

Abstract
Cell migration is of vital importance in many biological processes, including organismal development, immune response and development of vascular diseases. For instance, migration of vascular smooth muscle cells from the media to intima is an essential part of the development of atherosclerosis and restenosis after stent deployment. While it is well characterized that cells use actin polymerization at the leading edge to propel themselves to move on two-dimensional substrates, the migration modes of cells in three-dimensional matrices relevant to in vivo environments remain unclear. Intracellular tension, which is created by myosin II activity, fulfils a vital role in regulating cell migration. We note that there is compelling evidence from theoretical and experimental work that myosin II accumulates at the cell rear, either isoform-dependent or -independent, leading to three-dimensional migration modes driven by posterior myosin II tension. The scenario is not limited to amoeboid migration, and it is also seen in mesenchymal migration in which a two-dimensional-like migration mode based on front protrusions is often expected, suggesting that there may exist universal underlying mechanisms. In this review, we aim to shed some light on how anisotropic myosin II localization induces cell motility in three-dimensional environments from a biomechanical view. We demonstrate an interesting mechanism where an interplay between mechanical myosin II recruitment and biochemical myosin II activation triggers directional migration in three-dimensional matrices. In the case of amoeboid three-dimensional migration, myosin II first accumulates at the cell rear to induce a slight polarization displayed as a uropod-like structure under the action of a tension-dependent mechanism. Subsequent biochemical signalling pathways initiate actomyosin contractility, producing traction forces on the adhesion system or creating prominent motile forces through blebbing activity, to drive cells to move. In mesenchymal three-dimensional migration, cells can also take advantage of the elastic properties of three-dimensional matrices to move. A minor myosin isoform, myosin IIB, is retained by relatively stiff three-dimensional matrices at the posterior side, then activated by signalling cascades, facilitating prominent cell polarization by establishing front–back polarity and creating cell rear. Myosin IIB initiates cell polarization and coordinates with the major isoform myosin IIA-assembled stress fibres, to power the directional migration of cells in the three-dimensional matrix.
Keywords: myosin II, actomyosin tension, cell polarization, three-dimensional migration, matrix stiffness
1. Introduction
Cell migration is of vital importance in many biological processes, including organismal development, immune response and development of vascular disease [1–3]. Cell migration, a highly orchestrated multi-step process, begins with the transduction of the external signal through the stimulation of membrane receptors to a series of remodelling events in the cytoskeleton [4], giving the cell a polarized morphology. Most studies on cell migration have been focused on the biochemical signalling that functions to modulate cell migration significantly, while much less attention has been given to the role of mechanical factors [5]. In essence, mechanical factors associated with cellular behaviour, including actomyosin tension, external mechanical loading, such as shear stresses and cyclic stretch, and elastic properties of the matrix, fulfil a vital role in regulating cell migration through mechanical or biochemical pathways.
Among them, intracellular tension is one of the most prominent [6–8]. The tension is created in actomyosin structures at both leading and trailing edges of the cell, stress fibres of non-muscle cells and sarcomere-like structures in smooth muscle cells owing to the active interaction between actin filaments and myosin II aroused by either chemical or mechanical stimuli. Actomyosin tension is essential in translocating the cell body and disassembling of focal adhesions at the trailing edge of the cell during cell migration [9]. Cell experiments show mutant Dictyostelium in the absence of actomyosin tension migrate more slowly than wild-type strains [10]. It has been suggested that cells try to maintain a ‘tensional homoeostasis’ within the cell body in response to mechanical loading [11,12].
Myosin II activity fulfils its crucial role in cell migration by regulating adhesions and polarity [13]. In previous works, we demonstrated the important contribution to cell migration and adhesion by cytoskeletal reorganization associated with focal adhesions assembly when the cells overexpressed Id1 [14], stimulated by various concentrations of oxLDLs (oxidized low-density lipoprotein) at static conditions [15–17], placed on surfaces with different degrees of wettability [18,19] or LDL treatment under shear stress [20,21]. It is well established that cells use actin polymerization coupled with integrin-mediated adhesion to generate lamellipodial protrusions at the cell front to migrate on two-dimensional substrates [2]. Driven by polymerization of actin filaments, cells that migrate first become polarized and extend protrusive structures, thin sheet-like lamellipodia (0.1–0.2 μm) and thin finger-like filopodia (0.1–0.3 μm), at the leading edge of the cells towards chemical stimulus and mechanical cues [4,22]. Weak nascent adhesions are formed under the lamellipodium owing to the binding of integrins to the matrix, presumably to provide just enough resistance to traction forces applied to the matrix of this region [23,24]. Focal adhesions composed of integrins, kinases like focal adhesion kinase (FAK), and actin-binding proteins such as talin, vinculin, paxillin and α-actinin respond dynamically to external stimuli [25]. Myosin II is not necessarily involved in forming the nascent adhesions, but can impact the net rate of the protrusions [26–28]. Next, the nucleus and cell body are moved forward by the tension created by actomyosin structures, stress fibres, which span the whole cell body and are anchored by focal adhesions [22]. As a feedback mechanism, the nascent adhesions are further promoted by actomyosin tension and transformed into elongated mature focal adhesions, providing strong mechanical attachment points to propel the cell by more prominent traction forces [29,30]. Last, the cell retracts its trailing edge by destabilizing and releasing focal adhesions of this region. Thus, the whole migration process is accomplished. The full process of cell migration is depicted in figure 1.
Figure 1.

Cell migration is a highly orchestrated multi-step process. (a) Driven by polymerization of actin filaments, cells that migrate first become polarized and extend protrusive structures; (b) weak nascent adhesions are formed under the lamellipodium owing to the binding of integrins to the extracellular matrix; (c) the nucleus and cell body are moved forward by the tension created by stress fibres; and (d) the cell retracts its trailing edge by destabilizing and releasing focal adhesions of this region. (e) Integrins are linked to the actin cytoskeleton through multiple focal adhesion molecules, including kinases like FAK, and actin-binding proteins such as talin, vinculin, paxillin and α-actinin. (Online version in colour.)
Cells in vivo typically move in three-dimensional matrices and this environment poses serious challenges for the cells to migrate. Cells in three-dimensional matrices are encountered by strong mechanical matrix resistance [31]. They exhibit fewer stress fibres, weaker adhesion or multiple front pseudopods [2,32–34], sometimes display a round morphology without apparent front and rear, thus cannot support a broad front protrusion-driven migration mode seen in two-dimensional cases. The migration mechanism in the three-dimensional environment remains unclear. Cells migrating in three-dimensional matrices adopt either a mesenchymal or amoeboid mode [35]. In mesenchymal migration, which bears some resemblance to lamellipodia-driven two-dimensional migration, cells adopt elongated morphology and use percellular matrix proteolysis to extend front actin filaments-based protrusions to move themselves directionally ahead in an adhesion-dependent manner [36–38]. In amoeboid mode typically seen in invasive tumour cells, leucocytes [39] and fibroblasts [34], cells exhibit rounded morphologies and use different strategies, contraction-based membrane blebbing together with myosin II-enriched cell rear, to migrate [35,40–43]. Matrix degradation by protease is not needed in the movement. It has been suggested that mesenchymal and amoeboid migration are powered by actin polymerization and actomyosin contraction, respectively [42,44].
Various biomimetic three-dimensional matrices within which cells embed include gels based on native matrix components (Matrigel, fibril, collagen, etc.), synthetic materials (poly(ethylene glycol) and biological constructs (dermal explants). These materials assemble into various fibre structures differing in mechanical properties, such as stiffness, fibre diameter and pore size. Experimental investigations suggest that cells migrating in three-dimensional Matrigel or collagen gels displayed characteristic amoeboid modes and myosin II was recruited towards the cell rear under the action of a tension-dependent mechanism. A recent study by Petrie et al. [34] suggested that modes of three-dimensional migration were modulated by an association of actomyosin contractility and mechanical properties of the matrix. They found RhoA mediated high actomyosin tension together with linear elasticity of dermal explants or cell-derived matrix switched cells to a migration mode characterized by front blunt, cylindrical protrusions, e.g. lobopodia. Lobopodia-based migration did not require the canonical polarization of PIP3, Rac1 and Cdc42 activity typically seen in two-dimensional migration and was independent of lamellipodia formation. The migration mode was determined intrinsically by myosin II activity, consistent with previous actomyosin contractility-based three-dimensional migration modes [35,39]. However, the localization of myosin II remains to be elaborated.
Cells exhibit ‘wisdom’ to regulate their behaviour in response to external perturbations [45]. Cells of the same type use different modes to migrate depending on the elastic properties of the matrix and biochemical signalling factors [46,47]. It has been suggested that cell migration should be considered as continuous modes balanced among adhesion, contractility and mechanical properties of the matrix, rather than strictly separate mechanisms [40]. We note that there is compelling evidence from recent theoretical and experimental work that myosin II accumulates at the cell rear, either isoform-dependent or -independent, leading to directional three-dimensional migration driven by posterior myosin II contractility. The scenario is not limited to amoeboid migration, and it is also seen in mesenchymal migration in which lamellipodia-based motility is often expected, suggesting there may exist universal underlying mechanisms. In this review, we aim to shed some light on how anisotropic myosin II localization induces cell motility in three-dimensional environments from a biomechanical view. We demonstrate an interesting mechanism where the interplay between mechanical myosin II recruitment and biochemical myosin II activation triggers directional migration in the three-dimensional matrix.
2. Isoforms, structures and functions of myosin II
Actomyosin tension created by myosin II activity, one of the foremost sources of intracellular forces, plays a pivotal role in several fundamental cellular processes, including cell migration, division and morphogenesis [12,48–51]. Myosin II molecules can move towards the plus (barbed) end of actin filaments via the hydrolysis of adenosine triphosphate at the myosin head, propelling sliding between them and creating tension [13,50]. This well-established paradigm of tension creating by actomyosin (the protein complex composed of actin and myosin), termed cross-bridge cycling, is recognized as the fundamental contractile apparatus in almost all forms of muscle cells and non-muscle cells [50].
In addition to smooth muscle, cardiac and skeletal myosin II, there are three isoforms of non-muscle myosin II: myosin IIA (MIIA), myosin IIB (MIIB) and myosin IIC (MIIC). Both MIIA and MIIB are typically seen in most mammalian cells [12,13]. They fulfil different but overlapping functions in cell migration. MIIA is situated all through the cell, even in protrusions, but not including the leading edge. It plays a crucial role in retracting cell edges and adhesion maturation at the leading edge [26,52]. MIIB localizes in the central and rear regions, but not in protrusions [53]. It helps to determine the overall morphology of the cell as well as facilitate adhesion maturation as evident from experimental observations that cells with inhibited MIIB activity exhibit small adhesions, round morphology and extend multiple protrusions [26,54]. The release of adenosine diphosphate from MIIB is rather slow owing to its high affinity for adenosine diphosphate when compared with other myosin isoforms [55], making MIIB well suited for creating tension for longer duration and less energy cost than MIIA [13]. This may be an indication of MIIB's role in prolonged cell migration in chronic diseases like atherosclerosis. The intracellular localization of both isoforms depends on the self-assembly regions in the coiled-coil C terminus [13,56]. The function of MIIC remains to be explored. Unlike the other two non-muscle isoforms, this isoform is not expressed in fetal tissues [57].
A myosin II molecule is constituted of three parts: two myosin heavy chains (MHCs) of 230 kDa which are composed of head and tail domains with the latter appearing as a coiled-coil morphology, holding the two heavy chains together, containing four myosin light chains (MLCs); two 20 kDa regulatory light chains (RLCs), which bind the heavy chains in the neck region; and two 17 kDa essential light chains (ELCs), which function to stabilize the heavy chain structure. A representation of the myosin II structure is shown in figure 2. Previous studies have determined a relatively clear cloud of signalling cascades in regulating actomyosin activity. The actomyosin contraction system is regulated in at least two independent pathways, a Ca2+-dependent pathway and a Rho-dependent pathway that does not depend on Ca2+ [4,41,58]. The former leads to MLC kinase (MLCK) activation and subsequent MLC phosphorylation and the latter takes effect either by direct MLC phosphorylation or by blocking MLC phosphatase (MLCP) phosphorylation. Moreover, myosin II can also be regulated by other regulatory proteins like S100A4 and inhibitory phosphorylation at the heavy chains [12]. The signalling pathways by which myosin II activity is regulated are shown in figure 3.
Figure 2.
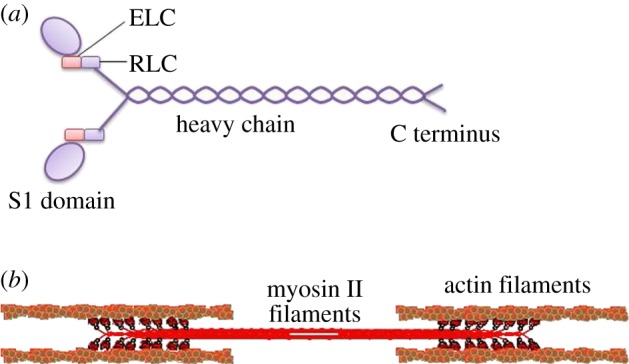
Molecular structure of myosin II and tension-generating mechanism. (a) Molecular constituents of myosin II: two 230 kDa MHCs, two 20 kDa RLCs and two 17 kDa ELCs. S1 domain is a fragment of myosin II that comprises the motor domain and neck. (b) Myosin II molecules walk along actin filaments and propel the sliding of actin filaments to generate tension. (Online version in colour.)
Figure 3.
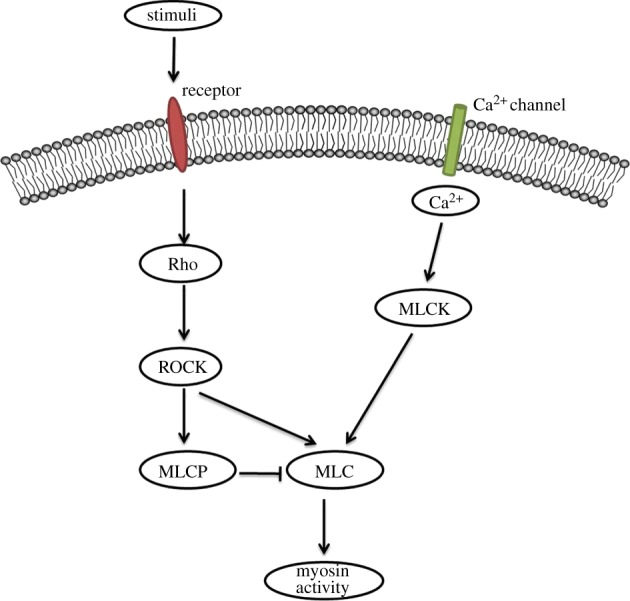
Myosin II is regulated through two independent pathways, a Ca2+-dependent pathway and a Rho-dependent pathway. The former leads to MLCK activation and subsequent MLC phosphorylation and the latter takes effect either by direct MLC phosphorylation or by blocking MLCP phosphorylation. (Online version in colour.)
3. Tension-dependent myosin II recruitment towards the cell rear
Myosin II functions as the most prominent intracellular tension contributor of actomyosin structures through biochemical signalling [12]. Recently, this well-recognized cloud has been enriched by recent findings that mechanical tension can feed back to regulate myosin II binding to actin filaments. Accumulated data suggest that myosin II motors prefer to bind to stretched actin filaments bearing large tension. Experimental examinations performed by Fernandez-Gonzalez et al. [59] showed that myosin II preferred to stabilize at the regions of the cortex in Drosophila cells bearing increased tension, and conversely relieving tension resulted in a rapid loss of myosin II. Ren et al. [60] and later Luo et al. [49] also reported this tension-dependent myosin II accumulation at Dictyostelium plasma membrane with the involvement of cortexillin I. Uyeda et al. [61] first demonstrated that green fluorescent protein-fused myosin II motor domain (S1) probes preferred to locate in the posterior portion and cleavage furrows of fixed fibroblasts, with actin filaments bearing more tension owing to the interactions with myosin II. Similar myosin II enrichment in the cortex can also be observed by applying external stretching forces to the portion using a microcapillary [61]. This may account for the affinity of myosin II filaments assembly to lamellipodia bundles and lateral concave arcs of cells, leading to increased myosin II accumulation and greater tension in these structures through subsequent biochemical activity [29].
Several studies suggest that this tension-dependent myosin recruitment is aroused by the correlated change in MLC phosphorylation. However, this hypothesis is inconsistent with the results that the level of MLC phosphorylation does not change [29,62,63] or even increases [64,65] in the presence of blebbistatin, which is an effective myosin II inhibitor. Previous experimental results also suggest that blebbistatin inhibited myosin II activity by inducing a swelling conformation in S1 instead of the classical MLC signalling cascades [66]. A more reasonable explanation has been given by Uyeda et al. [61]. They suggest that this tension-dependent myosin recruitment depends on mechanical cues rather than the signalling proteins. They demonstrate that, in the experiment, neither cortexillin I nor PTEN, two signalling proteins which have been reported in the recruitment of myosin II to stretched cortex [60,67], nor MLC phosphorylation participates in the process. The underlying mechanical mechanism given by them is: stretching causes the untwisting of the filament helix (demonstrated recently by molecular dynamics simulations [68]) and binding of skeletal S1 to actin filaments could slightly untwist the helix of the latter [69], making it plausible that S1 prefers to bind to stretched actin filaments. This explanation is supported in the theoretical work by Luo et al. [49]. Assuming that the binding of myosin II induces changes in twisting degree of the helix in actin filaments in Monte Carlo simulations, they capture the experimental observations of tension-dependent myosin II accumulation.
Mechanosensory focal adhesion components, such as talin, vinculin and α-actinin, are also expected to play a role in the tension-dependent myosin II recruitment. Cells orchestrate these adhesion molecules precisely to fulfil mechanotransduction events. It has been well established that integrins initially link to actin cytoskeleton through talin, which bears actomyosin tension aiding the formation of nascent focal adhesions [70,71]. Previous results found that stretching talin exposes new sites for vinculin binding, and this would support the idea that myosin II prefers to bind stretched actin filaments [72]. The scenario is enriched by a most recent work suggesting that α-actinin competes with talin to bind cytoplasmic domain of β3 integrins [73]. It was suggested that talin initially binds to β3 integrins and triggers the formation of nascent adhesions, then α-actinin replaces talin when these focal adhesions are mature.
The tension-dependent myosin II accumulation is supported by the latest findings by Iwadate et al. [74]. They found Dictyostelium cells accumulated myosin II, assembled into force-responding filamentous structures, at both stretching boundaries. Accumulated myosin II filaments induced directional migration of the cells, in which biochemical signalling-initiated actomyosin contraction was needed. The mechanism would enable cells to escape from external mechanical stimuli owing to enhanced contractility in the locally stretched portion of the cell cortex. Fragments of a fish keratocyte that contain the actomyosin machinery but lack essential cell components provide a remarkable platform to test this. When a discoid stationary fragment of a fish keratocyte was pushed by the tip of a microneedle, the stimulated fragment started to move unidirectionally away [75]. This may be owing to the stretch of the cell cortex enhancing the contractility at the site of deformation by the recruitment of myosin II filaments [61].
4. Myosin II's back accumulation commits cells into a polarized, moving state
The tension-dependent recruitment of myosin II makes a significant contribution to the molecules’ initial asymmetric accumulation in cells ready to move. Actin filaments at the leading and trailing edges of cells ready to move exhibit a difference in contact with membrane (front) or intracellular portion of focal adhesions (rear)—pushing versus pulling. Thus actin filaments at the posterior of the cells are more stretched than those at the opposite edge. Owing to myosin II's affinity to stretched actin filaments, myosin II resides preferentially at the rear of migrating cells, creating a slight anisotropic distribution of tension. This aligns well with the experimental observations that pre-existing polarization cues induce steady-state cortical flow and destabilize the cortex [76,77]. Anisotropic myosin II distribution is essential in establishing cell polarization, which is considered as a prerequisite of cell movement [40]. Various migration modes differ by the degree of polarization. In general, mesenchymal three-dimensional migration adopts a severe polarization with apparent cell front and rear, while amoeboid three-dimensional migration displays just slight polarization with uropod-like structure.
Cell polarity (referring to the front–back polarity in cell migration), the ability of a cell to form a vectorial axis [78], is crucial to directional cell migration. Almost all migrating cells exhibit a certain degree of cell polarity, which assists in initiating cell movement. Cells need to polarize themselves to commit to a moving state [2,40,75]. MIIB's accumulation at the cell rear is essential for polarization by creating a distinct rear [53]. Experimental investigations show that myosin II null mutant Dictyostelium cells cannot be polarized in shape on sticky substrates, although the cells extend pseudopod protrusions at the front side [6]. Myosin II exhibits an asymmetric accumulation in moving cells. It has been observed in experiments that myosin II resides preferentially at the rear of migrating cells in contrast to the uniform distribution of myosin II in stationary cells [75,79,80]. Local application of calyculin A at one side of migrating keratocytes makes this side become a rear rich in myosin II, increasing the frequency of motility initiation [81]. Myosin II activation via MLC phosphorylation results in the formation of an extended rear and increased directional migration in fibroblasts [53].
A recent mesoscopic mean-field simulation (a simulation method based on mean-field theory on scales in the range of nano- to micrometres) of models constituted of filaments, motors and cell boundaries provides a direct validation of myosin distribution in static and moving cells [82]. The results demonstrate that, in the static state, myosin II distribution is rather isotropic throughout the cell, and the distribution of actin filaments is rotationally symmetric and relatively flat. However, in the moving state, both myosin II and actin filaments are distributed with a peak density at the back of the cells, in comparison with a vanishingly small density of myosin II and flat distribution of actin filaments at the front. Myosin II accumulation at the back of the cells essentially directs the cells to commit to a polarized, moving state, which is in accordance with the suggestion in Hawkins et al. [83] that myosin II transport first induces cortical instability and then leads to motility in a three-dimensional environment. In the absence of myosin II, the cells are driven into a symmetric, static state. The calculation results obtained by Du et al. [82] clearly show that if the initial myosin II distribution is not exactly isotropic the resulting tension anisotropy applies asymmetric force on the boundary, destabilizing the static state and giving rise to the locomotion of the cell.
5. Tension-dependent myosin II accumulation plays a role in amoeboid three-dimensional migration
Cells in vivo often move in a three-dimensional environment, like an extracellular matrix or a tissue of cells. Cell polarization is often visible in migrating cells and is considered as a prerequisite in cell migration, as it determines the moving direction [40]. However, tumour cells exhibit a round morphology and membrane blebs with a slight polarization—uropod at the rear, instead of significant polarization in shape—an apparent front or back. Recent investigations have confirmed the uropod, which is enriched in myosin II and exhibit high actomyosin tension, plays a crucial role in amoeboid cancer migration in three-dimensional matrices. Round amoeboid migration of cancer cells takes at least three distinct rear myosin II activity-driven modes distinguished by bleb features: rear, multiple and large hemispherical blebs [40,43,46]. Note that these modes are distinct from previously established amoeboid migration mode on two-dimensional substrates, which shares some features with two-dimensional mesenchymal migration. In the mode, front protrusions determine the moving direction and posterior myosin II activity detaches the cell rear [44,84–86].
A recent experiment found that MDA-MB-231 human breast cancer cells migrated in three-dimensional Matrigel with a rounded morphology and with myosin II accumulating at the cell rear, while the cells display neither lamellipodia nor bleb extensions at the front side [43]. Silencing Arp2/3, the main actin polymerization nucleator in lamellipodia, did not affect the velocity of migration. Whereas, inhibiting myosin II activity almost completely abolished cell movement. Accumulation of phosphorylated MLCs and intensive blebbing activity were observed at the cell rear, indicating actomyosin tension at the site. Strong actomyosin tension in the uropod generated traction forces on adhesions at the side and rear to power the cells to migrate. It was suggested that the rear blebbing activity was caused by the strong actomyosin tension at the site and did not contribute to the cell movement. Another investigation reported A375 melanoma cells migrating through three-dimensional Matrigel and collagen exhibited similar rounded shape with many membrane blebs and uropod rich in errin, myosin II and other proteins [40]. The migration of the cells was also driven by rear actomyosin tension bearing a resemblance to amoeboid migration of MDA-MB-231 cells, but the details differ. In brief, actomyosin tension created by rear myosin II activity endowed the cells with traction forces through bleb activity and directly determined the migration direction. Myosin II accumulated and was then activated by MLC phosphorylation at the cell rear, creating high actomyosin tension at the cell boundaries. Thus, hydrostatic pressure was exhibited on the cytoplasm, releasing many membrane blebs through the membrane except the rear uropod. Each bleb generated a small force, creating a net force to drive the cells to migrate towards the side opposite to the uropod.
Considering that the blebs represent merely a result from cortical tension concentrating in the rear region [40,43,87], the mechanism behind the appearance of rear blebs in MDA-MB-231 cell migration and many blebs in A375 cells is also similar to the third round amoeboid migration mode featured by a large, hemispherical bleb [42,88,89]. In that case, high actomyosin tension is maintained through the actin cortex mostly owing to increasing myosin II activity at the cell rear, creating hydrostatic pressure to form growing blebs towards the opposite side of the rear [40]. In essence, migration represented by large blebs is powered by the rear myosin II activity rather than the front bleb.
Tension-dependent recruitment of myosin II is expected to make a significant contribution to its asymmetric accumulation in migrating cells. Actin cytoskeleton in anterior and posterior regions exhibits a difference in contact with membrane—pushing versus pulling—thus actin filaments at the posterior of the cells are more stretched than those at the opposite side. Owing to myosin II's affinity to stretched actin filaments, myosin II resides preferentially at the rear of migrating cells, creating an anisotropic distribution of tension. The expected tension-dependent mechanism in cell migration is illustrated in figure 4. This aligns well with the experimental observations that pre-existing polarization cues induce steady-state cortical flow and destabilize the cortex [76,77]. Note that the mechanism would be most effective to accumulate myosin II at the rear for cells in three-dimensional environments. Actin filaments at the posterior of cells embedded in three-dimensional matrices would be stretched along a wide curved surface along the rear membrane, in comparison with a narrow flat surface along the rear substrate in two-dimensional cases. Thus, actin filaments in contact with the rear membrane are stretched in a much wider space through adhesion system than that in the two-dimensional scenario. The front side of the cell is strongly pushed against by the surrounding matrix. Even in the case of low-adhesion three-dimensional amoeboid migration, myosin II still prefers to localize at the free rear rather than the strongly pushed opposite side. Thus, myosin II will be effectively recruited towards the cell rear, helping to increase the degree of polarization.
Figure 4.
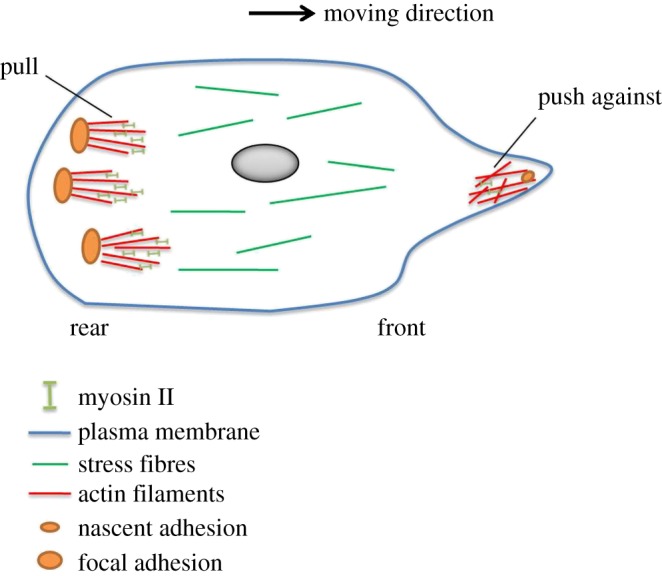
A tension-dependent myosin II recruitment at the cell rear. Asymmetric accumulation of myosin II at the leading and trailing edges. Actin cytoskeleton at the leading and trailing edges of cells ready to move exhibits a difference in contact with membrane (front) or intracellular portion of focal adhesions (rear)—pushing versus pulling—thus actin filaments at the posterior of the cells are more stretched than those at the opposite edge. Owing to myosin II's affinity to stretched actin filaments, actin cytoskeleton at the cell rear would recruit more myosin II motors and create larger tension to support large and mature focal adhesions, creating a slight anisotropic distribution of tension in front and rear regions. Dynamic and transient nascent adhesions can be found at the leading edge. The mechanism would be most effective to accumulate myosin II at the rear in three-dimensional matrices. In that scenario, actin filaments in contact with the rear membrane are stretched in a much wider space than that in two-dimensional cases. (Online version in colour.)
6. Isoform-dependent myosin II accumulation induces directional migration in three-dimensional matrices
6.1. Matrix stiffness affects three-dimensional migration
Matrix stiffness is a rather effective effector for cell behaviour and correlates with many physiological and pathological processes. It has been well established that cells sense the substrate stiffness by polarizing stress fibres and in doing so create strong cytoskeletal tension to propel the cell to move. Cells would readily polarize and migrate when cultured on a stiff matrix [90]. Native matrix is characterized by its intrinsic three-dimensional structure and low stiffness, which both affect cell migration significantly. Investigators have reported that the elastic properties of the matrix dictate the migration mode of cells in three-dimensional matrices [34]. It should be noted that the cytoskeletal polarization and migration fashion of cells on soft substrates and in three-dimensional matrices is distinct from the scenario typically seen on stiff two-dimensional substrates [91]. Polarization of stress fibres in soft two- or three-dimensional systems is always independent of the moving direction, leading to random migration without an obvious orientation [92,93]. Whereas cells cultured on a stiff two-dimensional substrate polarize stress fibres in the moving direction, exhibiting directional migration [91]. A matrix with stiffness gradient is needed to achieve directional migration, durotaxis, in a soft matrix system [93,94].
Cells often need to migrate directionally to fulfil physiological processes in response to directional cues, such as soluble molecules (chemotaxis), adhesion proteins (haptotaxis) and stiffness (durotaxis) [95]. Among these forms of directional migration, chemotaxis may be the most well understood and is most commonly seen in in vitro experiments, while haptotaxis and durotaxis are much less understood [65]. Durotaxis, the tendency of cells to migrate from soft to rigid matrix in the absence of any soluble biochemical stimuli, has been demonstrated in a large number of experimental observations [94,96–98]. Durotaxis seems to provide an excellent mechanical fashion for cells to move through the three-dimensional matrix in which they are trapped. It has been suggested that soft three-dimensional matrix microenvironment affects cell migration by the gradient in matrix stiffness [93]. Cells are able to migrate directionally if persistent spontaneous polarization is maintained, even if no external cues exist [99]. As we discussed in the previous sections, this can be fulfilled by inducing spontaneous polarization through accumulating myosin II at the cell rear, even if front protrusions are not so prominent as on two-dimensional surfaces.
6.2. Myosin IIB accumulation at the cell rear induces directional migration in three-dimensional matrices
In some cases like mesenchymal three-dimensional migration, cells take elongated morphology, and actin filaments are stretched at the cell rear within a narrow space. Tension-dependent mechanism plays a minor role in these cases while only a limited fraction of myosin II is recruited at the cell rear by the mechanism. This time myosin II exhibits isoform-dependent accumulation at the cell rear owing to the affection of self-assembly regions of the coiled-coil C terminus [13,56]. The elastic properties of the matrix have a significant impact on the migration mode in three-dimensional matrices. Experimental observations showed that as mesenchymal stem cells (MSCs) were deposited within three-dimensional matrices with stiffness gradient, the cells also exhibited durotaxis as seen in two-dimensional durotaxis [93]. The cells also migrated directionally in relatively stiff matrices, whereas this decayed to random migration [93]. MIIB only accumulates at the cell rear in relatively stiff three-dimensional matrices, yet is uniformly distributed within the cell body in soft three-dimensional matrices. It is most intriguing to note that anisotropic distribution of MIIB is promoted as matrix stiffness increases, in contrast with a slight anisotropic yet constant distribution of MIIA [93].
MSCs in three-dimensional matrices exhibit multiple lateral lamellipodia [93], which is indicative of random migration and a minor role of front protrusions in migration [2]. However, these cells still exhibit directional migration in a three-dimensional matrix, including stiff matrix and stiffness gradient cases [93]. Cells can move directionally to fulfil physiological roles even in a very soft three-dimensional matrix provided gradient stiffness exists in the matrix. It was suggested that the accumulation of minor isoform MIIB at the cell rear drives the cells to move directionally in three-dimensional matrices, including cases of stiff matrices and durotaxis [93]. Previous investigations have confirmed that MIIB functions to establish front–back polarity [26] and create cell rear [53]. MLC phosphorylation activates the accumulated MIIB, inducing prominent actomyosin tension at the cell rear, producing large, stable adhesions and defining the morphology of the rear [53]. Although showing no apparent accumulation at any side in three-dimensional matrices with variable stiffness, MIIA is also essential in cell migration by initiating the formation of stress fibres to which MIIB then binds and stabilizes [26]. Coordination between MIIA and MIIB is needed in directional three-dimensional migration.
Tension-dependent mechanism can make a contribution, at least a minor one, to the accumulation of both myosin II isoforms at the cell rear, although it seems difficult to distinguish the effect contributed by the tension-dependent mechanism from that contributed by structural features in the C terminus for MIIB accumulation at the cell rear. It has been observed that MIIA in MSCs in three-dimensional matrices exhibits a slight, persistent accumulation at the cell rear in the whole studied stiffness range [93]. Considering the MIIA back accumulation is stiffness-independent and is not intrinsically located there, it is the tension-dependent mechanism that may drive the slight MIIA accumulation.
7. Summary
It is said that cells have ‘wisdom’ to regulate their behaviour in response to external perturbations [45]. In the context of the three-dimensional environment, cells exhibit such wisdom to migrate and in doing so fulfil physiological and pathological roles. They use rear contraction apparatus to propel themselves ahead when the front protrusions typically seen on a two-dimensional surface are lacking. There is compelling evidence from theoretical and experimental work that myosin II accumulates at the cell rear, in either an isoform-dependent or a general way, leading to distinct three-dimensional migration modes dominated by myosin II contractility. As far as we are concerned, not much is known about the mechanism underlying these migration modes. Biomechanical factors, including a tension-dependent myosin II recruitment, the elastic properties of matrices and the instability and polarization of cells caused by anisotropic myosin II distribution, are expected to be key players in the process. In this paper, we attempt to decipher these migration modes from a biomechanical view by summarizing a large body of mathematical modelling and experimental work. Myosin II can be recruited at the cell rear under the action of a tension-dependent mechanism. Subsequent biochemical signalling pathways initiate actomyosin contractility, producing traction forces on the adhesion system or creating prominent motile forces through blebbing activity, to drive cells to move directionally. The full scenario is depicted in figure 5. This mechanism is most effective in amoeboid three-dimensional migration of tumour cells. These cells exhibit a rounded shape, with slight polarization displayed as a uropod-like structure. Abundant myosin II accumulates at the cell rear owing to actin filaments at the portion being stretched in a wide space in contact with the matrix through adhesions. Thus strong actomyosin tension is created by subsequent MLC phosphorylation to propel the cells to move towards the side opposite to the uropod. In mesenchymal three-dimensional migration, cells take an elongated morphology, only a limited fraction of myosin II is recruited at the cell rear and the tension-dependent mechanism plays a minor role. This time cells take advantage of the elastic properties of the three-dimensional matrix to recruit a certain minor isoform, MIIB, at the cell rear. MIIB activity induces prominent cell polarization by establishing front–back polarity and creating cell rear. This works in coordination with the polarization of MIIA-assembled stress fibres to direct the cells to commit to a polarized, moving state.
Figure 5.
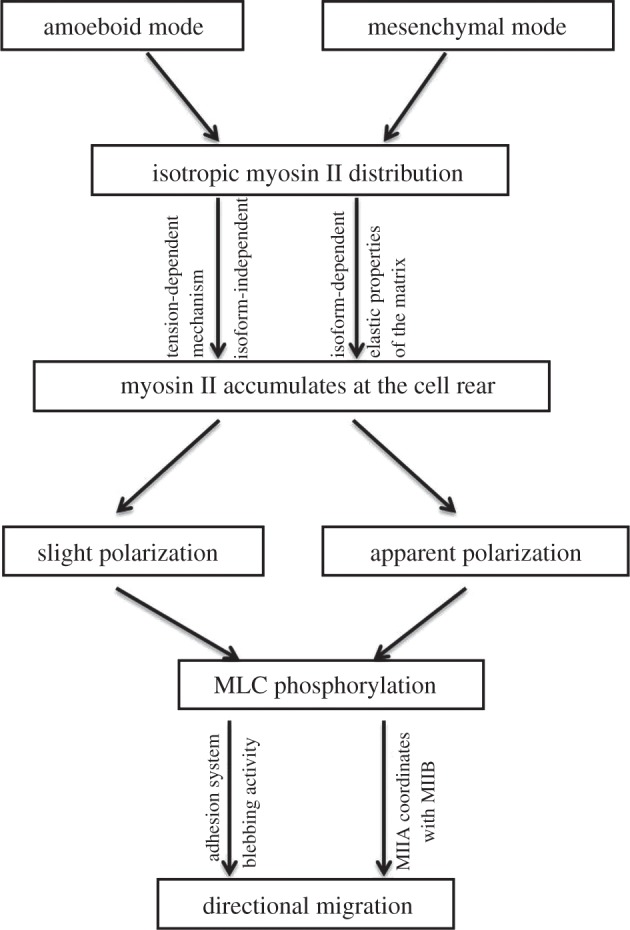
The full cloud of directional migration of cells in three-dimensional environments triggered by an interplay between mechanical myosin II recruitment and biochemical myosin II activation. In amoeboid three-dimensional migration, myosin II first accumulates at the cell rear to induce a slight polarization displayed as a uropod-like structure under the action of a tension-dependent mechanism. Subsequent biochemical signalling pathways initiate actomyosin contractility, producing traction forces on the adhesion system or creating prominent motile forces through blebbing activity, to drive cells to move. In mesenchymal three-dimensional migration, cells can also take advantage of the elastic properties of three-dimensional matrices to recruit myosin II. A minor myosin isoform, MIIB, is retained by relatively stiff three-dimensional matrices at the posterior side, then activated by MLC phosphorylation, creating apparent cell polarization. MIIB initiates cell polarization and coordinates with the major isoform MIIA-assembled stress fibres, to power the directional migration of cells in three-dimensional matrices.
Funding statement
This study was supported by grants from the National Natural Science Foundation of China (31370949, 11332003, 30970721) and the National Key Technology R&D Programme of China (2012BAI18B02) as well as the National ‘111 Plan’ Base (B06023) and the Public Experiment Center of State Bioindustrial Base (Chongqing), China.
References
- 1.Friedl P, Gilmour G. 2009. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445–457. ( 10.1038/nrm2720) [DOI] [PubMed] [Google Scholar]
- 2.Petrie RJ, Doyle AD, Yamada KM. 2009. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538–549. ( 10.1038/nrm2729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mogilner A. 2009. Mathematics of cell motility: have we got its number? J. Math. Biol. 58, 105–134. ( 10.1007/s00285-008-0182-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerthoffer WT. 2007. Mechanisms of vascular smooth muscle cell migration. Circ. Res. 100, 607–621. ( 10.1161/01.res.0000258492.96097.47) [DOI] [PubMed] [Google Scholar]
- 5.Keren K, Theriot JA. 2008. Biophysical aspects of actin-based cell motility in fish epithelial keratocytes. In Cell motility (ed. Lenz P.). Biological and Medical Physics, Biomedical Engineering, pp. 31–58. New York, NY: Springer. [Google Scholar]
- 6.Jay PY, Pham PA, Wong SA, Elson EL. 1995. A mechanical function of myosin II in cell motility. J. Cell Sci. 108, 387–393. [DOI] [PubMed] [Google Scholar]
- 7.Beningo KA, Hamao K, Dembo M, Wang Y-l, Hosoya H. 2006. Traction forces of fibroblasts are regulated by the Rho-dependent kinase but not by the myosin light chain kinase. Arch. Biochem. Biophys. 456, 224–231. ( 10.1016/j.abb.2006.09.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meili R, Alonso-Latorre B, del Alamo JC, Firtel RA, Lasheras JC. 2010. Myosin II is essential for the spatiotemporal organization of traction forces during cell motility. Mol. Biol. Cell 21, 405–417. ( 10.1091/mbc.E09-08-0703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. 2003. Cell migration: integrating signals from front to back. Science 302, 1704–1709. ( 10.1126/science.1092053) [DOI] [PubMed] [Google Scholar]
- 10.Wessels D, Soll DR, Knecht D, Loomis WF, De Lozanne A, Spudich J. 1988. Cell motility and chemotaxis in Dictyostelium amebae lacking myosin heavy chain. Dev. Biol. 128, 164–177. ( 10.1016/0012-1606(88)90279-5) [DOI] [PubMed] [Google Scholar]
- 11.Mizutani T, Haga H, Kawabata K. 2004. Cellular stiffness response to external deformation: tensional homeostasis in a single fibroblast. Cell Motil. Cytoskeleton 59, 242–248. ( 10.1002/cm.20037) [DOI] [PubMed] [Google Scholar]
- 12.Clark K, Langeslag M, Figdor CG, van Leeuwen FN. 2007. Myosin II and mechanotransduction: a balancing act. Trends Cell. Biol. 17, 178–186. ( 10.1016/j.tcb.2007.02.002) [DOI] [PubMed] [Google Scholar]
- 13.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. 2009. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10, 778–790. ( 10.1038/nrm2786) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu J, Wang G, Peng Q, Hu J, Luo X, Zheng Y, Teng Y, Tang C. 2011. Id1 induces tubulogenesis by regulating endothelial cell adhesion and cytoskeletal organization through beta 1-integrin and Rho-kinase signalling. Int. J. Mol. Med. 28, 543–548. ( 10.3892/ijmm.2011.741) [DOI] [PubMed] [Google Scholar]
- 15.Qiu J, Wang G, Zheng Y, Hu J, Peng Q, Yin T. 2011. Coordination of Id1 and p53 activation by oxidized LDL regulates endothelial cell proliferation and migration. Ann. Biomed. Eng. 39, 2869–2878. ( 10.1007/s10439-011-0382-6) [DOI] [PubMed] [Google Scholar]
- 16.Qiu J, et al. 2012. OxLDL stimulates Id1 nucleocytoplasmic shuttling in endothelial cell angiogenesis via PI3K pathway. Biochim. Biophys. Acta 1821, 1361–1369. ( 10.1016/j.bbalip.2012.07.016) [DOI] [PubMed] [Google Scholar]
- 17.Qiu J, Zheng Y, Hu J, Liao D, Gregersen H, Deng X, Fan Y, Wang G. 2014. Biomechanical regulation of vascular smooth muscle cell functions: from in vitro to in vivo understanding. J. R. Soc. Interface 11, 20130852 ( 10.1098/rsif.2013.0852) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen Y, Wang G, Huang X, Zhang Q, Wu J, Tang C, Yu Q, Liu X. 2012. Surface wettability of plasma SiOx:H nanocoating-induced endothelial cells’ migration and the associated FAK-Rho GTPases signalling pathways. J. R. Soc. Interface 9, 313–327. ( 10.1098/rsif.2011.0278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen Y, Ma Y, Gao M, Lai Y, Wang G, Yu Q, Cui FZ, Liu X. 2013. Integrins-FAK-Rho GTPases pathway in endothelial cells sense and response to surface wettability of plasma nanocoatings. ACS Appl. Mater. Interfaces 5, 5112–5121. ( 10.1021/am400973a) [DOI] [PubMed] [Google Scholar]
- 20.Wei D, Chen Y, Tang C, Huang H, Liu L, Wang Z, Li R, Wang G. 2013. LDL decreases the membrane compliance and cell adhesion of endothelial cells under fluid shear stress. Ann. Biomed. Eng. 41, 611–618. ( 10.1007/s10439-012-0677-2) [DOI] [PubMed] [Google Scholar]
- 21.Xie X, et al. 2013. In vitro and in vivo investigations on the effects of low-density lipoprotein concentration polarization and haemodynamics on atherosclerotic localization in rabbit and zebrafish. J. R. Soc. Interface 10, 20121053 ( 10.1098/rsif.2012.1053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mattila PK, Lappalainen P. 2008. Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 9, 446–454. ( 10.1038/nrm2406) [DOI] [PubMed] [Google Scholar]
- 23.Dimilla PA, Stone JA, Quinn JA, Albelda SM, Lauffenburger DA. 1993. Maximal migration of human smooth muscle cells on fibronectin and type IV collagen occurs at an intermediate attachment strength. J. Cell Biol. 122, 729–737. ( 10.1083/jcb.122.3.729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauffenburger DA, Wells A. 2001. Getting a grip: new insights for cell adhesion and traction. Nat. Cell Biol. 3, E110– E112 ( 10.1038/35074631) [DOI] [PubMed] [Google Scholar]
- 25.Billadeau DD, Nolz JC, Gomez TS. 2007. Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol. 7, 131–143. ( 10.1038/nri2021) [DOI] [PubMed] [Google Scholar]
- 26.Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. 2007. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 176, 573–580. ( 10.1083/jcb.200612043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. 2004. Two distinct actin networks drive the protrusion of migrating cells. Science 305, 1782–1786. ( 10.1126/science.1100533) [DOI] [PubMed] [Google Scholar]
- 28.Cai Y, et al. 2006. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys. J. 91, 3907–3920. ( 10.1529/biophysj.106.084806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shutova M, Yang C, Vasiliev JM, Svitkina T. 2012. Functions of nonmuscle myosin II in assembly of the cellular contractile system. PLoS ONE 7, e40814 ( 10.1371/journal.pone.0040814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peyton SR, Putnam AJ. 2005. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J. Cell. Physiol. 204, 198–209. ( 10.1002/jcp.20274) [DOI] [PubMed] [Google Scholar]
- 31.Ehrbar M, Sala A, Lienemann P, Ranga A, Mosiewicz K, Bittermann A, Rizzi SC, Weber FE, Lutolf MP. 2011. Elucidating the role of matrix stiffness in 3D cell migration and remodeling. Biophys. J. 100, 284–293. ( 10.1016/j.bpj.2010.11.082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S, et al. 2003. Genomic analysis of smooth muscle cells in 3-dimensional collagen matrix. FASEB J. 17, 97–99. ( 10.1096/fj.02-0256fje) [DOI] [PubMed] [Google Scholar]
- 33.Peyton SR, Kim PD, Ghajar CM, Seliktar D, Putnam AJ. 2008. The effects of matrix stiffness and RhoA on the phenotypic plasticity of smooth muscle cells in a 3D biosynthetic hydrogel system. Biomaterials 29, 2597–2607. ( 10.1016/j.biomaterials.2008.02.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrie RJ, Gavara N, Chadwick RS, Yamada KM. 2012. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J. Cell Biol. 197, 439–455. ( 10.1083/jcb.201201124) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahai E, Marshall CJ. 2003. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5, 711–719. ( 10.1038/ncb1019) [DOI] [PubMed] [Google Scholar]
- 36.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510–523. ( 10.1016/j.cell.2008.09.043) [DOI] [PubMed] [Google Scholar]
- 37.Sanz-Moreno V, Marshall CJ. 2010. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 22, 690–696. ( 10.1016/j.ceb.2010.08.020) [DOI] [PubMed] [Google Scholar]
- 38.Pollard TD, Borisy GG. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465. ( 10.1016/S0092-8674(03)00120-X) [DOI] [PubMed] [Google Scholar]
- 39.Laemmermann T, et al. 2008. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55. ( 10.1038/nature06887) [DOI] [PubMed] [Google Scholar]
- 40.Lorentzen A, Bamber J, Sadok A, Elson-Schwab I, Marshall CJ. 2011. An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J. Cell Sci. 124, 1256–1267. ( 10.1242/jcs.074849) [DOI] [PubMed] [Google Scholar]
- 41.Rottner K, Stradal TEB. 2011. Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 23, 569–578. ( 10.1016/j.ceb.2011.07.003) [DOI] [PubMed] [Google Scholar]
- 42.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker EB, Friedl P. 2003. Compensation mechanism in tumor cell migration: mesenchymal–amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 160, 267–277. ( 10.1083/jcb.200209006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poincloux R, Collin O, Lizárraga F, Romao M, Debray M, Piel M, Chavrier P. 2011. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc. Natl Acad. Sci. USA 108, 1943–1948. ( 10.1073/pnas.1010396108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laemmermann T, Sixt M. 2009. Mechanical modes of ‘amoeboid’ cell migration. Curr. Opin. Cell Biol. 21, 636–644. ( 10.1016/j.ceb.2009.05.003) [DOI] [PubMed] [Google Scholar]
- 45.Chien S. 2007. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am. J. Physiol. Heart Circ. Physiol. 292, H1209–H1224. ( 10.1152/ajpheart.01047.2006) [DOI] [PubMed] [Google Scholar]
- 46.Petrie RJ, Yamada KM. 2012. At the leading edge of three-dimensional cell migration. J. Cell Biol. 125, 5917–5926. ( 10.1242/jcs.093732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanz-Moreno V, et al. 2011. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 20, 229–245. ( 10.1016/j.ccr.2011.06.018) [DOI] [PubMed] [Google Scholar]
- 48.Robinson DN, Spudich JA. 2004. Mechanics and regulation of cytokinesis. Curr. Opin. Cell Biol. 16, 182–188. ( 10.1016/j.ceb.2004.02.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo T, Mohan K, Srivastava V, Ren Y, Iglesias PA, Robinson DN. 2012. Understanding the cooperative interaction between myosin II and actin cross-linkers mediated by actin filaments during mechanosensation. Biophys. J. 102, 238–247. ( 10.1016/j.bpj.2011.12.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gunst SJ, Zhang WW. 2008. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am. J. Physiol. Cell Physiol. 295, C576–C587. ( 10.1152/ajpcell.00253.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaneko-Kawano T, et al. 2012. Dynamic regulation of myosin light chain phosphorylation by Rho-kinase. PLoS ONE 7, e39269 ( 10.1371/journal.pone.0039269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. 2007. Myosin IIA regulates cell motility and actomyosin microtubule crosstalk. Nat. Cell Biol. 9, 299–309. ( 10.1038/ncb1540) [DOI] [PubMed] [Google Scholar]
- 53.Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, Horwitz AF. 2008. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 183, 543–554. ( 10.1083/jcb.200806030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. 2004. Nonmuscle myosin IIB is involved in the guidance of fibroblast migration. Mol. Biol. Cell 15, 982–989. ( 10.1091/mbc.E03-06-0359) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kovacs M, Thirumurugan K, Knight PJ, Sellers JR. 2007. Load-dependent mechanism of nonmuscle myosin 2. Proc. Natl Acad. Sci. USA 104, 9994–9999. ( 10.1073/pnas.0701181104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sandquist JC, Means AR. 2008. The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol. Biol. Cell 19, 5156–5167. ( 10.1091/mbc.E08-05-0533) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Golomb E, et al. 2004. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 279, 2800–2808. ( 10.1074/jbc.M309981200) [DOI] [PubMed] [Google Scholar]
- 58.Loirand G, Pacaud P. 2010. The role of Rho protein signaling in hypertension. Nat. Rev. Cardiol. 7, 637–647. ( 10.1038/nrcardio.2010.136) [DOI] [PubMed] [Google Scholar]
- 59.Fernandez-Gonzalez R, Simoes SdM, Roeper J-C, Eaton S, Zallen JA. 2009. Myosin II dynamics are regulated by tension in intercalating cells. Dev. Cell 17, 736–743. ( 10.1016/j.devcel.2009.09.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren Y, Effler JC, Norstrom M, Luo T, Firtel RA, Iglesias PA, Rock RS, Robinson DN. 2009. Mechanosensing through cooperative interactions between myosin II and the actin crosslinker cortexillin I. Curr. Biol. 19, 1421–1428. ( 10.1016/j.cub.2009.07.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uyeda TQP, Iwadate Y, Umeki N, Nagasaki A, Yumura S. 2011. Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS ONE 6, e26200 ( 10.1371/journal.pone.0026200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hale CM, Sun SX, Wirtz D. 2009. Resolving the role of actoymyosin contractility in cell microrheology. PLoS ONE 4, e7054 ( 10.1371/journal.pone.0007054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watanabe M, Yumoto M, Tanaka H, Wang HH, Katayama T, Yoshiyama S, Black J, Thatcher SE, Kohama K. 2010. Blebbistatin, a myosin II inhibitor, suppresses contraction and disrupts contractile filaments organization of skinned taenia cecum from guinea pig. Am. J. Physiol. Cell Physiol. 298, C1118–C1126. ( 10.1152/ajpcell.00269.2009) [DOI] [PubMed] [Google Scholar]
- 64.Goeckeler ZM, Bridgman PC, Wysolmerski RB. 2008. Nonmuscle myosin II is responsible for maintaining endothelial cell basal tone and stress fiber integrity. Am. J. Physiol. Cell Physiol. 295, C994–C1006. ( 10.1152/ajpcell.00318.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. 2012. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 148, 973–987. ( 10.1016/j.cell.2011.12.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang HH, et al. 2008. Blebbistatin inhibits the chemotaxis of vascular smooth muscle cells by disrupting the myosin II-actin interaction. Am. J. Physiol. Heart Circ. Physiol. 294, H2060–H2068. ( 10.1152/ajpheart.00970.2007) [DOI] [PubMed] [Google Scholar]
- 67.Pramanik MK, Iijima M, Iwadate Y, Yumura S. 2009. PTEN is a mechanosensing signal transducer for myosin II localization in Dictyostelium cells. Genes Cells 14, 821–834. ( 10.1111/j.1365-2443.2009.01312.x) [DOI] [PubMed] [Google Scholar]
- 68.Matsushita S, Inoue Y, Hojo M, Sokabe M, Adachi T. 2011. Effect of tensile force on the mechanical behavior of actin filaments. J. Biomech. 44, 1776–1781. ( 10.1016/j.jbiomech.2011.04.012) [DOI] [PubMed] [Google Scholar]
- 69.Tsaturyan AK, Koubassova N, Ferenczi MA, Narayanan T, Roessle M, Bershitsky SY. 2005. Strong binding of myosin heads stretches and twists the actin helix. Biophys. J. 88, 1902–1910. ( 10.1529/biophysj.104.050047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, Sheetz MP. 2008. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 10, 1062–1068. ( 10.1038/ncb1765) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang G, Giannone G, Critchley DR, Fukumoto E, Sheetz MP. 2003. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature 424, 334–337. ( 10.1038/nature01805) [DOI] [PubMed] [Google Scholar]
- 72.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. 2009. Stretching single talin rod molecules activates vinculin binding. Science 323, 638–641. ( 10.1126/science.1162912) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roca-Cusachs P, del Rio A, Puklin-Faucher E, Gauthier NC, Biais N, Sheetz MP. 2013. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc. Natl Acad. Sci. USA 110, E1361–E1370. ( 10.1073/pnas.1220723110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iwadate Y, Okimura C, Sato K, Nakashima Y, Tsujioka M, Minami K. 2013. Myosin-II-mediated directional migration of Dictyostelium cells in response to cyclic stretching of substratum. Biophys. J. 104, 748–758. ( 10.1016/j.bpj.2013.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Verkhovsky AB, Svitkina TM, Borisy GG. 1999. Self-polarization and directional motility of cytoplasm. Curr. Biol. 9, 11–20. ( 10.1016/S0960-9822(99)80042-6) [DOI] [PubMed] [Google Scholar]
- 76.Bray D, White JG. 1988. Cortical flow in animal cells. Science 239, 883–888. ( 10.1126/science.3277283) [DOI] [PubMed] [Google Scholar]
- 77.Mayer M, Depken M, Bois JS, Juelicher F, Grill SW. 2010. Anisotropies in cortical tension reveal the physical basis of polarizing cortical flows. Nature 467, 617–621. ( 10.1038/nature09376) [DOI] [PubMed] [Google Scholar]
- 78.Goehring NW, Grill SW. 2013. Cell polarity: mechanochemical patterning. Trends Cell Biol. 23, 72–80. ( 10.1016/j.tcb.2012.10.009) [DOI] [PubMed] [Google Scholar]
- 79.Kolega J. 2003. Asymmetric distribution of myosin IIB in migrating endothelial cells is regulated by a Rho-dependent kinase and contributes to tail retraction. Mol. Biol. Cell 14, 4745–4757. ( 10.1091/mbc.E03-04-0205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kolega J. 1998. Cytoplasmic dynamics of myosin IIA and IIB: spatial ‘sorting’ of isoforms in locomoting cells. J. Cell Sci. 111, 2085–2095. [DOI] [PubMed] [Google Scholar]
- 81.Yam PT, Wilson CA, Ji L, Hebert B, Barnhart EL, Dye NA, Wiseman PW, Danuser G, Theriot JA. 2007. Actin–myosin network reorganization breaks symmetry at the cell rear to spontaneously initiate polarized cell motility. J. Cell Biol. 178, 1207–1221. ( 10.1083/jcb.200706012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Du XX, Doubrovinski K, Osterfield M. 2012. Self-organized cell motility from motor-filament interactions. Biophys. J. 102, 1738–1745. ( 10.1016/j.bpj.2012.03.052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hawkins RJ, Poincloux R, Bénichou O, Piel M, Chavrier P, Voituriez R. 2011. Spontaneous contractility-mediated cortical flow generates cell migration in three-dimensional environments. Biophys. J. 101, 1041–1045. ( 10.1016/j.bpj.2011.07.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Charras G, Paluch E. 2008. Blebs lead the way: how to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 9, 730–736. ( 10.1038/nrm2453) [DOI] [PubMed] [Google Scholar]
- 85.Friedl P, Weigelin B. 2008. Interstitial leukocyte migration and immune function. Nat. Immunol. 9, 960–969. ( 10.1038/ni.f.212) [DOI] [PubMed] [Google Scholar]
- 86.Sanchez-Madrid F, del Pozo MA. 1999. Leukocyte polarization in cell migration and immune interactions. EMBO J. 18, 501–511. ( 10.1093/emboj/18.3.501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Estecha A, Sánchez-Martín L, Puig-Kröger A, Bartolomé RA, Teixidó J, Samaniego R, Sánchez-Mateos P. 2009. Moesin orchestrates cortical polarity of melanoma tumour cells to initiate 3D invasion. J. Cell Sci. 122, 3492–3501. ( 10.1242/jcs.053157) [DOI] [PubMed] [Google Scholar]
- 88.Blaser H, Reichman-Fried M, Castanon I, Dumstrei K, Marlow FL, Kawakami K, Solnica-Krezel L, Heisenberg CP, Raz E. 2006. Migration of zebrafish primordial germ cells: a role for myosin contraction and cytoplasmic flow. Dev. Cell 11, 613–627. ( 10.1016/j.devcel.2006.09.023) [DOI] [PubMed] [Google Scholar]
- 89.Keller HU, Bebie H. 1996. Protrusive activity quantitatively determines the rate and direction of cell locomotion. Cell Motil. Cytoskeleton 33, 241–251. () [DOI] [PubMed] [Google Scholar]
- 90.Prager-Khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, Kam Z, Geiger B, Bershadsky AD. 2011. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat. Cell Biol. 13, 1457–1465. ( 10.1038/ncb2370) [DOI] [PubMed] [Google Scholar]
- 91.Pelham RJ, Wang YL. 1997. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl Acad. Sci. USA 94, 13 661–13 665. ( 10.1073/pnas.94.25.13661) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Distel M, Hocking JC, Volkmann K, Koster RW. 2010. The centrosome neither persistently leads migration nor determines the site of axonogenesis in migrating neurons in vivo. J. Cell Biol. 191, 875–890. ( 10.1083/jcb.201004154) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raab M, Swift J, Dingal PC, Shah P, Shin JW, Discher DE. 2012. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell Biol. 199, 669–683. ( 10.1083/jcb.201205056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lo CM, Wang HB, Dembo M, Wang YL. 2000. Cell movement is guided by the rigidity of the substrate. Biophys. J. 79, 144–152. ( 10.1016/S0006-3495(00)76279-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim H-D, Peyton SR. 2012. Bio-inspired materials for parsing matrix physicochemical control of cell migration: a review. Integr. Biol. 4, 37–52. ( 10.1039/c1ib00069a) [DOI] [PubMed] [Google Scholar]
- 96.Engler A, Bacakova L, Newman C, Hategan A, Griffin M, Discher D. 2004. Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 86, 617–628. ( 10.1016/S0006-3495(04)74140-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang HB, Dembo M, Hanks SK, Wang YL. 2001. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl Acad. Sci. USA 98, 11 295–11 300. ( 10.1073/pnas.201201198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beningo KA, Lo CM, Wang YL. 2002. Flexible polyacrylamide substrata for the analysis of mechanical interactions at cell–substratum adhesions. Methods Cell Biol. 69, 325–339. ( 10.1016/S0091-679X(02)69021-1) [DOI] [PubMed] [Google Scholar]
- 99.Moissoglu K, Schwarz MA. 2006. Integrin signalling in directed cell migration. Biol. Cell 98, 547–555. ( 10.1042/BC20060025) [DOI] [PubMed] [Google Scholar]