalent Enzyme Inactivation. Methods Enzymol. 2013;528(C):129–154. doi: 10.1016/B978-0-12-405881-1.00008-2. [DOI] [PubMed] [Google Scholar]
- 15.Tanner JJ, Parson ZD, Cummings AH, Zhou H, Gates KS. Redox Regulation of Protein Tyrosine Phosphatases: Structural and Chemical Aspects. Antioxid. Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 16.LaButti JN, Gates KS. Biologically relevant properties of peroxymonophosphate (=O3POOH). Bioorg. Med. Chem. Lett. 2009;19:218–221. doi: 10.1016/j.bmcl.2008.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hecht D, Zick Y. Selective inhibition of protein tyrosine phosphatase activities by H2O2 and vanadate in vitro. Biochem. Biophys. Res. Commun. 1992;188:773–779. doi: 10.1016/0006-291x(92)91123-8. [DOI] [PubMed] [Google Scholar]
- 18.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 19.Weibrecht I, Boehmer S-A, Dagnell M, Kappert K, Oestman A, Boehmer F-D. Oxidatiion sensitivity of the catalytic cysteine of the protein tyrosine phosphatases SHP-1 and SHP-2. Free Rad. Biol. Med. 2007;43:100–110. doi: 10.1016/j.freeradbiomed.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 20.Woo HA, Yim SH, Shin DH, Kang D, Yu D-Y, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized hydrogen peroxide accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Chen C-Y, Willard D, Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry. 2009;48:1399–1409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 22.Sivaramakrishnan S, Cummings AH, Gates KS. Protection of a single-cysteine redox switch from oxidative destruction: on the functional role of sulfenyl amide formation in the redox regulated enzyme PTP1B. Bioorg. Med. Chem. Lett. 2010;20:444–447. doi: 10.1016/j.bmcl.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sivaramakrishnan S, Keerthi K, Gates KS. A chemical model for the redox regulation of protein tyrosine phosphatase 1B (PTP1B). J. Am. Chem. Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- 24.Rinna A, Torres M, Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B Free Rad. Biol. Med. 2006;41:86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vlamis-Gardikas A, Holmgren A. Thioredoxin and glutaredoxin isoforms. Methods Enzymol. 2002;347:286–296. doi: 10.1016/s0076-6879(02)47028-0. [DOI] [PubMed] [Google Scholar]
- 26.Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrett WC, DeGnore JP, König S, Fales HM, Keng Y-F, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 28.Andersen HS, Olsen OH, Iversen LF, Sørensen ALP, Mortensen SB, Christensen MS, Branner S, Hansen TK, Lau JF, Jeppesen L, Moran EJ, Su J, Bakir F, Judge L, Shahbaz M, Collins T, Vo T, Newman MJ, Ripka WC, Møller NPH. Discovery and SAR of a novel selective and orally bioavailable nonpeptide classical competitive inhibitor class of protein tyrosine phosphatase 1B. J. Med. Chem. 2002;45:4443–4459. doi: 10.1021/jm0209026. [DOI] [PubMed] [Google Scholar]
- 29.Hart GJ, O'Brien RD. Recording spectrophotometric method for determination of dissociation and phosphorylation constants for the inhibition of acetylcholinesterase by organophosphates in the presence of substrate. Biochemistry. 1973;12:2940–2945. doi: 10.1021/bi00739a026. [DOI] [PubMed] [Google Scholar]
- 30.Szajewski RP, Whitesides GM. Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J. Am. Chem. Soc. 1980;102:2011–2026. [Google Scholar]
- 31.van Montfort RLM, Congreeve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 32.Salmeen A, Anderson JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 33.Fava A, Iliceto A, Camera E. Kinetics of the Thiol-Disulfide Exchange. J. Am. Chem. Soc. 1957;79:833–838. [Google Scholar]
- 34.Senatore L, Ciuffari E, Fava A, Levita G. Nucleophilic substitution at sulfur. Effect of nucleophile and leaving group basicity as probe of bond formation and breaking. J. Am. Chem. Soc. 1973;95:2918–2922. [Google Scholar]
- 35.Bracher PJ, Snyder PW, Bohall BR, Whitesides GM. The relative rates of thiol-thioester exchange and hydrolysis for alkyl and aryl thioalkanoates in water. Orig. Life Evol. Biosph. 2011;41:399–412. doi: 10.1007/s11084-011-9243-4. [DOI] [PubMed] [Google Scholar]
- 36.Harris DC. Quantitative Chemical Analysis. 8th ed. W. H. Freeman and Co.; New York: 2010. [Google Scholar]
- 37.Whitesides GM, Lilburn JE, Szajewski RP. Rates of thiol disulfide interchange reactions between mono- and dithiols and Ellman's reagent. J. Org. Chem. 1977;42:332–338. [Google Scholar]
- 38.Loechler EL, Hollocher TC. Reduction of flavins by thiols. 3. The case for concerted N,S-acetal formation in attack and an early transition state in breakdown. J. Am. Chem. Soc. 1980;102:7328–7334. [Google Scholar]
- 39.Zhang Y-Z, Zhang Z-Y. Low-affinity binding determined by titration calorimetry using a high-affinity coupling ligand: a thermodynamic study of ligand binding to protein tyrosine phosphatase 1B. Anal. Biochem. 1998;261:139–148. doi: 10.1006/abio.1998.2738. [DOI] [PubMed] [Google Scholar]
- 40.Sarmiento M, Zhao Y, Gordon SJ, Zhang Z-Y. Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J. Biol. Chem. 1998;273:26368–26374. doi: 10.1074/jbc.273.41.26368. [DOI] [PubMed] [Google Scholar]
- 41.Montalibet J, Skorey K, McKay D, Scapin G, Asante-Appiah E, Kennedy BP. Residues Distant from the Active Site Influence Protein tyrosine Phosphatase 1B Inhibitor Binding. J. Biol. Chem. 2006;281 doi: 10.1074/jbc.M511546200. [DOI] [PubMed] [Google Scholar]
- 42.Pregel MJ, Storer AC. Active site titration of tyrosine phosphatases SHP-1 and PTP1B using aromatic disulfides. J. Biol. Chem. 1997;272:23552–23558. doi: 10.1074/jbc.272.38.23552. [DOI] [PubMed] [Google Scholar]
- 43.Gong J-X, Shen X, Yao L-G, Jiang H, Krohn K, Guo Y-W. Total synthesis of gymnorrhizol, an unprecedented 15-membered macrocyclic polydisulfide from the Chinese Mangrove Bruguiera gymnorrhiza. Org. Lett. 2007;9:1715–1716. doi: 10.1021/ol0703783. [DOI] [PubMed] [Google Scholar]
- 44.Burns JA, Butler JC, Moran J, Whitesides GM. Selective reduction of disulfides by tris(2-carboxyethyl)phosphine. J. Org. Chem. 1991;56:2648–2650. [Google Scholar]
- 45.Cline DJ, Redding SE, Brohawn SG, Psathas JN, Schneider JP, Thorpe C. New water-soluble phosphines as reductants of peptide and protein disulfide bonds: reactivity and membrane permeability. Biochemistry. 2004;43:15195–15203. doi: 10.1021/bi048329a. [DOI] [PubMed] [Google Scholar]
- 46.Getz EB, Xiao M, Chakrabarty T, Cooke R, Selvin PR. A comparison between the sulfhydryl reductants tris(2-carboxyethyl)phosphine and dithiothreitol for use in protein biochemistry. Anal. Biochem. 1999;273:73–80. doi: 10.1006/abio.1999.4203. [DOI] [PubMed] [Google Scholar]
- 47.Michalek RD, Nelson KJ, Holbrook BC, Yi JS, Stridiron D, Daniel LW, Fetrow JS, King SB, Poole LB, Grayson JM. The requirement of reversible cysteine sulfenic acid formation for T-cell activation and function. J. Immunol. 2007;179:6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 48.Loechler EL, Hollocher TC. Reduction of flavins by thiols. 1. Reaction mechanism from the kinetics of the attack and breakdown steps. J. Am. Chem. Soc. 1980;102:7312–7321. [Google Scholar]
- 49.Yokoe I, Bruice TA. Oxidation of thiophenol and nitroalkanes by an electron deficient isoalloxazine. J. Am. Chem. Soc. 1975;97:450–451. [Google Scholar]
- 50.Holden JW, Main L. The kinetics and mechanism of the oxidation of mercaptoethanol by riboflavin. J. Aust. Chem. 1977;30:1387–1391. [Google Scholar]
- 51.Rabenstein DL, Weaver KH. Kinetics and equilibria of the thiol/disulfide exchange reactions of somatostatin with glutathione. J. Org. Chem. 1996;61:7391–7397. doi: 10.1021/jo960917+. [DOI] [PubMed] [Google Scholar]
- 52.Samet JM, Tal TL. Toxicological disruption of signaling homeostasis: tyrosine phosphatases as targets. Annu. Rev. Pharmacol. Toxicol. 2010;50:215–235. doi: 10.1146/annurev.pharmtox.010909.105841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.