

Abstract
Canine progressive rod-cone degeneration (prcd) is a retinal disease previously mapped to a broad, gene-rich centromeric region of canine chromosome 9. As allelic disorders are present in multiple breeds, we used linkage disequilibrium (LD) to narrow the ∼6.4 Mb interval candidate region. Multiple dog breeds, each representing genetically isolated populations, were typed for SNPs and other polymorphisms identified from BACs. The candidate region was initially localized to a 1.5 Mb zero recombination interval between growth factor receptor-bound protein 2 (GRB2) and SEC14-like 1 (SEC14L). A fine-scale haplotype of the region was developed which reduced the LD interval to 106 Kb, and identified a conserved haplotype of 98 polymorphisms present in all prcd-affected chromosomes from 14 different dog breeds. The findings strongly suggest that a common ancestor transmitted the prcd disease allele to many of the modern dog breeds, and demonstrate the power of LD approach in the canine model.
Keywords: Disease Models, Animal, Genetic Diversity, Genetic Linkage, Genetic Markers, Genetic Predisposition to Disease, Genetic Variation, Retinal Degeneration
Introduction
Identification of the genetic loci responsible for inherited disorders has progressed rapidly using a combination of mapping strategies and candidate gene evaluation. The observation of homologous genes associated with phenotypically similar disease, e.g. hereditary retinal degenerations in more than one species, strongly helps incriminate mutations in these genes as disease-causative. The many loci for hereditary retinal degeneration implicated in model species, such as rodents and dogs, for which human disease homologues have not been identified yet, reinforces the prediction that many more causative genes remain to be discovered. Identification of novel loci responsible for autosomal recessive phenotypes in model species can be valuable for suggesting new candidate genes for human disorders. In the latter, pedigrees are often too small to allow identification of the causative locus by association or linkage studies, and more than half the loci responsible for human autosomal recessive hereditary retinal degenerations have yet to be identified (RetNet-Retinal Information System; http://www.sph.uth.tmc.edu/RetNet/).
Linkage Disequilibrium (LD) mapping has been shown to be a powerful tool for genomic studies in humans [1; 2], but it presents different advantages and problems that depend on the population structure. In isolate populations with a small number of founders, LD frequently extends over distances of several cM around the disease locus [3; 4; 5; 6], making recognition of the LD region practicable, but gene discovery difficult. Conversely, in the broader non-isolate human populations, estimates of the marker density necessary to identify an LD region are much higher [7; 8], but, once the region is identified, it is much smaller, and thus harbors a correspondingly smaller set of positional candidate genes.
Recent advances in canine genomics suggest that the use of LD mapping in the dog may also provide an excellent resource for gene discovery. Some advantages of the dog for disease-association studies include: 1) unique history of the dog population that, together with the “breed barrier” (no dog may become a registered member of a breed unless both its parents are registered members), has ensured a relatively closed genetic pool among dogs of each breed. Using microsatellites to assess interbreed differences, a recent study has shown that 99% of individuals tested could be assigned to the correct breed [9]; 2) LD within a dog breed is ∼100 times more extensive than in man, suggesting that fewer markers will be needed to map genes in the canine [10]; 3) dogs have more than 350 inherited disorders, many of which are homologues of common human diseases [11], and there is ready access to a large sample pool of animals with phenotypically ascertained and characterized diseases. These observations emphasize that the dog will become an important model organism for gene discovery and genomic studies.
A case in point of how LD in the dog can be utilized is found in progressive rod-cone degeneration (prcd), an inherited canine retinal disease that closely resembles adult onset forms of autosomal recessive retinitis pigmentosa (RP) [12]. The disease initially was described as a form of Progressive Retinal Atrophy (PRA) affecting Miniature and Toy poodles (MP, TP). A resource colony of mixed breed dogs derived from such poodles was developed, and used for mapping the locus to the centromeric end of canine chromosome 9 (CFA9) [13]. This large candidate region is particularly gene rich, and exhibits suppressed recombination typical of centromeric chromosomal regions [14], thus requiring alternative approaches appropriate for the study population to further reduce the search interval.
A particularly opportune feature of prcd is that it occurs in multiple dog breeds in which either allelic or identical mutations segregate, previously demonstrated by crossbreeding experiments involving TP and MP, English and American cocker spaniels (ECS, ACS), and Labrador retriever (LR) breeds [12; 15]. This observation raised the possibility of using Linkage Disequilibrium (LD) mapping [1; 2] to further reduce the candidate region, especially if additional independent populations could be identified. The goal was to characterize the broad LD region within multiple independent isolate populations, and then identify regions common to all. This approach would be especially suitable if the mapped disease arose in all such isolate populations from a common ancestral founder.
Initial mapping of prcd defined a candidate region on CFA9 between MYL4 and TK1, with no recombinations in 70 informative offspring [13]. At that time the canine genome sequence was unavailable, and, under the assumption of completely conserved synteny and gene order between CFA9 and HSA17q, this 3.9-5.1 cM map interval was estimated to correspond to over 30 Mb on HSA17q (HSA17: 42,641,426-73,681,775). Subsequently, this interval was characterized in detail, and, although there was conservation in gene content between CFA9 and HSA17q, several rearrangements in gene order were found between homologous regions [16]. In the dog, ITGB3 and MYL4 mapped proximal to the gene cluster containing SCN4A, PRKCA, RGS9 and PRKAR1A, placing MYL4 closer to TK1, a distance estimated as ∼6.4 Mb (CFA9: 5,956,925-12,343,152). The refined map also placed GRB2, a positional candidate within the zero recombination interval, between FDXR and GALK1, and created a 1.5 Mb physical map of the segment bounded by FDXR and SRP68 at the telomeric and centromeric ends, respectively [16].
In the present study, we have revised the physical map of the prcd region, initially by extending the search area to the GRB2-SEC14L interval, and subsequently by establishing a haplotype of the interval using SNPs and small indels that narrowed the candidate region. A single fine-scale affected haplotype located within a 106 Kb common LD region is identified as the disease transmitting haplotype. This 106 Kb region, and the specific disease haplotype, is common to all affected chromosomes within and among the multiple dog breeds affected with prcd. The shared haplotype suggests that prcd is an ancestral disease that arose in a founder to a diverse subset of modern dog breeds. Furthermore, we demonstrate the power and suitability of the highly structured canine breed populations for LD-based mapping to identify chromosomal regions and genes responsible for traits segregating in multiple, relatively well isolated subpopulations.
Results
Interbreed crosses identify new prcd breeds
To identify additional independent populations with prcd for use in the LD studies, a series of interbreed crosses were carried out using prcd-affected dogs from the reference colony. When bred to Basenji, Border collie (BC), or Italian greyhound (IG) dogs affected with retinal degeneration, all resultant progeny had morphologically normal retinas, thus excluding allelism with prcd (Figure 1, A, B). In contrast, a similar strategy used with retinal degenerate Australian cattle dog (ACD), Nova Scotia duck tolling retriever (NSDTR), or Portuguese water dog (PWD) demonstrated that all the progeny were affected. In these 3 breeds, the retinas showed mild disorganization and disorientation of the photoreceptor outer segments, the hallmark early lesions of prcd (Figure 1, C-E) [15], and confirmed allelism with prcd.
Figure 1.
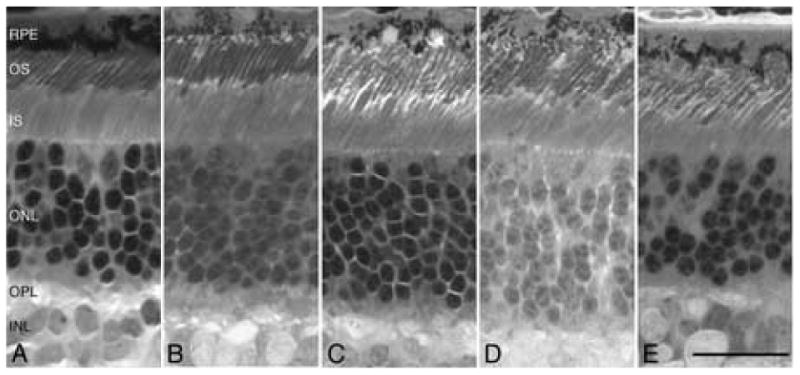
Retinal micrographs showing non-allelism with prcd for the crosses between Basenji (A, 17.4 wk) or Italian greyhound (B, 17.4 wk) and colony reference prcd-affected dogs; data for the Border collie cross is not illustrated. Retinal photoreceptors are normal. In contrast, crosses between the reference prcd-affected dogs and Australian cattle dog (C, 16.4 wk), Nova Scotia duck tolling retriever (D, 16.1 wk) and Portuguese water dog (E, 26 wk) show disorientation of the photoreceptor outer segments (OS) characteristic of the early stages of prcd. Calibration marker=25μm; RPE=retinal pigment epithelium, IS=inner segment, ONL=outer nuclear layer, OPL=outer plexiform layer, INL=inner nuclear layer.
Tiling path of the prcd interval
The previously published physical map [16] was extended with overlapping BAC clones 275K3, 33817, 262H18, 10M13, 36604; a sixth BAC, 10P17, that contained SEC14L but did not overlap the tiling path, was also included for analysis (Figure 2). To facilitate identification of SNPs for LD map construction, primers were designed from the 3.2X BAC sequence to amplify regions that did not include repetitive elements, and new markers were developed.
Figure 2.

Schematic representation of the prcd LD interval. Low-pass 3.2X sequence of ∼1.2 Mb from 6 BAC clones from the candidate region was analyzed. Ten affected haplotypes observed in different breeds are illustrated which reduced the LD to ∼106 Kb. Haplotypes 1-4 are common haplotypes found in specific affected breeds: Haplotype 1 in MP, TP, ECS, ACS, LR, PWD and CBR; Haplotype 2 in NSDTR; Haplotype 3 in ACD; Haplotype 4 in AE. Haplotypes 5-10 represent rare recombinant chromosomes observed in ACS (H5), NSDTR (H6), PWD (H7), LR (H8), MP and TP (H9) and TP (H10). Representative SNPs and indels show heterozygosity between the affected chromosomes. The final LD is boxed and contains 98 polymorphisms shared among all affected chromosome, and is represented here by 6 SNPs. Distances and recombination points are not drawn to scale. The 4 markers described in Table 1 (GRB2, AANAT, ST6GalNac2, SEC14L) are in bold letters. For the full data set see Supplementary Table 2A, B. Large black dots in Haplotypes 1 and 2 represent nucleotide deletions.
Broad scale analysis of the GRB2-SEC14L candidate region
Linkage analysis in the reference population placed prcd in a zero recombination interval between GRB2-SEC14L, a distance estimated at 1.5 Mb (Figure 2). This candidate region contains at least 40 known and hypothetical genes (May 2005 dog (Canis familiaris) whole genome shotgun (WGS) assembly v2.0; http://genome.ucsc.edu/cgi-bin/hgGateway). Four candidate genes (GRB2, AANAT, ST6GalNac2 and SEC14L) were initially evaluated to set the broad limits for developing an LD map. Polymorphisms were identified in the 4 genes (Supplementary Table 1: SNPs 1, 2, 3, and 4 in GRB2, SNP 20 in AANAT, SNP 116 in ST6GalNac2 and SNP 161 in SEC14L), and these co-segregated with the disease in the 70 informative colony dogs with no recombinants. Typing the 4 markers for >100 dogs/breed, both affected and unaffected relatives, was used to establish that different prcd-associated haplotypes segregated in the 10 different breeds or breed varieties (Table 1, A; MP/TP, ECS, ACS, NSDTR, PWD, ACD, LR, Chesapeake Bay retriever (CBR) and American eskimo (AE)).
Table 1. Selected haplotypes in the GRB2-SEC14L interval observed in different prcd affected breeds.
| Breed |
GRB2 allele |
AANAT allele |
ST6GalNac2 allele |
SEC14L allele |
|---|---|---|---|---|
| A. Common breed-specific haplotypes observed in homozygous state in affected dogs | ||||
| Poodles (Miniature and Toy) | H1 | A | A | A |
| English cocker spaniel | H1 | A | A | A |
| American cocker spaniel | H1 | A | A | A |
| Labrador retriever | H1 | A | A | A |
| Portuguese water dog | H1 | A | A | A |
| Chesapeake Bay retriever | H1 | A | A | A |
| Nova Scotia duck tolling retriever | H2 | A | A | G |
| Australian cattle dog | H2 | A | A | G |
| American eskimo | H3 | A | A | A |
| B. Rare haplotypes observed in affected dogsa | ||||
| Portuguese water dog (n=2) | H3 | A | A | A |
| Nova Scotia duck tolling retriever (n=1) | H4 | A | A | G |
| American cocker spaniel (n=1) | H4 | A | A | A |
| Labrador retriever-German origin (n=2) | H2 | A | A | A |
| Labrador retriever (n=2) | H3 | A | A | A |
| Toy poodle (n=2) | H2 | G | A | A |
| Miniature poodle (n=2) | H2 | G | A | A |
| Chesapeake Bay retriever (n=2) | H1 | A | A | G |
| Portuguese water dog (n=1) | H1 | A | A | G |
| Toy poodle (n=1) | H1 | A | A | G |
| Australian Cattle Dogs (n=1) | H2 | A | A | A |
the allele in bold is the one that differs from the breed-specific allele.
The haplotypes were different at the GRB2, AANAT and SEC14L loci, while the ST6GalNac2 “A” allele was the same in all haplotypes. At the GRB2 locus, 4 non-redundant polymorphisms defined 4 alleles, H1, H2, H3 and H4, that cosegregated with prcd in different populations (Table 1, Figure 2). H1 was the most common GRB2 allele on prcd-affected chromosomes, cosegregating in 7 breed/breed varieties (MP/TP, ECS, ACS, LR, CBR, PWD). H2 was associated with the affected chromosome in NSDTR, ACD, and small subsets of LR, TP and MP (Table 1B). H3 cosegregated with prcd in the AE, and in a subset of LR and PWD. H4 was only observed in the heterozygous state in 1 prcd-affected dog each of the ACS and NSDTR breeds (Table 1B).
At the SEC14L locus, the “G” allele was in phase with prcd in NSDTR and ACD, and the “A” allele in the remaining 8 breeds/breed varieties (Table 1A). Five affected dogs (2 CBR, 1 TP, 1 PWD and 1 ACD) were exceptions, with heterozygous status (A/G) for this allele (Table 1B). At AANAT, the “A” allele initially was in phase with all affected animals tested (see below). The finding of 8 different haplotypes in the affected population using a limited number of polymorphisms for broad-scale characterization of the ∼1.5 Mb interval (Table 1A, B) strongly suggested that the candidate region is within the interval flanked by GRB2 and SEC14L; moreover, because of interbreed specificity in the haplotypes, it appeared that this region could be reduced further by LD analysis of different breeds.
Fine-scale mapping of the LD interval
To test this hypothesis, a fine-scale haplotype of the LD interval was made. Three regions of the physical map were chosen for initial screening in 10 dogs from different breeds (Figure 2 and Supplementary Table 2A). Two prcd-affected (MP-NSDTR crossbred and ACD), and two carriers (MP-Beagle crossbred and LR) contributed 6 disease-associated chromosomes from 4 breeds. Six additional dogs from other breeds without prcd were used: BC, Basenji, English springer spaniel, Glen of Imaal terrier, English mastiff and Papillon. Together with the 2 normal chromosomes from the prcd carriers, a total of 14 normal chromosomes were examined from 8 different breeds. The screening of 20 chromosomes from 11 different breeds identified 47 SNPs; 23 of them (Supplementary Table 2A: SNPs 11 to 33) create a haplotype common to all affected chromosomes. Centromeric and telomeric to this region, the affected chromosomes from MP, ACD and NSDTR differ from each other, but the affected chromosomes of LR and MP are similar. Assuming the one founder hypothesis, this haplotype reduced the LD region for the tested breeds to approximately 664 Kb, and spanned 4 BACs (338A17, 262H18, 10M13 and 36604), and the region between BACs 36604 and10P17 which was not characterized or sequenced.
Further reduction of the LD region was sought by fine-scale analysis of the 664 Kb interval. The physical map locates BAC 10M13 in the middle of the candidate region; analysis of SNPs from flanking regions of normal (BC) and several prcd-affected chromosomes from different breeds (MP-NSDTR crossbred, ACD, CBR, PWD) was carried out. The purebred prcd-affected CBR and PWD were chosen because they were recombinant at SEC14L, an indication that they might be informative for recombinations closer to the disease locus. Twenty-five additional polymorphisms, 22 SNPs, 2 indels and 1 microsatellite, were identified, and heterozygosity was observed between affected chromosomes in the distal and proximal ends (Figure 2, Haplotypes 1, 2 and 3, and Supplementary Table 2B). This identifies a haplotype common to all affected chromosomes that spans an ∼184 Kb interval located between AANAT and ST6GalNac2. Outside of this interval, the NSDTR affected chromosome carried a different telomeric haplotype (Figure 2, Haplotype 2) compared with the MP (Figure 2, Haplotype 1), and the ACD (Figure 2, Haplotype 3) differed from both of those 2 breeds. The PWD and the CBR, for the most part, have the same haplotype as the MP.
A further reduction of the LD region was accomplished after 4 poodles, two closely related TP and two unrelated MP, were found to be affected with a retinal degeneration clinically compatible with prcd, but with a different genotype at AANAT. Two were homozygous G/G, and two were heterozygous A/G. The dogs were then typed for the SNPs within the LD interval, and were found to have the affected haplotype centromeric to AANAT (Table 1B, Figure 2, Haplotype 9). This historic recombination excludes AANAT from the LD region, and reduced the LD interval to 106 Kb.
Once the LD region was defined, a single fine-scale haplotype of the 106 Kb interval was assembled comprising 98 polymorphisms, and these were common to all prcd-affected chromosomes regardless of the breed (Supplementary Table 1). From this haplotype, a subset of 7 SNPs was used to test an additional 10 breeds of dogs with inherited retinal degeneration that was clinically compatible with prcd. Four additional breeds, Entlebucher mountain dog, Chinese crested, Silky terrier and Finnish Lapphund, were found to share the same haplotype for the screening SNP subset. This brings the number of breeds/breed varieties that share this common haplotype to 14.
Evaluation and exclusion of positional candidate genes
Prior to identifying ancestral recombinations between AANAT and the disease that reduced the LD interval to 106 Kb, we evaluated the 4 positional candidate genes in the 184 Kb candidate region: AANAT, RHBDL6, CYGB and ST6GalNac2. These were cloned (accession numbers: DQ336162, DQ336163, DQ336161, DQ336164), sequenced, and their retinal expression investigated. No differences were observed in retinal expression for the four genes (Figure 3). In addition, only one sequence variant was identified; this was the G616A transition in AANAT. The exclusion of the AANAT SNP from causal association with prcd also was confirmed in studies that bred a BC derived crossbred and a purebred LR, each A/G for the G616A transition in AANAT, to A/A prcd-affected dogs from the reference colony (data not shown). All A/A genotyped offspring had normal retinal structure when examined after the age of diagnosis. Together with the recombination results, the data confirm that the AANAT SNP is not the mutation, but rather a tightly linked benign polymorphism.
Figure 3.
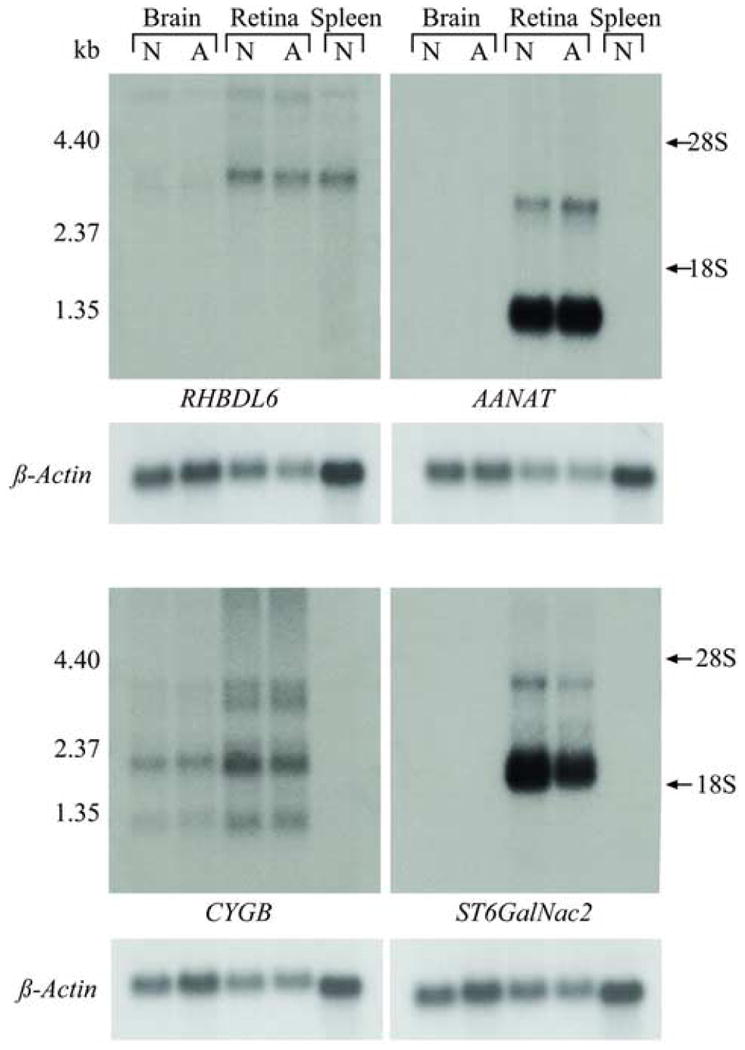
RNA expression of positional candidate genes (RHBDL6, CYGB, ST6GalNac2, AANAT) in the dog. Expression profile is shown for normal (N) and affected (A) brain and retina, and normal spleen. No difference in expression is observed between affected, and non-affected brain and retina. RHBDL6 shows equal expression in retina and spleen. AANAT and ST6GalNac2 are not expressed in brain or spleen, but are highly expressed in the retina, and have two variants: ∼1.3 kb (major transcript) and ∼3.0 kb for AANAT, and ∼2.2 kb (major transcript) and ∼4.0 kb for ST6GalNac2. CYGB is expressed in brain and retina, but not in spleen, and shows 4 different transcripts. Ribosomal RNA is indicated as 28S and 18S, and β-actin was used as a loading control.
We have continued examination of this interval, and have analyzed predicted exons of putative genes identified using a complementary EST project to characterize the canine retinome [17]. A G to A transition in codon 2 of a novel retinal expressed gene, provisionally termed PRCD, has been identified which changes the second amino acid from cysteine to tyrosine. The sequence change is present in all affected dogs from the different breeds/breed varieties with prcd. The positional cloning, validation and characterization of this novel retinal gene is detailed in the companion publication [18].
Phylogenetic analysis of prcd chromosomes
The genetic distance was calculated for nine affected chromosomes using 79 SNPs (see Methods for detail), and visualized as a bootstrapped neighbor joining cladogram (Figure 4). Both affected chromosomes present in the ACD are completely separated from the cluster combining chromosomes derived from PWD, CBR, Poodle and LR, while the chromosomes observed in the NSDTR remains isolated from either cluster at this level. The distinction of these clusters becomes more apparent when compared to normal chromosomes, in which case the NSDTR clusters more closely with the ACD than the other breeds (data not shown). This suggests that the affected chromosomes observed in the Poodles, LR, CBR and PWD separated more recently than the chromosomes derived from NSDTR and ACD, which are historically more isolated breeds. Thus, the original affected chromosome underwent at least two distinct paths of evolutionary history during the distribution of disease in the different prcd breeds examined, and this significantly helped in reducing the LD region.
Figure 4.
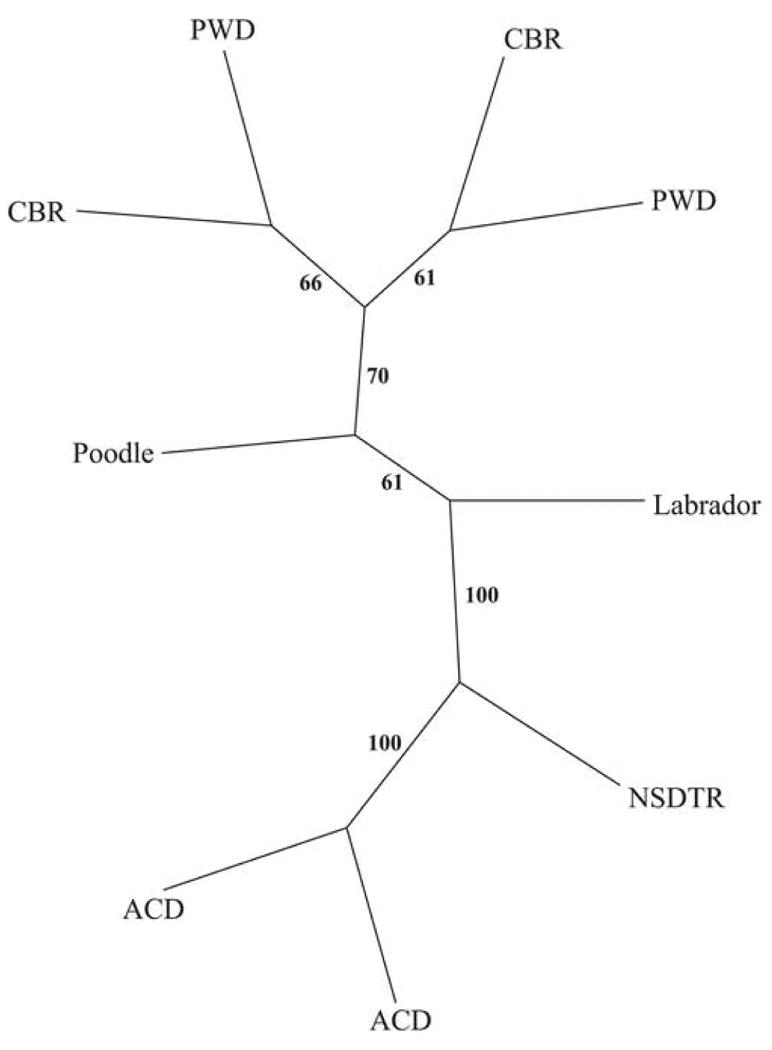
Genetic distance analysis between nine affected chromosomes (both chromosomes from affected ACD, PWD and CBR; single affected chromosomes from and Poodle-NSDTR crossbred and a heterozygous LR). Distances were calculated from 79 SNPs from the prcd candidate region (see Methods). Distances were calculated and clustered using the neighbor-joining method. Confidence in branching is inferred by bootstrap values (B=100). The individual haplotypes separate in one main cluster represented by Poodle, CBR and PWD. Affected chromosomes segregating in the NSDTR and ACD are clearly separated. Note that the PWD and CBR were selected because one chromosome from each was recombinant at SEC14L; the remainder of the haplotype was identical in both. NSDTR=Nova Scotia duck tolling retriever; ACD=Australian cattle dog; PWD=Portuguese water dog; CBR=Chesapeake Bay retriever.
Discussion
Nonrandom association of alleles, commonly referred to as LD, has become essential for studies of population structure, human evolution and gene localization. Although new technologies have improved the power for detecting association, e.g. the ‘HapMap’ project [8], the association of tightly linked markers and DNA polymorphisms has been recognized well since the early 1980's [19; 20]. LD mapping uses polymorphic markers to correlate a block or neighborhood of markers back to ancestral chromosomes. This approach is especially powerful when disease chromosomes are descended from a single founder mutation, and where the LD blocks are large. It also has been effective in identifying disease traits in presumably heterogeneous study populations, e.g. the CFH polymorphism in age-related macular degeneration [21; 22; 23]. Recent data support the theory that the extent of LD can vary dramatically among populations [24; 25; 26; 27; 28; 29; 30]. Understanding the pattern of LD blocks across the human genome in different populations is important for success of LD studies for both simple and complex diseases.
In this study we show the use of LD in the dog model to identify the chromosomal region harboring a disease trait of a previously mapped inherited retinal disease [13]. Under the assumption that all or most of the disease alleles in the populations segregating prcd shared a common disease founder, LD reduced the initial large interval of ∼6.4 Mb identified by conventional linkage analysis to 106 Kb. Progress in reducing the interval was facilitated by examining multiple breeds rather than single breeds. For example, it required a large screening effort of >100 dogs/breed to identify 17 dogs from 8 breeds that were discordant from the initially determined breed-specific disease haplotypes (compare Table 1A to 1B, see Figure 2). In contrast, comparing the affected breed-specific haplotypes across the different breeds yielded the same results with a much smaller sample size or screening effort. Post hoc analysis (Figures 2 and 4) shows that comparing only the ACD affected chromosome to either MP, LR, CBR or PWD would have been sufficient to lower the LD interval to 184 Kb. Additionally, haplotypes 1, 9 and 10 (Figure 2) observed in the TP reduced the prcd LD interval to 106 Kb. This suggests that breeds might not be as genetically homogenous as first expected. The ACD is a herding rather than retrieving breed, and was initially isolated geographically and, more recently, by quarantine restrictions. This may suggest that for this specific fragment of the genome, the ACD affected chromosome probably was split from the rest of the affected breeds early, and the conserved blocks observed in this study represent recombinations that had occurred many years ago. This is supported by the genetic distance analysis of prcd-affected chromosomes that shows the ACD and possibly NSDTR chromosomes form separate cluster from that of breeds such as MP, LR, PWD, and CBR. While the analysis clearly shows the relation of the disease-bearing interval, no inference can be made about breed origin, the ancestral population in which the mutation first occurred, or the time line for these separations.
Once the LD interval was identified, fine scale analysis identified a common haplotype consisting of 98 polymorphisms (Supplementary Table 1). This haplotype was shared by all prcd-affected chromosomes from the 10 different breed/breed varieties examined. A screening subset of these SNPs was used to identify prcd in 4 additional breeds (Chinese crested, Entlebucher mountain dog, Silky terrier and Finnish Lapphund).
At the time this study was initiated there was limited information on LD, haplotype sharing and diversity in dog breeds, information that recently has become available [10; 31]. For the five breeds examined in these studies, LD varied by breed, ranging from less than 1 Mb in Golden retrievers and LR to 3.2 Mb in Pekingese, and with low haplotype diversity in areas of LD [10]. However, for the overall dog population, i.e. across breeds, LD falls rapidly, reaching baseline levels for unlinked loci by ∼200 Kb; this is similar to what is observed in human populations, and emphasizes the importance of using multiple breeds for association studies of allelic disorders [31]. Within breeds, there were slight differences in LD between chromosomes and within chromosomes, details that have been very extensively studied in humans [32] and, to a less extent, in dog [31]. Our results support the observation, and show that across the breeds examined LD falls rapidly to a block of ∼100 Kb in size.
When this study began the 1.5X and 7.6X canine genome sequences were not available. To circumvent this limitation, we used the 3.2X BAC sequence from the minimal tiling path to examine the GRB2-SEC14L candidate region from several normal chromosomes, and identified 139 SNPs within the interval. Subsequent comparison of these 139 SNPs to those in the comparable region of the now-available canine SNP database (http://www.broad.mit.edu/ftp/pub/papers/dog_genome/snps_canfam2/) shows that only 25 are shared (18%). In particular, the most informative SNPs for LD mapping our study population are not represented in the canine SNP database. This observation has implication for future studies using a similar strategy with this resource. For example, all the SNPs, or a randomly selected subset spread evenly across CFA9, would be adequate to identify the prcd LD interval, but not sufficient to reduce it to the 106 Kb size identified in the current study. To refine and reduce the LD, it would be necessary to re-sequence the identified LD region in breeds/animals relevant to the disorder, to find those unique informative SNPs reported in this study.
The 98 polymorphisms identified in the 106 Kb prcd LD region, most of them SNPs, represent ∼1 SNP/1000 bp, a frequency similar to that reported for other regions of the dog genome [31]. Close analysis of a 29.7 Kb region in the middle of the LD interval (CFA9:7,199,890-7,170,231), identified 66 SNPs in several different breeds after re-sequencing at approximately 80% coverage. Together with 2 new additional SNPs from the Database for this region, the 68 SNPs give a frequency of 1 SNP/500 bp, a frequency that is two times higher then that reported for the dog genome [31]. This suggests that if more breeds were re-sequenced intensely, the SNP frequency across the dog genome will be higher then 1/1000.
Our findings suggest that prcd is an ancestral disease that originated from a dog that served as a common founder for a large variety of presumably distantly related modern dog breeds. Similar, but more limited, examples of founder effects in dogs have been shown previously for the breeds derived from the Collie lineage that have common mutations in both the canine multidrug resistance gene, MDR1, and the ocular disorder Collie Eye Anomaly (CEA) [33; 34](Acland et al., unpublished). In contrast, prcd is present in a diverse number of modern dog breeds whose distinct physical characteristics and functional use would suggest very different breed origins.
A close analysis of the 17 recombinant dogs shows that only one dog (Table 2B; NSDTR) had a denovo recombination while the other recombinant dogs present historical recombinations (data not shown). Pedigree analysis reveals no common grandparents to the rest of the tested dogs within their breed, and identifies a unique geographic location from which the recombinant dogs came. The recombinant LRs are of German (H2-A-A-A) or Dutch (H3-A-A-A) origin; the recombinant MP (H2-G-A-A) is of Russian origin; the recombinant TPs (H2-G-A-A) are from Canada. These observations might suggest that LD within a specific breed can also be reduced if affected dogs can be identified in the least related, geographically distinct subpopulations. Although these “pedigree outliers” might be rare in small breeds, and more common in larger breeds, once these dogs are found, they can be very useful. This was previously observed in Bedlington terriers, affected with copper toxicosis where Belgian and British origin dogs reduced the LD to ∼500 Kb [35; 36].
Most importantly, this study demonstrates that association mapping is now both practical and sufficiently powerful to study traits segregating in “real world” canine populations. This is in distinct contrast to the previous paradigm in which successful mapping relied on powerful informative pedigrees. Such informative pedigrees are rarely available from the natural population, and have usually been constructed, or at least significantly expanded, by experimental breedings [13; 34; 37]. These observations have significant implications for association mapping of a wide range of inherited canine traits. Numerous disorders are recognized that affect multiple breeds, e.g. hip and/or elbow dysplasia, posterior cortical cataracts, epilepsy, Addison's disease and others (see [11]; <http://www.upei.ca/∼cidd/intro.htm>; <http://www.angis.org.au/Databases/BIRX/omia>; <http://server.vet.cam.ac.uk:591/index.html>). But even traits that are private to single large breeds can be mapped using an appropriately structured approach. This significantly expands the range of biologically interesting and important questions that can be addressed in dogs that are relevant to both the dog and its human companion.
Materials and Methods
Study Animals
Several different populations of dogs were used that included:
prcd reference colony
The prcd strain of dogs is maintained as part of an NIH-sponsored project (EY-06855) at the Retinal Disease Studies Facility (RDSF) in Kennett Square, PA. This strain was derived from the original research colony of purebred MP in which the phenotype and inheritance of prcd were characterized [38]. Several prcd affected dogs were bred to homozygous normal unrelated MP, Beagles, and Beagle-crossbred dogs, and the heterozygous F1 progeny were then backcrossed to prcd-affected dogs to yield litters segregating the prcd phenotype. Nine related three-generation families from this colony with 70 prcd-informative progeny were studied [13]. Because the prcd reference colony was MP derived even though it is now highly outcrossed, we refer to them as MP. Additional independent lines derived from ACS and LR also are maintained separately.
Purebred and other dogs
Once allelism with prcd was established for ACD, NSDTR and PWD, a representative of each of these breeds was included for genotype analysis. In addition, selected privately owned dogs from breeds in which a form of retinal degeneration was segregating had DNA extracted from blood or tissue samples, and were typed for prcd-interval SNPs to test for association of markers with the disease (Supplementary Table 2). Ten further dogs were selected for re-sequencing to develop the initial prcd-interval haplotype. These included prcd-affected (MP-NSDTR crossbred, ACD) and carrier (MP-Beagle crossbred, LR) dogs, and, in addition, dogs that were known not be affected with prcd (BC, English mastiff, Basenji, English springer spaniel, Glen of Imaal terrier, Papillon). In addition, samples from four Red Wolves (Canis rufus) were similarly tested. Selected samples from a subset of the above dogs (MP-NSDTR crossbred, ACD, Basenji) plus additional samples from a prcd-affected CBR and a PWD were further re-sequenced to define the final fine scale haplotype map for the prcd interval.
Identification of prcd in new isolate populations
We selected 6 different breeds of dogs whose clinical retinal degeneration was clinically similar to prcd, and confirmed the disease in 3 of them. The breeds used for the allelism study included ACD, NSDTR, PWD, Basenji, IG and BC. Affected dogs from these breeds were mated to prcd-affected mix-breed colony dogs derived from MP or ECS lines. All dogs resulting from these matings were euthanatized with a barbiturate overdose after 14 weeks of age, and the retinas fixed and embedded in plastic for high-resolution optical microscopy [15].
Ascertainment of prcd status
Diagnosis of prcd was based on a combination of clinical examination, including indirect ophthalmoscopy and electroretinography, and retinal morphology using a combination of previously published ascertainment criteria for the disease [13; 15; 39]. For morphologic studies, hallmark retinal photoreceptor abnormalities are visible in animals 14 weeks of age and older using high-resolution optical microscopy [15; 38].
Blood collection and DNA extraction
DNA was extracted from whole blood samples collected from dogs with either citrate or EDTA using a standard phenol-chloroform-based purification protocol. In some cases the DNA was purified using Qiagen Mini Blood DNA kit (Qiagen,Valencia, CA) according to the manufacturer's protocol.
BAC library screening, sequencing and analysis
The physical map of the prcd interval [16] was extended to cover regions that include AANAT and SEC14L, two genes within the candidate region. The BAC library was probed with canine AANAT and SEC14L cDNA probes, and positive BACs were identified and purified as previously described [16]. BAC ends were sequenced, and BAC end-STSs were used to extend the BAC contig, and establish the minimal tiling pass. 3.2X sequence was generated for those BACs, and analyzed as previously described [16], and the order of the genes within that interval established.
Primer design, PCR amplification and sequencing
Primers were designed from the 3.2x consensus sequence of specific BAC clones for standardized amplification conditions selected for a Tm between 56°C and 63°C, and minimal risk of primer-dimer formation. 20ng of DNA were mixed with 1X PCR reaction Buffer (Invitrogen, Carlsbad, CA), 1.5mM MgCl2, 0.2mM dNTPs, 200μM forward and reverse primers, and 1 unit of Taq DNA polymerase (Invitrogen) in a final volume of 25μl. The DNA was then denatured at 96°C for 3 minutes, and 35 cycles of 94°C for 30 seconds, 55°C for 30 seconds and 72°C for 1 minute/1000bp were performed in a thermal cycler (MJ Research, Watertown, MA). An additional final extension time of 5 minutes at 72°C insured full length products. When necessary, PCR reactions were optimized by increasing the annealing temperature to 58°C or 60°C. For GC-rich amplicons, the Failsafe kit (Epicentre, Madison, WI) was used following the manufacturer's protocol. PCR products were run on 1.8% agarose, and stained with ethidium bromide (2μg/ml in a water bath). Single specific PCR products were extracted using the Qiagen PCR extraction kit (Qiagen), and eluted in 10mM Tris-HCl (pH= 7.5). If more than one amplification product was detected, the specific product was extracted from the gel using a Qiagen Gel extraction kit (Qiagen). 200ng/1000bp PCR product was mixed with 8pmole of either forward or reverse primer and DNA sequencing was performed using the Applied Biosystems Automated 3730 DNA Analyzer (Applied Biosystems, Foster City, CA). Each PCR product was sequenced with the forward and reverse primers. Sequences were then analyzed and compared using Sequencher® 4.2.2 Software (Gene Codes Corporation, Ann Arbor, MI).
GRB2 haplotypes
A GRB2 allele is composed of 4 polymorphisms that create a haplotype (Supplementary Table 1, polymorphisms number 1 to 4; amplicon IDs a, e, b and d). The different alleles are: H1 = [A-G-G-no deletion]; H2 = [G-A-A-no deletion]; H3 = [A-G-G-9 bases deleted]; H4 = [G-G-A- no deletion]
Northern analysis
10 μg of total RNA was mixed with 10μg/ml ethidium bromide and 3x of gel loading buffer (Ambion, Austin. TX) in a final volume of 10μl, heated at 65° C for 10 minutes, chilled on ice for 2-3 minutes and loaded on a 1% agarose-formaldehyde denaturing gel; 3μg of 0.24-9.5 kb RNA ladder was used as a size marker (Invitrogen, Carlbad, CA). The gel ran with continuously circulating 1xMOPS running buffer (Ambion) for 16 hours at 21 volts. After three 5 min washes in DEPC treated water, 20 min in 0.05N NaOH, and a 15 min soak in 10xSSC, transfer to a nylon-based membrane (GeneScreen Plus, NEN Life Science, Boston, MA) was done with 10xSSC buffer using a standard protocol. Full transfer was confirmed by exposing the gel to UV light. The membrane was washed in 2xSSC for 2 min, and RNA was cross-linked to the membrane (exposure=0.12 joules per cm2; Stratalinker UV Crosslinker, Stratagene, La Jolla, CA).
Northern probes were amplified from cDNA clones containing the respective genes (Accession DQ336162-DQ336165) with gene specific primers.
RHBDL6: F: CCTTCACCAGTGTCCGCTCTG; R: CGATGCCATACGTGCAAATCAC
AANAT: F: ATGTCCACACAGAGCGCACA; R: TCAGCAGCCGCTGTTCCTGC
CYGB: F: TGGAGCTGCTCATGGAGAAAG; R: GAACTCGGCCTTCTGCTCAAG
ST6GalNac2: F: AGCCAGCACAAAGCCCCCTACG; R: TCAGCGCTGGTACAGTTGAAGGAT
Probes were labeled with alpha- dCTP- P32 using RadPrime DNA labeling System (Invitrogen), and pre-hybridization (68°C for 30 minutes) and hybridization were carried out with ExpressHyb solution (Clontech, Mountain View, CA). The labeled probe was denatured at 95°C for 5 minutes, chilled on ice, and added to a fresh pre-warm ExpressHyb solution. The ExpressHyb solution was replaced with the fresh solution containing the radiolabled cDNA probe. Hybridization was carried out at 68°C for 16-18 hours, blots rinsed several times with 2xSSC, 0.05%SDS; the washes with the same solution were done twice with continuous agitation for 40 min. Then the blot was washed with 0.1xSSC and 0.1% SDS with continuous shaking at 50°C for 40 min with one change of fresh solution. Blots were exposed to x-ray film at −70°C for 24-96 hours with two intensifying screens. Loading control was achieved by hybridizing canine specific β-actin (Z70044) probe to the membranes under the same conditions, and exposure to x-ray film for 4 hours.
Phylogenetic analysis of prcd chromosomes
Individual chromosomes were assigned to the respective breed and transmittal of the affected phenotype according to pedigree information. Genetic distance between chromosomes was calculated from SNP data based on the Kimura 2-parameter with a transition/transversion ratio=2.0 [40; 41], and clustered under the neighbor-joining method [42] using the PHYLIP package [43; 44]. Confidence in the resulting branches was inferred by 100 bootstrap [45]; the consensus cluster was chosen based on the extended majority rule. The 79 SNP's used for analysis come from Supplementary Tables 1 (SNPs 30, 56, 65, 83, 88, 95, 98 and 116), 2A (46 SNPs and 3 GRB2 polymorphisms) and 2B (22 SNPs).
Supplementary Material
Acknowledgments
The authors thank John McElwee, Jacque Nelson, Julie Jordan, Amy Antosh, Brian Miller, Amanda Nickle, Gerri Antonini, and staff of RDS facility for excellent technical assistance; Weikuan Gu (Cornell University) for identification of the GRB2 polymorphisms used for constructing the different GRB2 haplotypes; Kunal Ray and Victoria Baldwin (Cornell University) for participating in the early phases of the project, and Maja Bucan (University of Pennsylvania) for manuscript review and helpful suggestions. Blood samples with pedigrees and clinical reports were provided by Drs. David Sargan (Finnish Lapphund), Keith Murphy (American eskimo subset; Texas A&M University) Simon Petersen-Jones (Entlebucher mountain dog; Michigan State University), The Seeing Eye Inc. (Labrador retriever subset), and Elaine Ostrander (many non-affected breeds; NCI, NIH), and many interested dog breeders and breed clubs.
Supported by: Morris Animal Foundation/The Seeing Eye, Inc., Foundation Fighting Blindness, NIH grants EY06855, EY13132, Van Sloun Fund for Canine Genetic Research, ONCE International Prize for Biomedicine and R&D for New Technologies for the Blind, and by targeted donations from many individuals and breed clubs.
Dog breed abbreviations
- ACD
Australian cattle dog
- ACS
American cocker spaniel
- AE
American eskimo
- BC
Border collie
- CBR
Chesapeake Bay retriever
- ECS
English cocker spaniel
- IG
Italian greyhound
- LR
Labrador retriever
- MP
Miniature Poodle
- NSDTR
Nova Scotia duck tolling retriever
- PWD
Portuguese water dog
- TP
Toy poodle
Footnotes
Conflict of Interest Statement: Authors J.S. Felix, G.M. Acland and G.D. Aguirre are co-owners of the company (OptiGen®, LLC, Ithaca, NY) that has licensed the technology for DNA testing of dogs with prcd from Cornell University.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maniatis N, Collins A, Xu CF, McCarthy LC, Hewett DR, Tapper W, Ennis S, Ke X, Morton NE. The first linkage disequilibrium (LD) maps: delineation of hot and cold blocks by diplotype analysis. Proc Natl Acad Sci U S A. 2002;99:2228–33. doi: 10.1073/pnas.042680999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Durrant C, Zondervan KT, Cardon LR, Hunt S, Deloukas P, Morris AP. Linkage disequilibrium mapping via cladistic analysis of single-nucleotide polymorphism haplotypes. Am J Hum Genet. 2004;75:35–43. doi: 10.1086/422174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Chapelle A, Wright FA. Linkage disequilibrium mapping in isolated populations: the example of Finland revisited. Proc Natl Acad Sci U S A. 1998;95:12416–23. doi: 10.1073/pnas.95.21.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peltonen L. Positional cloning of disease genes: advantages of genetic isolates. Hum Hered. 2000;50:66–75. doi: 10.1159/000022892. [DOI] [PubMed] [Google Scholar]
- 5.Varilo T, Paunio T, Parker A, Perola M, Meyer J, Terwilliger JD, Peltonen L. The interval of linkage disequilibrium (LD) detected with microsatellite and SNP markers in chromosomes of Finnish populations with different histories. Hum Mol Genet. 2003;12:51–9. doi: 10.1093/hmg/ddg005. [DOI] [PubMed] [Google Scholar]
- 6.Sheffield VC, Stone EM, Carmi R. Use of isolated inbred human populations for identification of disease genes. Trends Genet. 1998;14:391–6. doi: 10.1016/s0168-9525(98)01556-x. [DOI] [PubMed] [Google Scholar]
- 7.Kruglyak L. Genetic isolates: Separate but equal? Proc Natl Acad Sci USA. 1999;96:1170–1172. doi: 10.1073/pnas.96.4.1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altshuler D, Brooks LD, Chakravarti A, Collins FS, Daly MJ, Donnelly P. A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parker HG, Kim LV, Sutter NB, Carlson S, Lorentzen TD, Malek TB, Johnson GS, DeFrance HB, Ostrander EA, Kruglyak L. Genetic structure of the purebred domestic dog. Science. 2004;304:1160–1164. doi: 10.1126/science.1097406. [DOI] [PubMed] [Google Scholar]
- 10.Sutter NB, Eberle MA, Parker HG, Pullar BJ, Kirkness EF, Kruglyak L, Ostrander EA. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. 2004;14:2388–96. doi: 10.1101/gr.3147604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutter NB, Ostrander EA. Dog star rising: the canine genetic system. Nat Rev Genet. 2004;5:900–10. doi: 10.1038/nrg1492. [DOI] [PubMed] [Google Scholar]
- 12.Aguirre GD, Acland GM. Models, Mutants and Man: Searching for Unique Phenotypes and genes in the Dog Model of Inherited Retinal Degeneration. In: Ostrander EA, Giger U, Lindblad-Toh K, editors. The Dog and Its Genome. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2006. pp. 291–325. [Google Scholar]
- 13.Acland GM, Ray K, Mellersh CS, Gu W, Langston AA, Rine J, Ostrander EA, Aguirre GD. Linkage analysis and comparative mapping of canine progressive rod-cone degeneration (prcd) establishes potential locus homology with retinitis pigmentosa (RP17) in humans. Proceedings National Academy of Sciences, USA. 1998;95:3048–53. doi: 10.1073/pnas.95.6.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serre D, Nadon R, Hudson TJ. Large-scale recombination rate patterns are conserved among human populations. Genome Res. 2005;15:1547–52. doi: 10.1101/gr.4211905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguirre GD, Acland GM. Variation in retinal degeneration phenotype inherited at the prcd locus. Exp Eye Res. 1988;46:663–87. doi: 10.1016/s0014-4835(88)80055-1. [DOI] [PubMed] [Google Scholar]
- 16.Sidjanin DJ, Miller B, Kijas J, McElwee J, Pillardy J, Malek J, Pai G, Feldblyum T, Fraser C, Acland G, Aguirre G. Radiation hybrid map, physical map, and low-pass genomic sequence of the canine prcd region on CFA9 and comparative mapping with the syntenic region on human chromosome 17. Genomics. 2003;81:138–48. doi: 10.1016/s0888-7543(02)00028-9. [DOI] [PubMed] [Google Scholar]
- 17.Zangerl B, Sun Q, Pillardy J, Johnson JL, Schweitzer PA, Hernandez AG, Liu L, Acland GM, Aguirre GD. Development and characterization of a normalized canine retinal cDNA library for genomic and expression studies. Invest Ophthalmol Vis Sci. 2006;47:2632–2638. doi: 10.1167/iovs.05-1463. [DOI] [PubMed] [Google Scholar]
- 18.Zangerl B, Goldstein O, Philp AR, Lindauer SJ, Pearce-Kelling S, Graphodatzky AS, Ripoll D, Felix J, Stone EM, Acland GM, Aguirre GD. Identical Mutation in a Novel Retinal Gene Causes Progressive Rod-Cone Degeneration (prcd) in Dogs, and Retinitis Pigmentosa in Man. 2006 doi: 10.1016/j.ygeno.2006.07.007. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kazazian HH, Jr, Orkin SH, Boehm CD, Sexton JP, Antonarakis SE. beta-Thalassemia due to a deletion of the nucleotide which is substituted in the beta S-globin gene. Am J Hum Genet. 1983;35:1028–33. [PMC free article] [PubMed] [Google Scholar]
- 20.Arnheim N, Strange C, Erlich H. Use of pooled DNA samples to detect linkage disequilibrium of polymorphic restriction fragments and human disease: studies of the HLA class II loci. Proc Natl Acad Sci U S A. 1985;82:6970–4. doi: 10.1073/pnas.82.20.6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 23.Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 24.Kidd JR, Pakstis AJ, Zhao H, Lu RB, Okonofua FE, Odunsi A, Grigorenko E, Tamir BB, Friedlaender J, Schulz LO, Parnas J, Kidd KK. Haplotypes and linkage disequilibrium at the phenylalanine hydroxylase locus, PAH, in a global representation of populations. Am J Hum Genet. 2000;66:1882–99. doi: 10.1086/302952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goddard KA, Hopkins PJ, Hall JM, Witte JS. Linkage disequilibrium and allele-frequency distributions for 114 single-nucleotide polymorphisms in five populations. Am J Hum Genet. 2000;66:216–34. doi: 10.1086/302727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kidd KK, Pakstis AJ, Speed WC, Kidd JR. Understanding human DNA sequence variation. J Hered. 2004;95:406–20. doi: 10.1093/jhered/esh060. [DOI] [PubMed] [Google Scholar]
- 27.Jorde LB, Watkins WS, Kere J, Nyman D, Eriksson AW. Gene mapping in isolated populations new roles for old friends? Hum Hered. 2000;50:57–65. doi: 10.1159/000022891. [DOI] [PubMed] [Google Scholar]
- 28.Sawyer SL, Mukherjee N, Pakstis AJ, Feuk L, Kidd JR, Brookes AJ, Kidd KK. Linkage disequilibrium patterns vary substantially among populations. Eur J Hum Genet. 2005;13:677–86. doi: 10.1038/sj.ejhg.5201368. [DOI] [PubMed] [Google Scholar]
- 29.Evans DM, Cardon LR. A comparison of linkage disequilibrium patterns and estimated population recombination rates across multiple populations. Am J Hum Genet. 2005;76:681–7. doi: 10.1086/429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De La Vega FM, Isaac H, Collins A, Scafe CR, Halldorsson BV, Su X, Lippert RA, Wang Y, Laig-Webster M, Koehler RT, Ziegle JS, Wogan LT, Stevens JF, Leinen KM, Olson SJ, Guegler KJ, You X, Xu LH, Hemken HG, Kalush F, Itakura M, Zheng Y, de The G, O'Brien SJ, Clark AG, Istrail S, Hunkapiller MW, Spier EG, Gilbert DA. The linkage disequilibrium maps of three human chromosomes across four populations reflect their demographic history and a common underlying recombination pattern. Genome Res. 2005;15:454–62. doi: 10.1101/gr.3241705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, Mauceli E, Xie X, Breen M, Wayne RK, Ostrander EA, Ponting CP, Galibert F, Smith DR, Dejong PJ, Kirkness E, Alvarez P, Biagi T, Brockman W, Butler J, Chin CW, Cook A, Cuff J, Daly MJ, Decaprio D, Gnerre S, Grabherr M, Kellis M, Kleber M, Bardeleben C, Goodstadt L, Heger A, Hitte C, Kim L, Koepfli KP, Parker HG, Pollinger JP, Searle SM, Sutter NB, Thomas R, Webber C, Baldwin J, Abebe A, Abouelleil A, Aftuck L, Ait-Zahra M, Aldredge T, Allen N, An P, Anderson S, Antoine C, Arachchi H, Aslam A, Ayotte L, Bachantsang P, Barry A, Bayul T, Benamara M, Berlin A, Bessette D, Blitshteyn B, Bloom T, Blye J, Boguslavskiy L, Bonnet C, Boukhgalter B, Brown A, Cahill P, Calixte N, Camarata J, Cheshatsang Y, Chu J, Citroen M, Collymore A, Cooke P, Dawoe T, Daza R, Decktor K, Degray S, Dhargay N, Dooley K, Dooley K, Dorje P, Dorjee K, Dorris L, Duffey N, Dupes A, Egbiremolen O, Elong R, Falk J, Farina A, Faro S, Ferguson D, Ferreira P, Fisher S, Fitzgerald M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 32.Smith AV, Thomas DJ, Munro HM, Abecasis GR. Sequence features in regions of weak and strong linkage disequilibrium. Genome Res. 2005;15:1519–34. doi: 10.1101/gr.4421405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neff MW, Robertson KR, Wong AK, Safra N, Broman KW, Slatkin M, Mealey KL, Pedersen NC. Breed distribution and history of canine mdr1-1Delta, a pharmacogenetic mutation that marks the emergence of breeds from the collie lineage. Proc Natl Acad Sci U S A. 2004;101:11725–30. doi: 10.1073/pnas.0402374101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe JK, Kukekova AV, Kirkness EF, Langlois MC, Aguirre GD, Acland GM, Ostrander EA. Linkage mapping of the primary disease locus for collie eye anomaly. Genomics. 2003;82:86–95. doi: 10.1016/s0888-7543(03)00078-8. [DOI] [PubMed] [Google Scholar]
- 35.van de Sluis B, Peter AT, Wijmenga C. Indirect molecular diagnosis of copper toxicosis in Bedlington terriers is complicated by haplotype diversity. J Hered. 2003;94:256–9. doi: 10.1093/jhered/esg030. [DOI] [PubMed] [Google Scholar]
- 36.van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet. 2002;11:165–73. doi: 10.1093/hmg/11.2.165. [DOI] [PubMed] [Google Scholar]
- 37.Jónasdóttir TJ, Mellersh CS, Moe L, Heggebo R, Gamlem H, Ostrander EA, Lingaas F. Genetic mapping of a naturally occurring hereditary renal cancer syndrome in dogs. PNAS. 2000;97:4132–4137. doi: 10.1073/pnas.070053397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguirre G, Alligood J, O'Brien P, Buyukmihci N. Pathogenesis of progressive rod-cone degeneration in miniature poodles. Invest Ophthalmol Vis Sci. 1982;23:610–30. [PubMed] [Google Scholar]
- 40.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 41.Jin L, Nei M. Limitations of the evolutionary parsimony method of phylogenetic analysis. Mol Biol Evol. 1990;7:82–102. doi: 10.1093/oxfordjournals.molbev.a040588. [DOI] [PubMed] [Google Scholar]
- 42.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 43.Felsenstein J. PHYLIP -- Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 44.Felsenstein J. Distributed by the author, Department of Genetics. University of Washington; Seattle: 1993. PHYLIP (Phylogeny Inference Package) version 3.5c. [Google Scholar]
- 45.Felsenstein J. Confidence-limits on phylogenies-an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.