

Abstract
Activation of Wnt/β-catenin signaling can result in up-regulation of mTORC1 signaling in cancer cells. The low density lipoprotein receptor-related protein-6 (LRP6) is an essential Wnt co-receptor for Wnt/β-catenin signaling. We found that rottlerin, a natural plant polyphenol, suppressed LRP6 expression and phosphorylation, and inhibited Wnt/β-catenin signaling in HEK293 cells. Furthermore, the inhibitory effects of rottlerin on LRP6 expression/phosphorylation and Wnt/β-catenin signaling were confirmed in human prostate cancer PC-3 and DU145 cells and breast cancer MDA-MB-231 and T-47D cells. Mechanistically, rottlerin promoted LRP6 degradation, but had no effects on LRP6 transcriptional activity. In addition, rottlerin-mediated LRP6 down-regulation was unrelated to activation of 5′-AMP-activated protein kinase (AMPK). Importantly, we also found that rottlerin inhibited mTORC1 signaling in prostate and breast cancer cells. Finally, we demonstrated that rottlerin was able to suppress the expression of cyclin D1 and survivin, two targets of both Wnt/β-catenin and mTORC1 signaling, in prostate and breast cancer cells, and displayed remarkable anticancer activity with IC50 values between 0.7 and 1.7 μM for prostate cancer PC-3 and DU145 cells and breast cancer MDA-MB-231 and T-47D cells. The IC50 values are comparable to those shown to suppress the activities of Wnt/β-catenin and mTORC1 signaling in prostate and breast cancer cells. Our data indicate that rottlerin is a novel LRP6 inhibitor and suppresses both Wnt/β-catenin and mTORC1 signaling in prostate and breast cancer cells, and that LRP6 represents a potential therapeutic target for cancers.
Keywords: LRP6, Wnt signaling, mTORC1 signaling, rottlerin, cancer, drug discovery
1. Introduction
The low-density lipoprotein receptor-related protein 6 (LRP6) is a member of the low density lipoprotein receptor family, and acts as a co-receptor for Wnt ligands, which interact with both the seven transmembrane receptor of the Frizzled (Fzd) family and LRP6 to activate the Wnt/β-catenin signaling pathway [1–3]. β-Catenin is an essential transcriptional co-activator of the Wnt/β-catenin signaling pathway. In the absence of Wnt ligands, β-catenin level is efficiently regulated by a supramolecular complex containing adenomatous polyposis coli (APC), axin, and glycogen synthetase kinase 3β (GSK3β). This complex promotes phosphorylation of β-catenin by casein kinase 1 (Ck1) and GSK3β. Phosphorylated β-catenin becomes multi-ubiquitinated and degraded by the 26S proteasome. The action of this complex is inhibited upon the binding of Wnt to its receptors on the cell surface. As a result, β-catenin protein is stabilized and then enters the nucleus to form a complex with transcription factors of the T-cell factor/lymphoid enhancing factor (TCF/LEF) family to activate transcription of Wnt target genes that regulate cancer progression, including tumor initiation, tumor growth, cell senescence, cell death, differentiation and metastasis [1–3].
The mammalian target of rapamycin complex 1 (mTORC1) is a heterotrimeric protein kinase that consists of the mTOR catalytic subunit and two associated proteins, raptor (regulatory associated protein of mTOR) and mLST8 (mammalian lethal with sec-13). The PI3K-Akt pathway is a major upstream regulator of mTORC1 signaling. The activation of mTORC1 induces phosphorylation of P70 S6 kinase (P70S6K) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1), leading to enhanced translation of a subset of mRNAs that are critical for cell growth and metabolism [4]. Being downstream of AKT, mTORC1 has been described as the most essential effector in driving cell proliferation and susceptibility to oncogenic transformation [4–6].
There is a crosstalk between Wnt/β-catenin and mTORC1 signaling, and mounting evidence indicates that activation of Wnt/β-catenin signaling up-regulates mTORC1 signaling in cancer cells [7–15]. It has been showed that Wnt proteins activated mTORC1 signaling by a mechanism that involves the inhibition of that glycogen synthase kinase-3 (GSK-3) [7], and that GSK-3 regulated both Wnt/β-catenin and mTOR signaling in mouse hematopoietic stem cells [9]. In addition, Wnt/β-catenin signaling could contribute to mTORC1 activation through the increased level of mTOR protein in colorectal cancer cell lines and in intestinal polyps of the ApcΔ716 heterozygous mutant mouse, a model for human familial adenomatous polyposis [8]. Interestingly, it was reported that LRP6+/− mice displayed an impaired activity of mTORC1 pathway in brown adipose tissue [14], and that knockdown of LRP6 decreased mTORC1 signaling in prostate cancer cells [15], suggesting that LRP6 is involved in Wnt protein-induced activation of mTORC1 signaling.
Rottlerin is a natural plant polyphenol, and appears to have great potentiality for being used in chemotherapy because it affects several cell machineries involved in survival, apoptosis, autophagy, and invasion [16, 17]. In the present study, we demonstrated that rottlerin is a novel LRP6 inhibitor by enhancing the receptor degradation, and inhibits both Wnt/β-catenin and mTORC1 in prostate and breast cancer cells.
2. Materials and Methods
2.1. Materials
Rottlerin and niclosamide were purchased from Sigma. Plasmid pCS-Myc-hLRP6 containing the full-length human LRP6 cDNA was provided by Dr. Christof Niehrs (Deutsches Krebsforschungszentrum, Heidelberg, Germany), and plasmid pGST-E-cadherin was provided by Dr. Gail Johnson (University of Rochester). LRP6 and actin promoter reporter constructs were purchased from SwitchGear Genomics. The Super8XTOPFlash luciferase construct was provided by Dr. Randall T. Moon (University of Washington, Seattle). A β-galactosidase-expressing vector was from Promega. Polyclonal anti-LRP6 was from Santa Cruz Biotechnology. Monoclonal anti-phospho-LRP6, anti-axin2, anti-S6, anti-phospho-S6, anti-AMPKα and anti-phospho-AMPKα and polyclonal anti-p70S6K, anti-phospho-p70S6K were purchased from Cell Signaling Technology. Monoclonal anti-β-catenin was from BD Biosciences. Monoclonal anti-actin was from Sigma. Peroxidase labeled anti-mouse antibody and ECL system were purchased from Amersham Life Science. The luciferase and β-galactosidase assay systems were from Promega. Tissue culture media, fetal bovine serum (FBS), and plastic-ware were obtained from Life Technologies, Inc. Proteinase inhibitor cocktail Complete™ was obtained from Boehringer Mannheim.
2.2. Cell culture and conditioned media
All cell lines were obtained from ATCC and grown under standard cell culture conditions at 37°C in a humidified atmosphere with 5% CO2. Human fibrosarcoma cancer HT1080 cells stably transfected with HA-tagged LRP6 have been described before [18]. The prostate cancer PC-3 and DU145 cells were cultured in RPMI-1640 medium containing 10% FBS, 2 mM of L-glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Wnt3A-secreting L cells, control L cells, HEK293 cells, HT1080 cells and breast cancer MDA-MB-231 and T-47D cells were cultured in DMEM medium containing 10% of FBS, 2 mM of L-glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Wnt3A-conditioned medium (Wnt3A CM) and L cell control CM were prepared according to manufacturer’s specifications.
2.3. Western blotting
Cells in 6-well plates were lysed in 0.5 ml of lysis buffer (phosphate-buffered saline containing 1% Triton X-100 and 1 mM PMSF) at 4°C for 10 min. Equal quantities of protein were subjected to SDS-PAGE under reducing conditions. Following transfer to immobilon-P transfer membrane, successive incubations with a primary antibody, and a horseradish peroxidase-conjugated secondary antibody were carried out for 60–120 min at room temperature. The immunoreactive proteins were then detected using the ECL system. Films showing immunoreactive bands were scanned by Hp Scanjet 5590.
2.4. Cytosolic free β-catenin analysis with GST-E-cadherin binding assay
The GST-E-cadherin binding assay was carried out exactly as previously described [19]. Uncomplexed cytosolic free β-catenin present in 100 μg of total cell lysate was subjected to SDS-PAGE and detected using the monoclonal antibody to β-catenin.
2.5. Real-time RT-PCR
Real-time RT-PCR for LRP6 expression was performed as described before [20]. Brefiely, total RNA was isolated from cell cultures using RNA-Bee (Tel-Test), and first-strand cDNA synthesis was performed using ProSTARTM Ultro HF RT-PCR Kit (Strategene) primed with oligo(dT) primer in a 20 μl reaction mixture containing 1 μg total RNA. For analysis of LRP6 mRNA levels, real-time RT-PCR was performed with the specific LRP6 and GAPDH primers purchased from SABioscience/Qiagene. The LRP6 mRNA level was normalized to the GAPDH mRNA level.
2.6. Luciferase reporter assay for Wnt/β-catenin signaling
Cancer cells were plated into 24-well plates. After overnight culture, the cells were transiently transfected with the Super8XTOPFlash luciferase construct and β-galactosidase-expressing vector along with LRP6 plasmid. After 24 h incubation, cells were treated with rottlerin at the indicated concentrations. Cells were then lysed 24 h later and both luciferase and β-galactosidase activities were determined. The luciferase activity was normalized to the β-galactosidase activity.
2.7. Luciferase reporter assay for LRP6 promoter activity
LRP6 promoter activity was examined as described before [21]. Brefiely, cells were plated into 24-well plates. After overnight culture, the cells were transiently transfected with the LRP6 promoter reporter construct or the actin promoter reporter plasmid. After 24 h incubation, cells were treated with rottlerin. Cells were then lysed 24 h later and the luciferase activities were determined. The LRP6 promoter luciferase activity was normalized to the actin promoter luciferase activity.
2.8. Cell viability assay
Cells were seeded into 96-well tissue culture treated microtiter plates at a density of 5000 cells/well. RPMI-1640 containing 10% FBS was used as assay media for PC-3 and DU-145 cells, while DMEM containing 10% FBS was used as assay media for MDA-MB-231 and T-47D cells. After 24h incubation, the cells were treated with rottlerin at the indicated concentrations for 72 h. Cell viability was measured by the Cell Titer Glo Assay, which is a luminescent assay that is an indicator of live cells as a function of metabolic activity and ATP content.
2.9. Statistics
Statistical analyses were performed using Student’s unpaired t-test. Data were presented as mean ± SD. Differences at P < 0.05 were considered statistically significant.
3. Results
3.1. Rottlerin blocks Wnt/β-catenin signaling induced by Wnt3A and LRP6 in HEK293 cells
To test whether rottlerin is an inhibitor of Wnt/β-catenin signaling, we performed a Wnt/β-catenin signaling reporter assay in HEK293 cells transfected with LRP6 along with Wnt/β-catenin signaling reporter Super8XTOPFlash, and treated with rottlerin in the presence or absence of Wnt3A CM. As shown in Fig. 1A & 1B, LRP6 expression increased the Super8XTOPFlash activity in HEK293 cells, which was further enhanced when the LRP6-expressing cells were treated with Wnt3A CM. Importantly, the increased Super8XTOPFlash activity induced by LRP6 or LRP6 plus Wnt3A CM was blocked by rottlerin in a concentration dependent manner (Fig. 1A & 1B). Notably, rottlerin was able to significantly block the Super8XTOPFlash activity induced by LRP6 in HEK293 cells at a concentration as low as 0.25 μM (Fig. 1A), suggesting that rottlerin is a potent inhibitor of Wnt/β-catenin signaling.
Fig. 1.
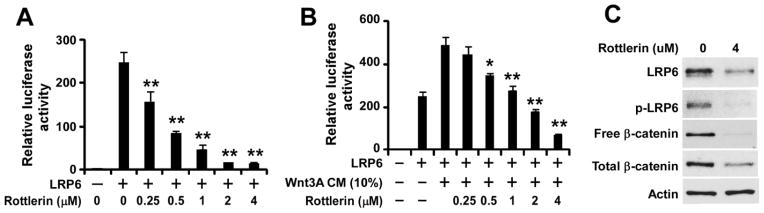
Effects of rottlerin on Wnt3A and LRP6-induced Wnt/β signaling in HEK293 cells. (A, B) HEK293 cells in 24-well plates were transiently transfected with LRP6 plasmid or the corresponding control vector, along with Super8XTOPFlash construct and β-galactosidase-expressing vector in each well. After being incubated for 24 h, cells were treated with rottlerin (A) or rottlerin plus Wnt3A CM (B) at indicated concentrations for 24 h. The luciferase activity was then measured 24 h later with normalization to the activity of the β-galactosidase. Values are averages of three determinations with the standard deviations indicated by error bars. **P < 0.01 compared to the control cells without niclosamide treatment. (C) HEK293 cells in 6-well plates were treated with rottlerin (4 μM) for 24 h. The levels of cytosolic free human β-catenin (92 kDa), total cellular human β-catenin (92 kDa), LRP6 (210 kDa for mature form) and phospho-LRP6 (p-LRP6, 210 kDa)) were examined by Western blotting. All the samples were also probed with anti-human actin antibody to verify equal loading.
Uncomplexed cytosolic β-catenin (free β-catenin) can translocate to the cell nucleus and bind transcription factors, such as TCF or LEF, leading to the transcription of Wnt target genes. As shown in Fig. 1C, rottlerin greatly suppressed levels of cytosolic free β-catenin and total cellular β-catenin in HEK293 cells, confirming that rottlerin is a novel inhibitor of Wnt/β-catenin signaling.
3.2. Rottlerin inhibits Wnt/β-catenin signaling in prostate and breast cancer cells
To determine whether rottlerin blocks Wnt/β-catenin signaling in cancer cells, we examined the level of cytosolic free β-catenin after rottlerin treatment in prostate cancer PC-3 cells. We found that cytosolic free β-catenin levels in PC-3 cells were significantly reduced after rottlerin treatment (Fig. 2A). Furthermore, expression of axin2, a specific transcriptional target of the Wnt/β-catenin signaling pathway [22–25], was significantly deceased after rottlerin treatment in PC-3 cells (Fig. 2A).
Fig. 2.
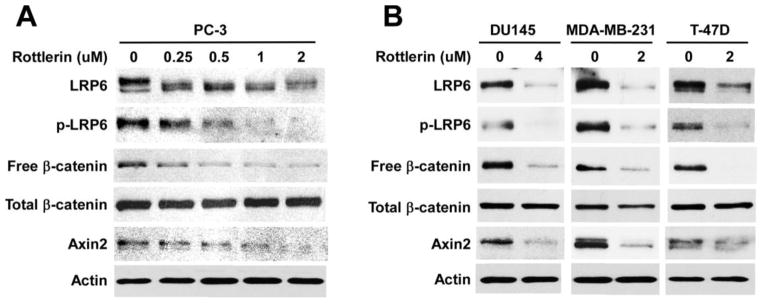
Effects of rottlerin on Wnt/β-catenin signaling in cancer cells. Prostate cancer PC-3 and DU145 cells and breast cancer MDA-MB-231 and T-47D cells in 6-well plates were treated with rottlerin at the indicated concentrations for 24 h. The levels of cytosolic free human β-catenin (92 kDa), total cellular human β-catenin (92 kDa), human LRP6 (210 kDa for mature form), phospho-LRP6 (p-LRP6, 210 kDa)) and axin2 (95 and 98 kDa) were examined were examined by Western blotting. All the samples were also probed with anti-human actin antibody to verify equal loading.
To confirm the effects of rottlerin on Wnt/β-catenin signaling in cancer cells, we repeated the experiments in prostate tumor cell line DU145 and breast cancer cell lines MDA-MB-231 and T-47D. As expected, rottlerin significantly reduced the levels of cytosolic free β-catenin, and inhibited axin2 expression in all three cancer cell lines tested (Fig. 2B).
3.3. Rottlerin suppressing LRP6 expression
LRP6 is an essential Wnt co-receptor for the Wnt/β-catenin signaling pathway, and LRP6 phosphorylation is critical for Wnt/β-catenin signaling activation induced by Wnt proteins [1–3]. To explore the molecular mechanism underlying Wnt/β-catenin signaling inhibition by rottlerin, we examined LRP6 phosphorylation and LRP6 expression after rottlerin treatment. As shown in Fig. 1C, treatment of rottlerin markedly inhibited endogenous LRP6 phosphorylation in HEK293 cells. Importantly, we also found that the total cellular level of endogenous LRP6 was greatly decreased after rottlerin treatment in HEK293 cells (Fig. 1C). Moreover, rottlerin suppressed LRP6 expression and LRP6 phosphorylation in all four cancer lines tested (Fig. 2).
3.4. Rottlerin suppresses LRP6 expressing by inducing LRP6 degradation
To define the mechanism underlying rottlerin-mediated on LRP6 down-regulation, we studied LRP6 turnover. PC-3 cells were treated with rottlerin at 2 μM for 0, 3, 6, 10 and 24h in the presence of cycloheximide, a protein synthesis inhibitor. As shown in Fig. 3, rottlerin significantly enhanced LRP6 turnover in PC-3 cells.
Fig. 3.
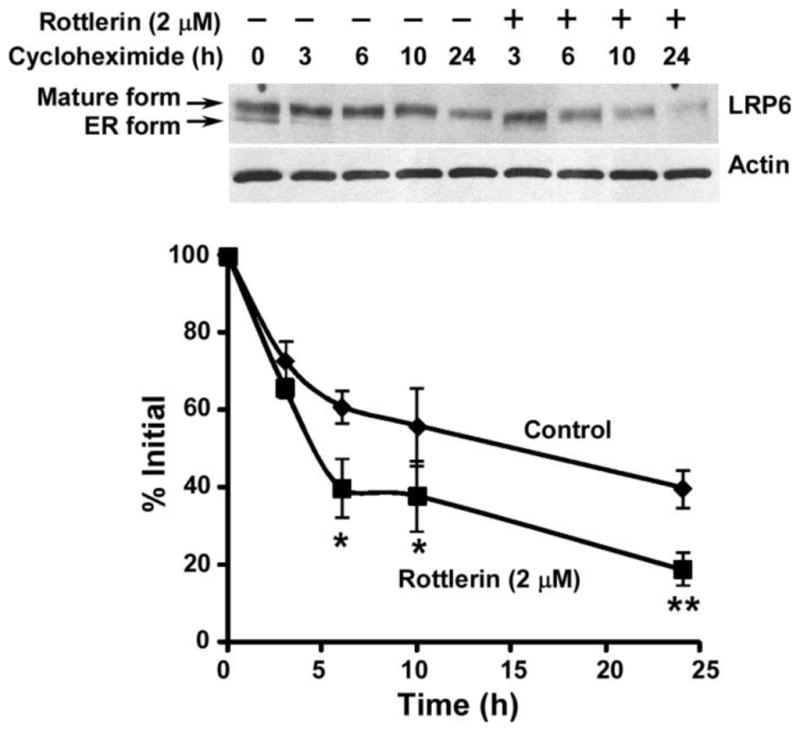
Effects of rottlerin on endogenous LRP6 degradation. PC-3 cells were incubated with 10 μg/ml of cycloheximide in the presence of rottlerin (2 μM) or vehicle for 0, 3, 6, 10 or 24 h. Cells were then harvested, and the level of endogenous human LRP6 (210 kDa for mature form) was examined by Western blotting. Samples were also probed with anti-actin antibody to verify equal loading. Lower panel: the pixels for each band were measured, normalized and plotted. Data are mean values of three independent experiments with the SD values indicated by error bars. *P < 0.05, **P < 0.01 versus corresponding control value.
Niclosamide, an Food and Drug Administration-approved antihelminthic drug, is an inhibitor of Wnt/β-catenin signaling by promoting LRP6 degradation in prostate and breast cancer cells [20]. We found that rottlerin, like niclosamide, was able to inhibit exogenous HA-LRP6 expression driven by CMV promoter in human fibrosarcoma cancer HT1080 cells (Fig. 4), indicating that rottlerin and niclosamide share a similar mechanism of action on LRP6 expression.
Fig. 4.
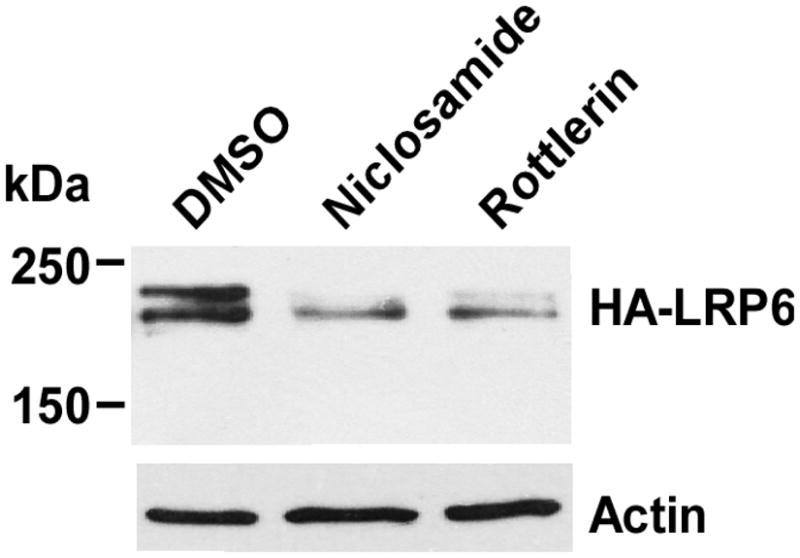
Effects of rottlerin on enforced LRP6 expression in HT1080 cells. HT1080 cells stably transduced with human LRP6 with HA tag in 6-well plates were treated with rottlerin (1 μM) or niclosamide (1.2 μM) for 24 h. The cells were then harvested and levels of HA-LRP6 (210 kDa for mature form) were examined by Western blotting with anti-HA antibody. All the samples were also probed with anti-actin antibody to verify equal loading.
To test whether rottlerin regulates LRP6 expression at the transcription level too, we examined LRP6 mRNA levels by real-time RT-PCR. As shown in Fig. 5A, LRP6 mRNA levels were not significantly changed after rottlerin treatment in prostate cancer PC-3 cells and breast cancer T-47D cells. In addition, rottlerin treatment had no effects on the activity of LRP6 promoter in both PC-3 and T-47D cells (Fig. 5B). Therefore, rottlerin-induced LRP6 suppression is mainly due to the enhanced LRP6 degradation.
Fig. 5.

Rottlerin suppresses LRP6 expression at the transcription level. (A) Prostate cancer PC-3 cells and breast cancer T-47D cells were treated with rottlerin at the indicated concentrations for 24 h. Total RNA was extracted from the indicated cell lines, and LRP6 mRNA levels were determined by real-time RT-PCR and normalized to the message levels of GAPDH mRNA. All the values are the average of triple determinations with the s.d. indicated by error bars. (B) Cancer cells were transiently transfected with the LRP6 promoter plasmid, or the actin promoter reporter plasmid. After being incubated for 24 h, cells were treated with rottlerin at the indicated concentrations. The luciferase activities were then measured 24 h later, and the LRP6 promoter luciferase activity was normalized to the actin promoter luciferase activity. All the values are the average of triplicate determinations.
3.5. Rottlerin-mediated LRP6 down-regulation is unrelated to AMPK activation in prostate and breast cancer cells
It is well recognized that rottlerin is a mitochondrial uncoupler, which depolarizes the mitochondrial membrane potential, reduces cellular ATP levels, activates 5′-AMP-activated protein kinase (AMPK) and affects mitochondrial production of reactive oxygen species [16]. Rottlerin can enhance AMPK phosphorylation in 3T3-L1 adipocytes, vascular smooth muscle cells and prostate cancer stem cells [26–28]. To test whether AMPK is involved in rottlerin-mediated LRP6 degradation, we examined AMPK activation in cancer cells. It was found that AMPK phosphorylation was increased in prostate cancer PC-3 cells but decreased in prostate cancer DU145 cells after rottlerin treatment (Fig. 6). Furthermore, AMPK phosphorylation in breast cancer MDA-MB-231 cells was increased with rottlerin treatment at a low concentration (2 μM), but unchanged at a high concentration (20 μM) (Fig. 6). In addition, AMPK phosphorylation was unchanged in breast cancer T-47D cells with rottlerin treatment at both low and high concentrations (Fig. 6). As expected, the level of LRP6 expression was significantly decreased in all tested cell lines with rottlerin treatment at both low and high concentrations (Fig. 6). Together, these results indicate that AMPK activation is not associated with rottlerin-mediated LRP6 down-regulation in prostate and breast cancer cells.
Fig. 6.
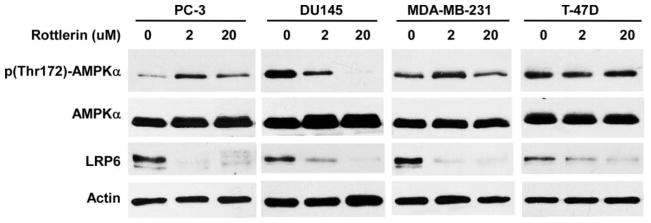
Effects of rottlerin on AMPK activation in cancer cells. Prostate cancer PC-3 and DU145 cells and breast cancer MDA-MB-231 and T-47D cells in 6-well plates were treated with rottlerin at the indicated concentrations for 24 h. The levels of human AMPKα (62 kDa), phospho-AMPKα (62 kDa) and LRP6 (210 kDa for mature form) were examined by Western blotting. All the samples were also probed with anti-actin antibody to verify equal loading.
3.6. Rottlerin inhibits mTORC1 signaling in prostate and breast cancer cells
Inhibition of Wnt/β-catenin can result in down-regulation of mTORC1 signaling in cancer cells [7–15]. It was reported that LRP6 deficiency caused inhibition of mTORC1 signaling in prostate cancer PC-3 cells [15]. Therefore, we tested effects of rottlerin on mTORC1 signaling in cancer cells. As shown in Fig. 7A, rottlerin suppressed phosphorylation of P70-S6K and S6 in a dose dependent manner in PC-3 cells. Furthermore, rottlerin also significantly inhibited P70-S6K and S6 phosphorylation in prostate cancer DU145 cells and breast cancer MDA-MB-231 and T-47D cells (Fig. 7B).
Fig. 7.
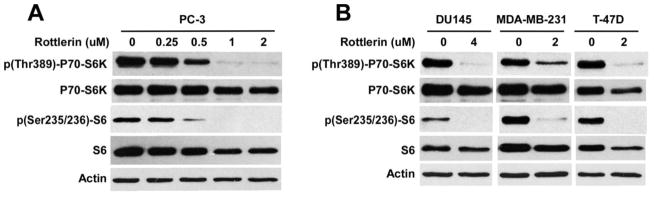
Effects of rottlerin on mTORC1 signaling in cancer cells. Prostate cancer PC-3 and DU145 cells and breast cancer MDA-MB-231 and T-47D cells in 6-well plates were treated with rottlerin at the indicated concentrations for 24 h. The levels of human P70-S6K (70 kDa), phospho-P70-S6K (70 kDa), S6 (32 kDa) and phospho-S6 (32 kDa) were examined by Western blotting. All the samples were also probed with anti-actin antibody to verify equal loading.
3.7. Rottlerin inhibits cyclinD1 and surviving expression in prostate and breast cancer cells
Cyclin D1 is critical for cancer cell proliferation, and is a transcriptional target of both Wnt/β-catenin signaling [29, 30] and mTORC1 signaling [31]. Survivin is a dual regulator of cancer cell proliferation and cell death, and its expression is also regulated by both Wnt/β-catenin signaling [32, 33] and mTORC1 signaling [34]. As expected, rottlerin treatment resulted in down-regulation of cyclin D1 and survivin expression in a dose dependent manner in prostate cancer PC-3 cells (Fig. 8A). Moreover, rottlerin also significantly inhibited cyclin D1 and survivin expression in prostate cancer DU145 cells and breast cancer MDA-MB-231 and T-47D cells (Fig. 8B).
Fig. 8.
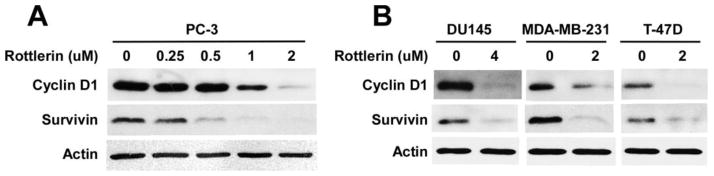
Effects of rottlerin on levels of of cyclin D1 and survivin expression in cancer cells. Prostate cancer PC-3 and DU145 cells and breast cancer MDA-MB-231 and T-47D cells in 6-well plates were treated with rottlerin at the indicated concentrations for 24 h. The levels of human cyclin D1 (36 kDa) and survivin (16 kDa) were examined by Western blotting. All the samples were also probed with anti-actin antibody to verify equal loading.
3.8. Rottlerin inhibits cancer cell proliferation
Given that rottlerin can block Wnt/β-catenin and mTORC1signaling and inhibit cyclin D1 and surviving expression in prostate and breast cancer cells, we then examined the effect of rottlerin on cancer cell proliferation. As shown in Fig. 9, rottlerin inhibited cancer cell proliferation with IC50 values between 0.73 and 1.70 μM for the four tested cell lines. The IC50 values are comparable to those shown to suppress LRP6 expression and inhibit the activities of Wnt/β-catenin and mTORC1 signaling in 4 tested cancer cell lines, suggesting that suggest that the inhibitory effects of rottlerin on prostate and breast cancer cell proliferation may involve the suppression of Wnt/β-catenin and mTORC1 signaling.
Fig. 9.
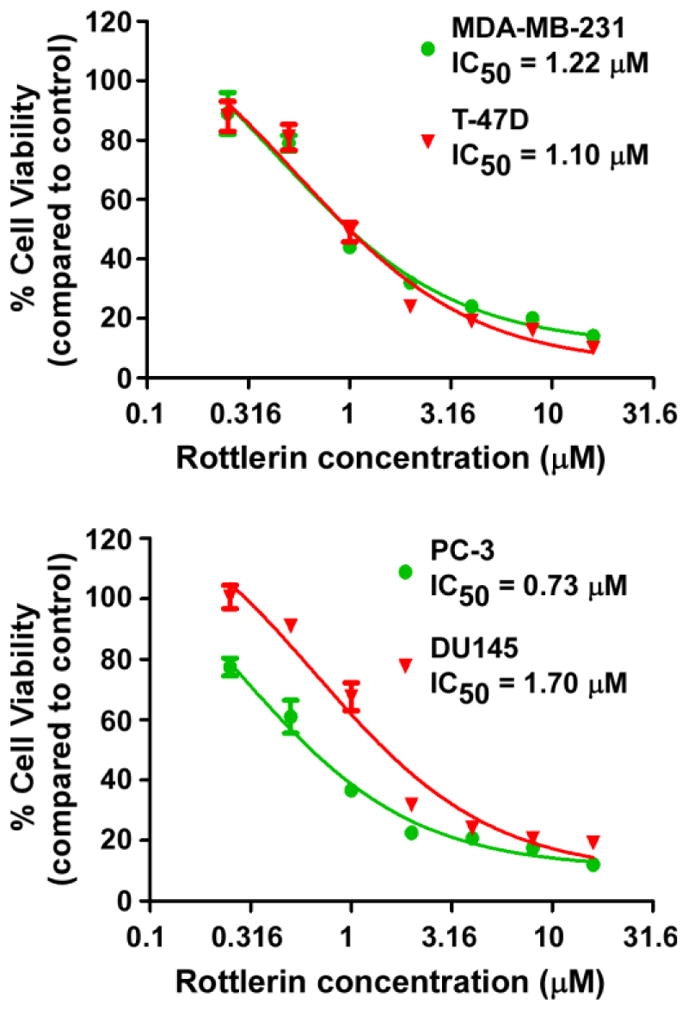
Effects of rottlerin on cancer cell proliferation. Prostate cancer PC-3 and DU145 cells and breast cancer MDA-MB-231 and T-47D cells in 96-well plates were treated with niclosamide for 72 h. Cell viability was measured by the Cell Titer Glo Assay system. All the values are the average of triplicate determinations with the s.d. indicated by error bars.
4. Discussion
Wnt/β-catenin signaling plays an important role in embryonic development and can lead to tumor formation when aberrantly activated [1–3]. LRP6 is expressed in human cancer cell lines and up-regulated in human malignant tissues [35]. Wnt/β-catenin signaling activation, as defined by β-catenin nuclear expression and overexpression of the Wnt/β-catenin target cyclin D1, was associated with a poorer prognosis in breast cancer patients [36]. Recent studies have further demonstrated that the Wnt/β-catenin signaling pathway is preferentially activated in triple negative breast cancer (TNBC), and that LRP6 is up-regulated in human TNBC [37–39]. LRP6 expression is also significantly up-regulated in prostate patients with metastatic disease compared to those without metastasis, and is associated with a significantly increased risk of recurrent disease [40]. As an essential Wnt co-receptor to activate Wnt/β-catenin signaling, LRP6 is a promising therapeutic target for the development of novel anticancer drugs [35]. Blocking Wnt/β-catenin signaling by N-myc downstream regulated gene-1 (NDRG1), a tumor metastasis suppressor which interacts with LRP6 and represses Wnt/β-catenin signaling, led to drastic suppression of metastatic phenotypes of mammary tumor cells in vitro and in vivo [40]. In addition, transcriptional knockdown of LRP6 in human TNBC MDA-MB-231 cells significantly decreased Wnt/β-catenin signaling, cell proliferation, and tumor growth in vivo [38]. Moreover, small molecule LRP6 inhibitors were able to inhibit human breast and prostate cancer cell proliferation [20, 21]. We recently demonstrated that the recombinant Mesd protein and its C-terminal region peptide, two universal inhibitors of LRP6, markedly inhibited Wnt/β-catenin signaling in prostate and breast cancer cells, and suppressed cancer cell proliferation in vitro and tumor growth in vivo [41–43]. In the present study, we demonstrated for the first time that rottlerin is a potent inhibitor of Wnt/β-catenin signaling by inducing LRP6 degradation in prostate and breast cancer cells. We also found that effects of rottlerin on LRP6 expression occurred at concentrations comparable to those required for inhibiting cancer cell proliferation. Our results indicate that the anti-cancer activity of rottlerin is associated with its inhibitory effects on Wnt/LRP6 signaling.
It was recently demonstrated that the cell-surface transmembrane E3 ubiquitin ligase zinc and ring finger 3 (ZNRF3) and its homologue ring finger 43 (RNF43) are negative feedback regulators of Wnt/β-catenin signaling, and that ZNRF3 is associated with the Wnt receptor complex, and inhibits Wnt/β-catenin signaling by promoting the turnover of Fzd and LRP6 [44, 45]. Furthermore, Park et al. reported that Rap2, a member of the Ras family of small GTP-binding proteins, is essential for the stabilization of LRP6 [46]. In addition, the transmembrane proteins Kremen1 and Kremen2 are able to form a ternary complex with LRP6 and the Wnt inhibitor Dickkopf-1 to induce rapid endocytosis and removal of LRP6 from the cell surface [47]. In the present study, we showed that rottlerin suppressed LRP6 expression through promoting LRP6 degradation. However, the exact mechanism underlying rottlerin-mediated LRP6 turnover remains to be elucidated in the future studies.
Deregulation of the mTORC1 signaling pathway has been found in a variety of human cancers [4–6]. Activation of Wnt/β-catenin signaling can enhance mTORC1 signaling [7–15]. Particularly, LRP6+/− mice displayed a diminished Wnt-dependent mTORC1 activity in adipose tissues [14]. Moreover, LRP6 depletion in prostate cancer cells suppressed mTORC1 signaling in prostate cancer cells [15]. In the present study, we demonstrated that rottlerin is a potent inhibitor of mTORC1 signaling in prostate and breast cancer cells. Our results are consistent with earlier studies, which demonstrated that rottlerin was able to stimulate autophagy and inhibited mTORC1 signaling [48, 49]. The inhibitory effect of rottlerin on mTORC1 signaling in prostate and breast cancer cells could be associated with its effect on LRP6 expression. However, more studies are required to dissect molecule mechanism underlying rottlerin-mediated inhibition of mTORC1 signaling in details in the future.
Natural compounds may represent effective candidate molecules for drug discovery. Rottlerin is a polyphenolic compound isolated from Mallotus philipinensis (Euphorbiaceae). Rottlerin displays its anti-cancer activities by affecting several cell machineries involved in survival, apoptosis, autophagy, and invasion [50–57]. Furthermore, rottlerin is able to potentiate the cytotoxicity towards tumor cells of different anticancer agents, including sorafenib, imatinib camptothecin, and tumor necrosis factor-related apoptosis-inducing ligands, in several cancer models [58–62]. Rottlerin was original reported as a protein kinase C-δ (PKCδ) inhibitor. However, it has now been unequivocally believed that rottlerin does not inhibit this kinase [16]. Rottlerin can trigger anticancer activities through PKCδ-independent pathways [16, 17]. In the present study, we showed that rottlerin is a potent dual inhibitor of Wnt/β-catenin and mTORC1 signaling in prostate and breast cancer cells, and exhibits an excellent anti-cancer activity with IC50 values around 1 μM. Rottlerin is not an FDA approved drug but it displays a low toxicity profile in rodents [63]. All together, these studies provide evidence for a potential value of rottlerin in development of new therapeutic strategies for cancer treatment.
Highlights.
Rottlerin blocks Wnt/β-catenin signaling induced by Wnt3A and LRP6 in HEK293 cells.
Rottlerin suppresses LRP6 expression and phosphorylation in cancer cells.
Rottlerin enhances LRP6 degradation.
Rottlerin blocks both Wnt/β-catenin and mTORC1 signaling in cancer cells.
The anti-cancer activity of rottlerin is linked to its effects on LRP6 signaling.
Acknowledgments
We are grateful to Dr. Christof Niehrs (Deutsches Krebsforschungszentrum, Heidelberg, Germany) for providing LRP6 cDNA, Dr. Gail Johnson (University of Rochester) for providing GST-E Cadherin cDNA, and Dr. Randall T. Moon (University of Washington) for providing the Super8XTOPFlash luciferase construct. This work was supported by grants from the National Institutes of Health RO1CA124531 and R21CA182056 (to Y.L.).
Abbreviations
- 4E-BP1
eukaryotic initiation factor 4E binding protein 1
- AMPK
5′-AMP-activated protein kinase
- APC
adenomatous polyposis coli
- CK1
casein kinase 1
- CM
conditioned medium
- eIF4E
eukaryotic initiation factor 4E
- GSK3β
glycogen synthase kinase-3β
- FBS
fetal bovine serum
- Fzd
Frizzled
- LRP6
the low-density lipoprotein receptor-related protein-6
- mTORC1
the mammalian target of rapamycin complex 1 (mTORC1)
- P70S6K
P70 S6 kinase
- PKCδ
protein kinase C-δ
- TCF/LEF
T-cell factor/lymphoid enhancing factor
- TNBC
triple negative breast cancer
- ZNRF3
E3 ubiquitin ligase zinc and ring finger 3
Footnotes
Disclosures
No conflicts to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clevers H, Nusse R. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Polakis P. EMBO J. 2012;31:2737–2746. doi: 10.1038/emboj.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anastas JN, Moon RT. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 4.Guertin DA, Sabatini DM. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Garcia JA, Danielpour D. Mol Cancer Ther. 2008;7:1347–1354. doi: 10.1158/1535-7163.MCT-07-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dancey J. Nat Rev Clin Oncol. 2010;7:209–219. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 7.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BQ, Guan KL. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 8.Fujishita T, Aoki K, Lane HA, Aoki M, Taketo MM. Proc Natl Acad Sci U S A. 2008;105:13544–13549. doi: 10.1073/pnas.0800041105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J, Zhang Y, Bersenev A, O’Brien WT, Tong W, Emerson SG, Klein PS. J Clin Invest. 2009;119:3519–3529. doi: 10.1172/JCI40572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castilho RM, Squarize CH, Chodosh LA, Williams BQ, Gutkind JS. Cell Stem Cell. 2009;5:279–289. doi: 10.1016/j.stem.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, Fu W, Zhang J, Wu W, Zhang X, Chen YG. Nat Cell Biol. 2010;12:781–790. doi: 10.1038/ncb2082. [DOI] [PubMed] [Google Scholar]
- 12.Hagenmueller M, Malekar P, Fieger C, Weiss CS, Buss SJ, Wolf D, Katus HA, Hardt SE. FEBS Lett. 2010;584:74–80. doi: 10.1016/j.febslet.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 13.Kwan HT, Chan DW, Cai PC, Mak CS, Yung MM, Leung TH, Wong OG, Cheung AN, Ngan HY. PLoS One. 2013;8:e53597. doi: 10.1371/journal.pone.0053597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu W, Singh R, Choi CS, Lee HY, Keramati AR, Samuel VT, Lifton RP, Shulman GI, Mani A. J Biol Chem. 2012;287:7213–7223. doi: 10.1074/jbc.M111.286724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahir SA, Yang G, Goltsov A, Song KD, Ren C, Wang J, Chang W, Thompson TC. Cancer Res. 2013;73:1900–1911. doi: 10.1158/0008-5472.CAN-12-3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soltoff SP. Trends Pharmacol Sci. 2007;28:453–458. doi: 10.1016/j.tips.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Maioli E, Torricelli C, Valacchi G. Scientific World Journal. 2012;2012:350826. doi: 10.1100/2012/350826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Lu W, He X, Schwartz AL, Bu G. Oncogene. 2004;23:9129–9135. doi: 10.1038/sj.onc.1208123. [DOI] [PubMed] [Google Scholar]
- 19.Lu W, Kim KA, Liu J, Abo A, Feng X, Cao X, Li Y. FEBS Lett. 2008;582:643–650. doi: 10.1016/j.febslet.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 20.Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. PLoS One. 2011;6:e29290. doi: 10.1371/journal.pone.0029290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu W, Lin C, King TD, Chen H, Reynolds RC, Li Y. Cell Signal. 2012;24:2291–2296. doi: 10.1016/j.cellsig.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan D, Wiesmann M, Rohan M, Chan V, Jefferson AB, Guo L, Sakamoto D, Caothien RH, Fuller JH, Reinhard C, Garcia PD, Randazzo FM, Escobedo J, Fantl WJ, Williams LT. Proc Natl Acad Sci U S A. 2001;98:14973–14978. doi: 10.1073/pnas.261574498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER. J Biol Chem. 2002;277:21657–21665. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- 25.Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bazuine M, van der Zon GC, van de Ven R, van den Broek PJ, Antonie Maassen J. Biochem Pharmacol. 2004;68:105–112. doi: 10.1016/j.bcp.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 27.Kojima K, Motoshima H, Tsutsumi A, Igata M, Matsumura T, Kondo T, Kawashima J, Ichinose K, Furukawa N, Inukai K, Katayama S, Goldstein BJ, Nishikawa T, Tsuruzoe K, Araki E. Biochem Biophys Res Commun. 2008;376:434–438. doi: 10.1016/j.bbrc.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Kumar D, Shankar S, Srivastava RK. Cancer Lett. 2014;343:179–189. doi: 10.1016/j.canlet.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. Proc Natl Acad Sci U S A. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tetsu O, McCormick F. Nature. 1999;39:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 31.Averous J, Fonseca BD, Proud CG. Oncogene. 2008;27:1106–1113. doi: 10.1038/sj.onc.1210715. [DOI] [PubMed] [Google Scholar]
- 32.Kim PJ, Plescia J, Clevers H, Fearon ER, Altieri DC. Lancet. 2003;362:205–209. doi: 10.1016/S0140-6736(03)13910-4. [DOI] [PubMed] [Google Scholar]
- 33.Ma H, Nguyen C, Lee KS, Kahn M. Oncogene. 2005;24:3619–3631. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- 34.Vaira V, Lee CW, Goel HL, Bosari S, Languino LR, Altieri DC. Oncogene. 2007;26:2678–2684. doi: 10.1038/sj.onc.1210094. [DOI] [PubMed] [Google Scholar]
- 35.King TD, Suto MJ, Li Y. J Cell Biochem. 2012;113:13–18. doi: 10.1002/jcb.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC. Proc Natl Acad Sci U S A. 2000;97:4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindvall C, Zylstra CR, Evans N, West RA, Dykema K, Furge KA, Williams BO. PLoS One. 2009;4:e5813. doi: 10.1371/journal.pone.0005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu CC, Prior J, Piwnica-Worms D, Bu G. Proc Natl Acad Sci U S A. 2010;107:5136–5141. doi: 10.1073/pnas.0911220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC, Deng X, Chen L, Kim CC, Lau S, Somlo G, Yen Y. Oncogene. 2011;30:4437–4446. doi: 10.1038/onc.2011.145. [DOI] [PubMed] [Google Scholar]
- 40.Liu W, Xing F, Iiizumi-Gairani M, Okuda H, Watabe M, Pai SK, Pandey PR, Hirota S, Kobayashi A, Mo YY, Fukuda K, Li Y, Watabe K. EMBO Mol Med. 2012;4:93–108. doi: 10.1002/emmm.201100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu W, Liu CC, Thottassery JV, Bu G, Li Y. Biochemistry. 2010;49:4635–4643. doi: 10.1021/bi1001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin C, Lu W, Zhai L, Bethea T, Berry K, Qu Z, Waud WR, Li Y. FEBS Lett. 2011;585:3120–3125. doi: 10.1016/j.febslet.2011.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin C, Lu W, Zhang W, Londoño-Joshi AI, Buchsbaum DJ, Bu G, Li Y. PLoS One. 2013;8:e58102. doi: 10.1371/journal.pone.0058102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, Mao X, Ma Q, Zamponi R, Bouwmeester T, Finan PM, Kirschner MW, Porter JA, Serluca FC, Cong F. Nature. 2012;485:195–200. doi: 10.1038/nature11019. [DOI] [PubMed] [Google Scholar]
- 45.Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, Clevers H. Nature. 2012;488:665–669. doi: 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- 46.Park DS, Seo JH, Hong M, Choi SC. Role of the Rap2/TNIK kinase pathway in regulation of LRP6 stability for Wnt signaling. Biochem Biophys Res Commun. 2013;43:338–343. doi: 10.1016/j.bbrc.2013.05.104. [DOI] [PubMed] [Google Scholar]
- 47.Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. Nature. 2002;417:664–667. doi: 10.1038/nature756. [DOI] [PubMed] [Google Scholar]
- 48.Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR, Roberge M. PLoS One. 2009;4:e7124. doi: 10.1371/journal.pone.0007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh BN, Kumar D, Shankar S, Srivastava RK. Biochem Pharmacol. 2012;84:1154–1163. doi: 10.1016/j.bcp.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 50.Kim EH, Kim SU, Choi KS. Oncogene. 2005;24:838–849. doi: 10.1038/sj.onc.1208241. [DOI] [PubMed] [Google Scholar]
- 51.Song KS, Kim JS, Yun EJ, Kim YR, Seo KS, Park JH, Jung YJ, Park JI, Kweon GR, Yoon WH, Lim K, Hwang BD. Autophagy. 2008;4:650–658. doi: 10.4161/auto.6057. [DOI] [PubMed] [Google Scholar]
- 52.Lim JH, Park JW, Kim SH, Choi YH, Choi KS, Kwon TK. Apoptosis. 2008;13:1378–1385. doi: 10.1007/s10495-008-0264-z. [DOI] [PubMed] [Google Scholar]
- 53.Lim JH, Park JW, Choi KS, Park YB, Kwon TK. Carcinogenesis. 2009;30:729–736. doi: 10.1093/carcin/bgn265. [DOI] [PubMed] [Google Scholar]
- 54.Ohno I, Eibl G, Odinokova I, Edderkaoui M, Damoiseaux RD, Yazbec M, Abrol R, Goddard WA, 3rd, Yokosuka O, Pandol SJ, Gukovskaya AS. Liver Physiol. 2010;298:G63–73. doi: 10.1152/ajpgi.00257.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin CJ, Lin CY, Chen Y, Huang SH, Wang SM. J Cell Biochem. 2010;110:428–437. doi: 10.1002/jcb.22555. [DOI] [PubMed] [Google Scholar]
- 56.Valacchi G, Pecorelli A, Sticozzi C, Torricelli C, Muscettola M, Aldinucci C, Maioli EE. Chem Biol Drug Des. 20111;77:460–470. doi: 10.1111/j.1747-0285.2011.01121.x. [DOI] [PubMed] [Google Scholar]
- 57.Lim JH, Woo SM, Min KJ, Park EJ, Jang JH, Seo BR, Iqbal T, Lee TJ, Kim SH, Choi YH, Kwon TK. Chem Biol Interact. 2012;197:1–7. doi: 10.1016/j.cbi.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 58.Tillman DM, Izeradjene K, Szucs KS, Douglas L, Houghton JA. Cancer Res. 2003;63:5118–5125. [PubMed] [Google Scholar]
- 59.Jane EP, Premkumar DR, Pollack IF. J Pharmacol Exp Ther. 2006;319:1070–1080. doi: 10.1124/jpet.106.108621. [DOI] [PubMed] [Google Scholar]
- 60.Kurosu T, Tsuji K, Kida A, Koyama T, Yamamoto M, Miura O. Oncogene. 2007;26:2975–2987. doi: 10.1038/sj.onc.1210117. [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, Liu N, Liu S, Liu Y, Zheng D. J Cell Biochem. 2005;96:522–532. doi: 10.1002/jcb.20535. [DOI] [PubMed] [Google Scholar]
- 62.Hsu JL, Ho YF, Li TK, Chen CS, Hsu LC, Guh JH. Biochem Pharmacol. 2012;84:59–67. doi: 10.1016/j.bcp.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG. J Pharmacol Expt Ther. 2007;322:913–922. doi: 10.1124/jpet.107.124669. [DOI] [PubMed] [Google Scholar]