

Abstract
Protein folding has been a major area of research for many years. Nonetheless, the mechanisms leading to the formation of an active biological fold are still not fully apprehended. The huge amount of available sequence and structural information provides hints to identify the putative fold for a given sequence. Indeed, protein structures prefer a limited number of local backbone conformations, some being characterized by preferences for certain amino acids. These preferences largely depend on the local structural environment. The prediction of local backbone conformations has become an important factor to correctly identifying the global protein fold. Here, we review the developments in the field of local structure prediction and especially their implication in protein fold recognition.
Keywords: bioinformatics, protein fold recognition, local structure prediction, structural alphabets, threading, sequence annotation
1. Introduction
A detailed understanding of the function of a protein can be achieved by studying its three-dimensional structure. Insights into the molecular details of protein interactions and enzyme activities are obtained from the three-dimensional structure and the impact of protein structure-based drug design in pharmaceutical development has been impressive [1–3]. Although significant advances have been made in the field of experimental structure determination [4,5], the difficulty and cost associated with this limit the rate at which protein structures are solved. The sequence information, on the other hand, has grown immensely with the help of high-throughput sequencing techniques [6–11]. The huge gap between the sequence and structure space is formed by sequences whose three-dimensional structures are unknown.
Computational approaches provide effective ways for filling this gap. The methods used for structural annotation assign a probable three-dimensional fold for a given sequence. Applications of computational structure modelling have been enormous [12]. The modelled structures are useful in studying protein–protein [13] and protein–ligand interactions [12–14]. They are also used as search models in experimental structure determination [15] and for characterizing structures of huge assemblies [16–18]. Computational model-based drug design [19,20] and studies for understanding protein dynamics based on modelled structures [12,21,22] have been successful. Large-scale proteome-wide structural annotation studies have also been designed upon computational approaches for fold identification [23,24].
Modelling protein structures based on a template fold (comparative/homology modelling) is so far the most reliable technique for generating three-dimensional structure for a given sequence [25]. The most critical step in comparative modelling is the identification of the correct template fold. The possible number of protein folds are estimated to be limited in number and the currently available structures can cover most of it [26,27]. The ability to relate sequences to the correct folds forms the basis for protein fold recognition. The simplest means for deriving structural information is by direct comparison with other related sequences with known structures [28–30] (figure 1a). The tertiary structure of a protein is highly conserved across different species, when compared with its sequence [32]. Figure 1b shows the direct relationship existing between structural similarity in terms of global distance test (GDT) [31] and sequence identity. This analysis has been performed on a randomly chosen subset of about 500 SCOP domain, which were structurally aligned [33,34] with iPBA superimposition method [35,36]. Thus, if the sequence similarity (usually quantified as the percentage amino acid identity) is high, the structures are likely to be similar. Comparative modelling can generate reliable structural models (1–3 Å root mean squared deviation (RMSD) with the real structure), especially when the sequence identity is above 30% [25,37] (figure 1a(i)).
Figure 1.

‘Homology’ detection with variation in sequence identity. (a) Schematic demonstrating the use of sequence comparison for detecting structural homology. The sequence alignments are indicated with ‘X’ representing any amino acid. Same amino acids at equivalent positions are highlighted in red, similar ones are in green. At sequence identity levels above 30% (i), simple sequence alignments are largely sufficient to detect similar folds. Below this similarity threshold, the alignments are less accurate and thus less efficient in detecting genuine relationships. (ii) Between 20 and 30%, the correct fold is not often detected as the top hit. (iii) At very low sequence identities, simple sequence alignments are not very useful. (b) Variation of structural similarity (quantified in terms of GDT_TS score) [31] with change in sequence identity. Even at low sequence identities (less than 30%, highlighted in grey background) significant structural similarity could be observed. (Online version in colour.)
Nonetheless, the sequences of homologous protein structures can diverge beyond the point where pairwise sequence comparison methods fail. Figure 1a highlights the success in detecting the correct fold declines with decrease in sequence identity. The match between the sequence of interest (target) and the known folds is obtained as an alignment of sequences. Below 30% sequence identity (twilight zone, figure 1a(ii)), the accuracy of alignments falls significantly which results in a low sensitivity in fold recognition. At very low sequence identities (below 15%, figure 1a(iii)), the sequence alignments are not very informative. The values provided in the figure are just indicative of the sequence identity ranges, and no fixed thresholds have been characterized [38]. The lectin superfamily is a classic example of this case where the sequence identities fall below 10% but they are known to adopt a jelly-roll-sugar binding fold [39], and a random alignment is expected to have about 12% identity.
In this review, we focus mainly on the scenario where the target has only remote homology with a protein of known fold (template). In such cases, a clear understanding of the association of amino acid sequences with the three-dimensional structure (sequence–structure relationship) is the key factor in determining the success to recognize correct folds. Firstly, a general outline of the methods for fold recognition based on the detection of remote homology is presented. Then the role of local structure prediction in fold recognition is emphasized, followed by a discussion on the importance of careful extraction of sequence–structure relationships.
2. Fold recognition by remote similarity detection
In the absence of closely related proteins of known structure, it is difficult to assign the correct fold using simple sequence alignments (figure 1a). Additional information is required to enhance the sensitivity to detect more distant relationships. The knowledge on the evolutionary and structural constraints associated with the target sequence forms the major contributing factor in detecting distant relationships.
2.1. Evolutionary information: profile–profile alignments
The protein sequence databases nowadays contain homologous sequences from different species. A multiple sequence alignment (MSA) can be used to trace the extent of evolutionary divergence among the related sequences. As against a single sequence, MSA consists of information about amino acid propensities at each position in the sequence which characterizes a sequence ‘profile’. This information is highly valuable in differentiating conserved and variable positions, and these position-specific amino acid propensities are more effective in identifying a protein that is evolutionarily related to this group. Thus, the use of MSAs as sequence profiles has a huge impact on homology detection (figure 2a,b) [40,41].
Figure 2.

Different strategies for protein fold recognition. The fold space is highlighted by the blue background and the lengths of the black arrows joining the target sequence (space) and fold space give an idea of the distance of relationship. (a) Close relationships are often detected by simple sequence alignment techniques. (b) Addition of evolutionary information using sequence profiles derived from MSAs helps in detecting more distantly related folds. When the sequence-based alignments are not informative, sequence–structure matching needs to be carried out. (c) The target sequence can be threaded on to the known folds to check the compatibility. The compatibility is usually quantified based on the global interaction or energy potential. Obtaining an optimal alignment between the sequence and a fold is however difficult and computationally quite expensive. (d) The other alternative is to carry out prediction of different structural features like local backbone conformation, solvent accessibility or contact order and then matching the predicted features with that found in the known fold. (Online version in colour.)
A sequence profile is often described as a position-specific scoring matrix (PSSM), which holds information on amino acid propensities for each position in the sequence, derived based on an MSA. PSI-BLAST and RPS-BLAST [42,43] are widely used sensitive PSSM-based search algorithms for remote homology detection. The search for related sequences is usually carried out iteratively where the profile (PSSM) gets updated at each step with new sequence information. These new sequences, which can be termed as ‘intermediates’, could also be used as new targets to trace the linkage to the distant homologue [44,45]. Recently, a method named HangOut has been proposed to improve the sensitivity of PSI-BLAST searches especially for domains with long insertions [46].
Several approaches have been proposed to improve the search for related proteins based on sequence profiles. An MSA can be used to generate hidden Markov models (HMMs), which can also be used as efficient search tools [47–50]. HMM-based profile comparison tools are reported to have a higher accuracy than PSSM-based methods [45,51]. To improve the accuracy of profiles generated, efficient methods that can weigh conserved sequence regions have also been developed [52–56]. Amino acid substitution probabilities that drive the alignment scores are influenced by the local structural [57] or sequence neighbourhood [58,59] in the protein. CSI-BLAST [60] uses mutation probabilities from context-specific profiles based on 13-residue windows while DELTA-BLAST [61] incorporates frequencies from longer profiles corresponding to conserved domain sequences [62]. Two iterations of CSI-BLAST [60] are more sensitive than five iterations of PSI-BLAST, whereas DELTA-BLAST [61] is more sensitive than both CS-BLAST and PSI-BLAST in detecting relatively similar and remote homologues. Profile–profile comparison methods that optimize local alignments are also reported to be more effective in remote homology detection [55,63,64].
Fast and efficient HMM profile-based iterative search approaches similar to PSI-BLAST marked further advancements in this field. JACKHMMER [65] relies on a series of database filtering steps to reduce search time, whereas HHBLITS implements HMM–HMM comparisons with approximation of profile columns using 219 extended alphabets [66]. The sensitivity of HHBLITS in detecting remote homologues was about 50–100% more than that of PSI-BLAST.
Increasing amount of evidence of sequence permutations, duplications or fusion events [67–69] raises the need for developing methods that can detect homologues independent of the sequence order. A classic example is the methyl transferase family where gene fusion and permutation events are observed in the evolutionary process resulting in differences in substrate recognition and catalytic activity [70,71]. Interesting developments have also been made in this direction. A few alignment-free techniques were developed for this purpose [72,73], while the majority of methods that employ sequence-order-independent comparisons are optimized for multi-domain proteins [74–77].
However, at a sequence identity well below 20%, the chances of detecting true templates even with profile-based approaches remain low [78]. It is possible that the fold which the sequence adopts is already known but the sequence-based searches fail to trace such relationship. Such cases could be addressed if one can verify whether this protein sequence is compatible with any of the available protein folds. The question is how to derive this compatibility with known folds, given a sequence target.
2.2. Structure information: fold recognition by threading
Two proteins sharing the same fold could have diverged significantly in terms of sequence and it is often difficult to detect the relationship based on sequence- or profile-based methods. Some well-known examples of these are the oligonucleotide/oligosaccharide binding fold, cupins, TIM barrels, serine proteases, etc. Hence it becomes necessary to relate target sequence with the known folds. To assess the fit of a sequence on a template protein fold, it is necessary to quantify the preference for amino acids to occur in the structural environment of the template. This measure can enable us to generate an alignment between the sequence and the template structure (threading). Two major strategies have been used to score the sequence–structure compatibility.
2.2.1. Threading based on global potentials
The more direct approach would be to fit each residue from the query sequence onto each position in the template backbone and check whether it is energetically compatible (figure 2c). The energy could be quantified as pairwise interaction potentials [79,80] or solvation energy [81] which were used by many of the earlier methods for fold identification [82–84]. The non-local regions of the target residue could have significant difference when compared with that of the template. This means that the equivalent regions (or residues) have to be known a priori to calculate the compatibility score. Hence the target–template alignment has to be generated in advance and this makes the problem extremely hard.
An easier solution is the ‘frozen approximation’ [81], where each residue in the target is scored against the structural environment formed by the neighbouring template residues. However, as mentioned earlier, the target and the template often have different environments involving variation in the amino acid residues. Hence this method is frequently prone to accumulate errors that affect the final estimation of compatibility. Approximate solutions to identify residue neighbours could be derived using Monte Carlo simulations [85] or double-dynamic programming [83]. The divide-and-conquer approach used in Prospect [86] aims to solve several sub-structure alignments and integrate them in an optimal way. The complexity associated with fold identification purely based on global interaction potentials was alleviated by combining local structural information [87] and the local structural context is observed to have the major role in obtaining an optimal template alignment [88].
2.2.2. Threading based on local scoring functions
The second category of methods adopts a more localized approach for quantifying the compatibility of the target sequence on a template. They score the preference for each residue to occur in a local environment of the template structure. The preference for an amino acid to occur in a local structural environment can be referred to as the sequence–structure relationship. As mentioned earlier, several structural features can be used to describe the local structural environment of a protein. The most commonly used features are secondary structure, solvent accessibility, residue depth, backbone conformation, potential for hydrogen bonding or hydrophobic interactions [57,89]. A combination of some or all of these features can define one local environment. The issue is to choose the right set of features and weigh each feature to derive the combination.
Two different ways in which the preference of an amino acid for a local structural environment can be calculated are: (i) the amino acid preferences associated with a local structural environment can be extracted as a one- to three-dimensional substitution matrix which gives the score for assigning each amino acid in this environment [57,90–92]. These methods are based on the premise that the amino acid substitution patterns vary based on their structural environments [57]. (ii) The other strategy adopted by most of the recent methods is to predict the structural features separately based on the target sequence and then combine the predictions carefully to search for related structures (figure 2d) [93]. The use of predicted local structural features as strings can bring down the problem of sequence–structure comparison to that of a sequence–sequence comparison. As in the case of classical sequence alignment procedures, dynamic programming algorithm can be used to achieve this task [94]. Recognition of protein folds based on predicted local structural features is competitive with methods that consider non-local structural details [95–98].
The efficiency of different profile comparison methods is coupled with the predicted structural information to improve the quality of profile-based fold recognition [54,95,99–106]. The successful approaches for fold recognition use profile comparisons to obtain a limited set of alignments which are then evaluated and refined using the energy-based potentials [101,102,107,108]. An interesting recent observation is that the accuracy of fold recognition improves with the use of artificially evolved sequences compatible with the template folds [109]. These sequences are optimized to stabilize a given structure by simulated annealing based on a variety of potentials, similar to those commonly used in protein threading.
In the following sections, we discuss the developments in the field of local structure prediction which has been the most essential component of prediction-based threading methods.
2.2.2.1. Secondary structure prediction
The protein backbone prefers mainly a limited number of stable conformations constrained by the backbone dihedral angles and hydrogen bonds. The two important regularities seen in the local conformation of the backbone constitute α-helices and β-strands [110]. The rest of the backbone is usually considered as coils which represent that not assigned to one of these two conformations. These three states (helix, strand and coil) constitute the classical definition of secondary structures. This description has been used either to complement sequence-based searches or to identify the best structural relative from the results of sequence-based searches. It has been a strategic step in most of the fold recognition approaches [89,95,108,111,112]. The fold recognition methodologies like PHYRE [95], MUSTER [113], SPARKS [114] and SP5 [105] incorporate methods for predicting secondary structure of the target sequence and then comparing that with the secondary structure assigned for the templates.
Efficient machine learning methods like artificial neural networks (ANNs) [115] or support vector machines (SVMs) [116] are being developed to predict secondary structure from a protein sequence. They are trained to learn the amino acid preferences associated with different secondary structures and make predictions for new sequences. Most of the successful and recent approaches use the information on amino acid propensities derived from sequence profiles [115,117–122]. The use of multiple sequences (as a profile) adds details on amino acid variability at each position in the sequence. Studying the association of relatively large fragments of about 15–20 residues with a secondary structural state helped to incorporate the effect of long-range interactions to a certain extent [123].
It must be noted that ‘the machine learning algorithms predict what they learn’. The secondary structure assignments for known structures are made using a non-redundant dataset of protein structures and then the sequence association with each secondary structural unit is learnt. There are several methods available for assigning secondary structures, based on local backbone dihedral angles or hydrogen bonds. It is interesting to note that the assignments made by different methods do not agree to a significant extent [124–126]. Figure 3 presents the comparison of secondary structure assigned based on the three-dimensional structure of methyglyoxal synthase [127] with that of predictions made by different methods. The ambiguities are generally seen in predicting the short repetitive structures and especially boundaries of secondary structures. The underlying cause is that the capping regions do not necessarily follow the same rules set for defining the backbone conformation of helices and strands [132].
Figure 3.

Comparison of secondary structure prediction methods. For a recently solved structure of methylglyoxal synthase [PDB ID 2X8W] [127], the assigned secondary structure by (a) DSSP [128] and predicted ones are shown. The α-helices are shown in red, β-strands in yellow and coils in green. Different secondary structure prediction methods are shown: (b) PSIPRED [115], (c) PROF [129], (d) SSPRO [130] and (e) YASPIN [131]. The predictions are also shown as sequence alignment in (f). Helices, strands and coils are indicated by H, E and C, respectively. (Online version in colour.)
Taking into account the variation of secondary structure assignments among different conformations of the same structure (inherent flexibility) and between close structural homologues, a theoretical limit of 88% was proposed for secondary structure prediction accuracy [123]. The most popular and not very recent methods like PSIPRED [115], PROF [122,123] and SSPRO [130] could already achieve a prediction accuracy close to 80%. PHD [129] and PSIPRED [115] use two levels of neural network predictions where the initial predictions are refined at the second step. SSPRO [130] implements a recursive single neural network instead of multiple layers. The use of dual layer SVMs also gave performance comparable to these methods [116,119]. A few improvements or alternative methods have been proposed in the last few years [116–118,120,121,131,133–136] but the increase in prediction accuracy is minimal. Use of consensus approaches based on the results of multiple predictions [133,136] gave accuracy above 80%. The use of a dictionary of words of amino acid stretches associated with a secondary structure state also resulted in predictions comparable to the best machine learning methods available [137].
A recent assessment of available secondary structure prediction methods suggests that a prediction accuracy of about 82% could be reached [138]. The exposed coils were predicted higher than helices and strands. The accuracies of prediction for residues in 310 helices and β-bridges were less than 50% and 48%, respectively [138]. Complementing profile–profile comparisons with predicted secondary structure resulted in a significant improvement in the efficiency of fold recognition [95,108,139,140]. The prediction of non-local structural details mainly based on the predicted local features has also reported some success. The prediction of protein contact order which reflects the average length of sequence separating contacting residues, with the aid of predicted secondary structure information is reported to achieve a correlation of about 0.85 [141].
2.2.2.2. Beyond secondary structures
In the absence of reasonable sequence similarity, the secondary structure prediction makes a huge contribution to remote homology detection. However, proteins with similar secondary structure topology need not necessarily have the same tertiary fold. Figure 4 gives an example of one such case where a wrong fold is chosen purely based on the similarity in secondary structure. This is often true for proteins with repeating secondary structural elements, i.e. helix or strand repeats. The three-state secondary structural information (helix, sheet and coil) does not give precise details of the backbone conformation for the complete protein backbone [111]. More than 50% of the residues in the available protein structures are assigned to the coil state which by definition corresponds to irregular backbone conformations. The loss of information caused by considering such a large majority of residues as coils is a main contributor to the recognition of wrong folds [112,144].
Figure 4.

Going beyond three-state secondary structure. The structures related to methylglyoxal synthase (a) [PDB ID 2X8W] are identified purely based on the secondary structure, using SSEA server [142]. A different fold (b) (Response Regulator, PDB ID 1M5T) [143] was obtained as a top hit based on the secondary structure content. The secondary structure alignment (c) shows that the structures are close based on the secondary structure; however, the folds are different. The equivalent helices and strands are highlighted in the same colour in the two structures (a,b). A more precise description of the backbone conformation was obtained using PB (d). The assignment of PB instead of the three-state secondary structures highlights many differences between the two structures, which were otherwise masked by the secondary structure definition. The segments assigned as coils (indicated by ‘C’) are highlighted in red in the PB alignment. Other differences in the regular secondary structures are in blue. (Online version in colour.)
On the contrary, the coil state is not strictly irregular. Next to helices and strands, turns characterize another frequent regularity in the protein backbone [145]. β-Turns account for about 25% of the residues in proteins [146]. Other favourable local structures involve PolyProline II helices [147], hairpin loops, corner motifs, β-bulges, etc. [148]. Most of these repeating units are associated with strong sequence–structure relationships. Thus, a major portion of protein backbone can be associated with repeating local structural elements.
Attempts have been made to generate a minimum set of local backbone conformations, a combination of which can be used to represent most or all of the protein backbone conformation. A set of prototypes of local structures that can be used to approximately define the conformation of a protein backbone is called as a structural alphabet (SA) [149]. The number and size of these fragments vary depending on the approach used. Apart from their use in modelling, structural alignment and functional analysis, these libraries also help to extract precise information on the amino acid preferences associated with local structures.
Several approaches have been developed for the prediction of local structures based on fragment libraries [150–157]. The underlying methods used for prediction are based on both probabilistic [150,153,158] and machine learning algorithms [152,159–162]. Bystroff and Baker generated a library by clustering frequently observed short sequence patterns (I-sites) in protein structures. An efficient local structure prediction method was developed based on this library [150,163]. As the number of prototypes increase, the prediction accuracy was found to be lower. However, the regions predicted with high confidence add fine details on structural motifs and local backbone conformation. The use of secondary structure information is proved to improve the accuracy of local structure prediction. However, the repetitive structures are sometimes over-predicted in place of other local conformations [164].
Protein blocks (PBs) are a widely used SA composed of 16 pentapeptide conformations characterized by the series of backbone dihedral angles [153]. These conformations are characterized by strong sequence–structure relationships determined by the high predictability. Figure 4d gives an example of two different protein folds having the same secondary structure topology. It highlights that the PB description is more informative when compared to the three-state secondary structure, especially for the coil regions. It also underlines the significance of precise local structural information in fold recognition. A series of methods have been tested for the prediction of PBs from sequence data. Bayesian prediction models gave an accuracy ranging from 34 to 48% [153,164–166]. Dual layer ANNs and SVMs improved the prediction rate to about 58.5% [159], 61% [167] and 67% [160]. Combining information on secondary structure and solvent accessibility further enhanced the prediction rate of PBs [168]. Longer fragments based on PBs were predicted with better accuracy [166,169]. Frequently observed 11 residue fragments described based on the dihedrals were predicted with a significant accuracy of about 63% using SVMs [170]. Prediction of structural flexibility based on local structure preferences has also been successful [171]. Table 1 gives a list of popular and more recent methods for local structure prediction, based on fragment libraries.
Table 1.
Fragment-based local structure prediction methods. The table gives the list of methods for predicting local backbone conformation based on a library of fragments (prototypes). The length, number of prototypes and the distance measure used to generate the library are also mentioned. The reported prediction rates from the original publication are also listed. MDA, maximum deviation in torsion angles; ANN, artificial neural networks; SVM, support vector machines.
| research team | fragment length | distance measure | prediction method | prototype number | prediction rate (%) |
|---|---|---|---|---|---|
| Rooman et al. | 4, 5, 6, 7 | Cα RMSD | statistical mechanics (mean force potential) | 4 | 40–47 |
| Bystroff & Baker | 3–19 | sequence profiles, RMSD, MDA | profile–profile matching | 13 (later updated to 16) | 50 |
| de Brevern et al. | 5 | dihedral angles | Bayesian | 16 | 34.4 |
| Hunter & Subramaniam | 7 | hypercosine Cα | Bayesian | 28–16336 | 40 |
| Yang & Wang | 9 | dihedral angles | sequence profile matching | 138604 | 79 |
| Etchebest et al. | 5 | dihedral angles | Bayesian (simulated annealing) |
16 [153] | 49 |
| Benros et al. | 11 | Cα RMSD, PB based | hybrid protein model | 120 | 51.2 |
| Sander et al. | 7 | Cα distance | decision trees, SVM, random forest | 28 | 23–36 |
| Dong et al. | 7 | Cα distance | ANN | 28 | 45.6 |
| Dong et al. | 5 | dihedral angles | ANN | 16 [153] | 58.5 |
| Zimmermann et al. | 5 | dihedral angles | SVM | 16 [153] | 61 |
| Chen & Johnson | 9 | Cα distance | SVM | 800 | 72 |
| Bornot et al. | 11 | Cα RMSD, PB based | hybrid protein model | 120 | 63.1 |
| Rangwala et al. | 5 | dihedral angles | SVM | 16 [153] | 67 |
| Yu et al. | 7–19 | dihedral angles | Bayesian | 82 | 62 |
In recent years, a few attempts have been carried out to predict the φ/ψ backbone dihedral angles associated with each residue, based on the preferences observed in the Ramachandran map [172]. The highly preferred regions of dihedral angle pairs define the predicted classes. Both ANN- and SVM-based predictions [173,174] have been carried out, with a special focus on the coil regions. Prediction accuracy of above 80% was reported. Another approach predicting the probable dihedral angle associated with the coil residues has been developed [175]. The favoured dihedral region in the Ramachandran plot was divided into bins (30 × 30) and the probability of occurrence in these bins is predicted. Significant improvement could be achieved over random prediction and about 80% accuracy is reported for the prediction of the top 20 populated bins. Prediction of dihedral angles also helped in improving the accuracy of secondary structure prediction [135]. In this approach based on SVMs, an accuracy of prediction of about 54–57% was obtained by clustering the φ/ψ space into seven regions based on the population distribution.
Absolute values of φ and ψ angles are also predicted by several methods mainly based on ANNs [176–178]. Predicted secondary structure and/or solvent accessibility were combined with the sequence or profile information, as inputs for these learning methods. The ANGLOR method [177] was assessed to provide an error of 28° and 46° for the φ and ψ dihedral angles, respectively. These values were reduced to 22° and 36° with the improved Real-SPINE method [179].
Local structure prediction beyond the three-state secondary structures has been shown to improve recognition of remote homologues [180]. Prediction of absolute values of the dihedral angles has been incorporated in many recent fold recognition tools [105,113]. More than 5% increase in efficiency of detection of homologous folds was reported, with the addition of dihedral angle predictions [105]. A recent study also demonstrated that the use of local structure preference information significantly improves the quality of fold recognition [181].
2.2.3. Fragment-based fold recognition
The local regions of strong sequence–structure relationships are extracted as fragments and are assembled to generate the fold. The recognition of related folds by I-TASSER is mainly based on a profile–profile alignment method [98]. The efficiency of the profile search (see §2.1) is enhanced by the addition of secondary structure information. Various strategies involving HMMs [182], PSI-BLAST profiles [42], global [29] and local [30] dynamic programming algorithms are employed in the profile-based search of I-TASSER. The continuous fragments aligned with regions of multiple templates are reassembled into complete models [183]. The unaligned regions without structurally similar segments in the template are built by ab initio modelling, i.e. generating structural models based on physico-chemical and mechanical properties, without the use of a template [184].
In the absence of a template of similar fold, the target protein could still have local structural motifs observed in the available protein structures [185]. Though a homologous fold cannot be identified reliably, a probable model could be generated from a collection of such local motifs. This forms the basis of ROSETTA that generates models from fragments compatible with the sequence [186–188].
Hybrid template-based approaches associate fragment conformations for the sequence and detect distant fold similarities based on the fragment similarities. BBSP (building blocks structure predictor) [189] is one such algorithm that gave better accuracy when compared with COMPASS [190], HHpred [51] and PHYRE2 [95] on a dataset of 100 targets. Local backbone conformations from fragments are also used along with coarse-grained force fields to yield an energy optimized final model [191].
The generation of models by the combination of fragments is a remarkable development which can also enhance de novo structure prediction. SAs or local structure libraries can contribute significantly in this aspect and several modelling approaches based on such fragment libraries have been developed [163,192–196]. A combination of fragment assembly and lattice-based folding has been adopted to achieve fast conformational sampling and refine the models generated [108,197,198].
The length of fragments and the fragment insertion length can be chosen separately for different structural class of proteins; the α-helical proteins are modelled better with large fragment lengths and insertion size, whereas β-strand-rich proteins require shorter fragment insertions [199].
2.2.4. Refining local structure prediction
As mentioned earlier, the residue preferences for a local structure are strongly influenced by the structural environment [57,200–204]. Thus, the prediction of local structures could be enhanced with the information of other structural features. As the amino acid preferences for secondary structures differ between buried and solvent-exposed regions of protein structure [205–207], solvent accessibility is sometimes predicted prior to secondary structure prediction [205]. Addition of accessibility information resulted in the improvement of some probabilistic secondary structure prediction methods [208]. It was also reported that the use of residue-specific accessibility cut-offs and multi-state accessibilities improves the prediction of secondary structures. Discarding active site residues with functional constraints from the learning set also resulted in an improvement in the sensitivity of one- to three-dimensional substitution tables with representing amino acid preferences for local structural environments [209].
The stability of a local structural fragment might be determined by interactions not necessarily constrained within the fragment [203,210–212]. Such cases reflect weak sequence–structure relationships. Considering long sequence windows (15–20 residues) for prediction of local structures helps to include the effects of non-local interactions to a large extent [123]. However, the accuracy of local structure prediction is still limited by the inability to include long-range interactions [213,214]. A systematic identification of weak signals of sequence–structure relationships can help in refining the local structure prediction methodologies. Studies on chameleon sequences characterized by locally unstable structures have been carried out to explore such weak relationships [200,215]. Prediction of local regions of flexibility is gaining equal importance [216,217] and the use of flexibility information can aid in refining the available fold recognition protocols by complementing the local conformation predictions. Figure 5 highlights the high confidence predictions of secondary structures, flexible and disordered regions on glutamate mutase [218]. Structural dynamic profiles may also be used as another feature that can be used for fold detection.
Figure 5.
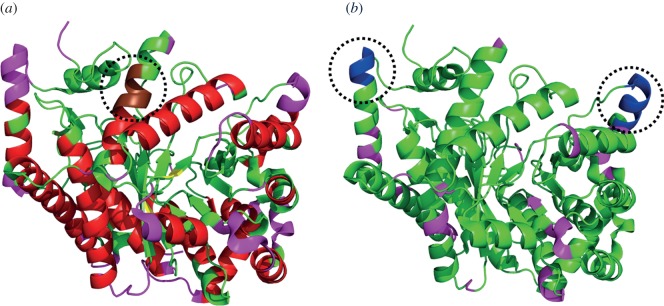
Prediction of local conformational rigidity and flexibility. Structure of glutamate mutase [PDB ID: 1CCW, chain B] [218] (a) highlighting the predicted secondary structures [219]: helices (red) and strands (yellow), flexible regions [217] (pink) and a discordant helix with high strand propensity (brown, highlighted within the dotted circle) [215]. (b) The flexible regions predicted (probability > 0.5 and confidence > 10) by PredyFlexy method [216] (pink) and the predicted disordered region [220] is shown in blue (highlighted within the dotted circles). (Online version in colour.)
2.2.5. In search of specificities
The knowledge of sequence–sequence and sequence–structure relationships is gained on the premise that it is applicable to all proteins. Even though this is often true, some specificities have also been identified among certain sets of proteins.
The amino acid composition varies between different proteomes. Depending on the organism, the variation could be sometimes quite significant such that the standard substitution matrices may fail to find the right homologues. Based on this perspective, substitution matrices adjusted for the background composition are derived [221]. These matrices are reported to be quite efficient in detecting homologues from the species. Species-specific variations are also observed with respect to amino acid propensities for local protein structures [222]. This observation is quite interesting and the specificities could be exploited in improving the accuracy of approaches for local structure prediction. A study conducted in this direction showed promising results with Plasmodium falciparum, Arabidopsis thaliana and Saccharomyces cerevisiae [223]. Considering the compositions corresponding to both query and template improves profile comparisons [224]. The amino acid preferences for local structures also show variations among different structural classes of proteins [225]. These specific preferences could be carefully exploited to enhance the accuracy of local structure prediction. The information on the specific amino acid preferences has been used to develop fold recognition approaches dedicated for specific classes of proteins [226,227].
2.3. Some successful methods
In order to assess the performance of different methods for protein structure prediction, Moult and co-workers introduced a public experiment called critical assessment of techniques for protein structure prediction (CASP). The participants are provided with protein sequences for which the structures have not been released. The I-TASSER server [108] gave the best performance in the recent CASP assessments (CASP 8, 9 and 10) of fold prediction servers [228]. The other servers in the top category were Robetta [186], Quark [229], HHpred [140], pmodeller [230], RAPTOR [87], pro-sp3 TASSER [231] and FALCON [232].
HHpred uses a novel HMM-based profile comparison method with enhanced accuracy by incorporating information on predicted secondary structure [51]. FALCON employs a fragment-HMM approach [232] where both the preferred dihedral angles of each residue and sequence–structure relationships from fragment libraries are integrated to predict structural models for a sequence.
It has become a common strategy to combine different predicted structural features along with amino acid sequences to improve the accuracy of profile-based fold detection [55,106,177,181,233]. Weighted matching of multiple profiles (sequence and structural features) has also been employed to enhance the accuracy of recognizing homologous folds [105,234].
2.3.1. Combining different approaches
One of the main limitations of the available fold recognition procedures is that they often fail to pick the correct template as the top hit. However in most cases, the right fold occurs among the top 10 or 20 hits [235]. The specificity can be improved by assessing the results of several different methods. This idea has led to the development of many meta-servers which run different fold recognition approaches and report the probable folds based on a consensus approach [98,183,236–241]. They also integrate methods for prediction of local structural features and scoring functions to assess and refine the results.
Hybrid methods generate models by combining structures of multiple templates which are either a set of initial predicted models or those obtained from different fold recognition methods [89,198,231,242–245]. Pro-sp3-TASSSER [231] is one such method that identifies template fold based on PROSPECTOR [102] and SP3 [106]. They also involve realignment of target–template in the uncertain regions [197,246,247]. Pcons [230,248] generates structural models based on the target–template alignments generated by different approaches and has been quite competitive in the recent CASP experiments. Nevertheless, a consensus approach may also suppress the true result, if only one of the underlying methods is successful [249].
The recent IntFOLD-TS approach [250] is one of these successful consensus methods; it integrates multiple sequence–structure alignments from methods such as SP3 [106], SPARKS [114], HHsearch [140] and COMA [251] and generates many models with multiple and single templates and scores them. The combination of these methods resulted in a better accuracy in generating models for 117 CASP targets when compared with the component methods individually. eThread [252] is another meta-threading approach that uses a combination of 10 methods and generates consensus alignments using machine learning techniques. This approach is reported to be more sensitive than any of the component methods and nearly 50% of the models were of high quality (TMscore > 0.5) [253].
3. Conclusion
The favourable local backbone conformations and the associated amino acid preferences can be carefully extracted to predict the conformation of a new sequence. Several methods for the efficient prediction of local backbone conformations have been proposed in recent years. It is becoming increasingly clear that these methods can contribute significantly to improve the accuracy of identifying related folds. Careful extraction of sequence–structure relationships is important for reducing the extent of false predictions. The knowledge of preferred local conformations is also used in generating structural models by the assembly of fragments. Strong signals of sequence–structure preferences need to discriminate from weak associations like chameleon sequences, flexible and disordered regions. The use of predicted flexibility profiles of a protein structure may also add to the accuracy of recognizing correct folds. The sequence–structure relationships learnt can be further refined based on specific proteomes or based on the structural class of proteins. Preliminary studies with available data point towards an improvement in prediction accuracy with such dataset-specific learning. Hence an efficient integration of local structural preferences with the global features helps significantly in our efforts for recognizing the fold that an amino acid sequence will adopt.
Acknowledgements
We thank Dr Adam Wilkins (Frontiers in Life Sciences doctorate school (FDV Paris, France), and Biochemistry, Biotherapy, Molecular Biology, Infectiology—B3MI doctorate school (Univ Paris Diderot and Univ Paris Descartes, Paris, France)) for useful discussions and suggestions. The authors also thank Jean-Christophe Gelly, Catherine Etchebest, N. Srinivasan, R. Sowdhamini, R.M. Bhaskara, S. Mahajan and G. Agarwal for fruitful discussions.
Funding statement
This work was supported by grants from the Ministry of Research (France), Univ Paris Diderot (France), Sorbonne Paris Cité (France), the National Institute of Blood Transfusion (INTS, France) and the National Institute for Health and Medical Research (INSERM, France). A.P.J. received a grant from CEFIPRA number 3903-E, and acknowledges NCBS for the post-doctoral fellowship. A.B. acknowledges CEFIPRA for a collaborative grant (no. 3903-E).
References
- 1.Anderson AC. 2003. The process of structure-based drug design. Chem. Biol. 10, 787–797. ( 10.1016/j.chembiol.2003.09.002) [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Morrow JK, Tran HT, Phatak SS, Du-Cuny L, Zhang S. 2013. From laptop to benchtop to bedside: structure-based drug design on protein targets. Curr. Pharm. Des. 18, 1217–1239. ( 10.2174/138161212799436386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verlinde CL, Hol WG. 1994. Structure-based drug design: progress, results and challenges. Structure 2, 577–587. ( 10.1016/S0969-2126(00)00060-5) [DOI] [PubMed] [Google Scholar]
- 4.Berman HM, et al. 2009. The protein structure initiative structural genomics knowledgebase. Nucleic Acids Res. 37, D365–D368. ( 10.1093/nar/gkn790) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandonia JM, Brenner SE. 2006. The impact of structural genomics: expectations and outcomes. Science 311, 347–351. ( 10.1126/science.1121018) [DOI] [PubMed] [Google Scholar]
- 6.Drmanac R, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81. ( 10.1126/science.1181498) [DOI] [PubMed] [Google Scholar]
- 7.International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945. ( 10.1038/nature03001) [DOI] [PubMed] [Google Scholar]
- 8.Lander ES. 2011. Initial impact of the sequencing of the human genome. Nature 470, 187–197. ( 10.1038/nature09792) [DOI] [PubMed] [Google Scholar]
- 9.Metzker ML. 2010. Sequencing technologies—the next generation. Nat. Rev. Genet. 11, 31–46. ( 10.1038/nrg2626) [DOI] [PubMed] [Google Scholar]
- 10.Morey M, Fernandez-Marmiesse A, Castineiras D, Fraga JM, Couce ML, Cocho JA. 2013. A glimpse into past, present, and future DNA sequencing. Mol. Genet. Metab. 110, 3–24. ( 10.1016/j.ymgme.2013.04.024) [DOI] [PubMed] [Google Scholar]
- 11.Parkhill J. 2013. What has high-throughput sequencing ever done for us? Nat. Rev. Microbiol. 11, 664–665. ( 10.1038/nrmicro3112) [DOI] [PubMed] [Google Scholar]
- 12.Petrey D, Honig B. 2005. Protein structure prediction: inroads to biology. Mol. Cell 20, 811–819. ( 10.1016/j.molcel.2005.12.005) [DOI] [PubMed] [Google Scholar]
- 13.Zhang QC, et al. 2012. Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature 490, 556–560. ( 10.1038/nature11503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kauffman C, Rangwala H, Karypis G. 2008. Improving homology models for protein-ligand binding sites. Comput. Syst. Bioinform. Conf. 7, 211–222. [PubMed] [Google Scholar]
- 15.Bibby J, Keegan RM, Mayans O, Winn MD, Rigden DJ. 2012. AMPLE: a cluster-and-truncate approach to solve the crystal structures of small proteins using rapidly computed ab initio models. Acta Crystallogr. D Biol. Crystallogr. 68, 1622–1631. ( 10.1107/S0907444912039194) [DOI] [PubMed] [Google Scholar]
- 16.Alber F, et al. 2007. The molecular architecture of the nuclear pore complex. Nature 450, 695–701. ( 10.1038/nature06405) [DOI] [PubMed] [Google Scholar]
- 17.Lasker K, Phillips JL, Russel D, Velazquez-Muriel J, Schneidman-Duhovny D, Tjioe E, Webb B, Schlessinger A, Sali A. 2010. Integrative structure modeling of macromolecular assemblies from proteomics data. Mol. Cell. Proteomics 9, 1689–1702. ( 10.1074/mcp.R110.000067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Topf M, Lasker K, Webb B, Wolfson H, Chiu W, Sali A. 2008. Protein structure fitting and refinement guided by cryo-EM density. Structure 16, 295–307. ( 10.1016/j.str.2007.11.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2007. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. Ch. 2, Unit 2.9 ( 10.1002/0471140864.ps0209s50) [DOI] [PubMed] [Google Scholar]
- 20.Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, Sali A. 2011. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc. Natl Acad. Sci. USA 108, 15 810–15 815. ( 10.1073/pnas.1106030108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen J, Zhang W, Fang H, Perkins R, Tong W, Hong H. 2013. Homology modeling, molecular docking, and molecular dynamics simulations elucidated alpha-fetoprotein binding modes. BMC Bioinformatics 14(Suppl. 14), S6 ( 10.1186/1471-2105-14-S14-S6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tseng CY, Gajewski M, Danani A, Tuszynski JA. In press. Homology and molecular dynamics models of toll-like receptor 7 protein and its dimerization. Chem. Biol. Drug Des. ( 10.1111/cbdd.12278) [DOI] [PubMed] [Google Scholar]
- 23.Drew K, et al. 2011. the proteome folding project: proteome-scale prediction of structure and function. Genome Res. 21, 1981–1994. ( 10.1101/gr.121475.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller A, MacCallum RM, Sternberg MJ. 2002. Structural characterization of the human proteome. Genome Res. 12, 1625–1641. ( 10.1101/gr.221202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y. 2008. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 18, 342–348. ( 10.1016/j.sbi.2008.02.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kihara D, Skolnick J. 2003. The PDB is a covering set of small protein structures. J. Mol. Biol. 334, 793–802. ( 10.1016/j.jmb.2003.10.027) [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Hubner IA, Arakaki AK, Shakhnovich E, Skolnick J. 2006. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl Acad. Sci. USA 103, 2605–2610. ( 10.1073/pnas.0509379103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. ( 10.1016/S0022-2836(05)80360-2) [DOI] [PubMed] [Google Scholar]
- 29.Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453. ( 10.1016/0022-2836(70)90057-4) [DOI] [PubMed] [Google Scholar]
- 30.Smith TF, Waterman MS. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147, 195–197. ( 10.1016/0022-2836(81)90087-5) [DOI] [PubMed] [Google Scholar]
- 31.Zemla A. 2003. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 31, 3370–3374. ( 10.1093/nar/gkg571) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chothia C, Lesk AM. 1986. The relation between the divergence of sequence and structure in proteins. EMBO J. 5, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balaji S, Sujatha S, Kumar SS, Srinivasan N. 2001. PALI—a database of Phylogeny and ALIgnment of homologous protein structures. Nucleic Acids Res. 29, 61–65. ( 10.1093/nar/29.1.61) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murzin AG, Brenner SE, Hubbard T, Chothia C. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540. ( 10.1016/S0022-2836(05)80134-2) [DOI] [PubMed] [Google Scholar]
- 35.Gelly JC, Joseph AP, Srinivasan N, de Brevern AG. 2011. iPBA: a tool for protein structure comparison using sequence alignment strategies. Nucleic Acids Res. 39, W18–W23. ( 10.1093/nar/gkr333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joseph AP, Srinivasan N, de Brevern AG. 2011. Improvement of protein structure comparison using a structural alphabet. Biochimie 93, 1434–1445. ( 10.1016/j.biochi.2011.04.010) [DOI] [PubMed] [Google Scholar]
- 37.Chakravarty S, Godbole S, Zhang B, Berger S, Sanchez R. 2008. Systematic analysis of the effect of multiple templates on the accuracy of comparative models of protein structure. BMC Struct. Biol. 8, 31 ( 10.1186/1472-6807-8-31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rost B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12, 85–94. ( 10.1093/protein/12.2.85) [DOI] [PubMed] [Google Scholar]
- 39.Emsley J, White HE, O'Hara BP, Oliva G, Srinivasan N, Tickle IJ, Blundell TL, Pepys MB, Wood SP. 1994. Structure of pentameric human serum amyloid P component. Nature 367, 338–345. ( 10.1038/367338a0) [DOI] [PubMed] [Google Scholar]
- 40.Ohlson T, Wallner B, Elofsson A. 2004. Profile–profile methods provide improved fold-recognition: a study of different profile–profile alignment methods. Proteins 57, 188–197. ( 10.1002/prot.20184) [DOI] [PubMed] [Google Scholar]
- 41.Rychlewski L, Jaroszewski L, Li W, Godzik A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 9, 232–241. ( 10.1110/ps.9.2.232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. ( 10.1093/nar/25.17.3389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. 2001. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005. ( 10.1093/nar/29.14.2994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Pio F, Pawlowski K, Godzik A. 2000. Saturated BLAST: an automated multiple intermediate sequence search used to detect distant homology. Bioinformatics 16, 1105–1110. ( 10.1093/bioinformatics/16.12.1105) [DOI] [PubMed] [Google Scholar]
- 45.Park J, Karplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C. 1998. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 284, 1201–1210. ( 10.1006/jmbi.1998.2221) [DOI] [PubMed] [Google Scholar]
- 46.Kim BH, Cong Q, Grishin NV. 2010. HangOut: generating clean PSI-BLAST profiles for domains with long insertions. Bioinformatics 26, 1564–1565. ( 10.1093/bioinformatics/btq208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14, 755–763. ( 10.1093/bioinformatics/14.9.755) [DOI] [PubMed] [Google Scholar]
- 48.Eddy SR. 2009. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211. [PubMed] [Google Scholar]
- 49.Hughey R, Krogh A. 1996. Hidden Markov models for sequence analysis: extension and analysis of the basic method. Comput. Appl. Biosci. 12, 95–107. [DOI] [PubMed] [Google Scholar]
- 50.Madera M. 2008. Profile Comparer: a program for scoring and aligning profile hidden Markov models. Bioinformatics 24, 2630–2631. ( 10.1093/bioinformatics/btn504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soding J. 2005. Protein homology detection by HMM–HMM comparison. Bioinformatics 21, 951–960. ( 10.1093/bioinformatics/bti125) [DOI] [PubMed] [Google Scholar]
- 52.Morgenstern B, Werner N, Prohaska SJ, Steinkamp R, Schneider I, Subramanian AR, Stadler PF, Weyer-Menkhoff J. 2005. Multiple sequence alignment with user-defined constraints at GOBICS. Bioinformatics 21, 1271–1273. ( 10.1093/bioinformatics/bti142) [DOI] [PubMed] [Google Scholar]
- 53.Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217. ( 10.1006/jmbi.2000.4042) [DOI] [PubMed] [Google Scholar]
- 54.O'Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C. 2004. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J. Mol. Biol. 340, 385–395. ( 10.1016/j.jmb.2004.04.058) [DOI] [PubMed] [Google Scholar]
- 55.Sadreyev RI, Tang M, Kim BH, Grishin NV. 2007. COMPASS server for remote homology inference. Nucleic Acids Res. 35, W653–W658. ( 10.1093/nar/gkm293) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramanian AR, Weyer-Menkhoff J, Kaufmann M, Morgenstern B. 2005. DIALIGN-T: an improved algorithm for segment-based multiple sequence alignment. BMC Bioinformatics 6, 66 ( 10.1186/1471-2105-6-66) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Overington J, Johnson MS, Sali A, Blundell TL. 1990. Tertiary structural constraints on protein evolutionary diversity: templates, key residues and structure prediction. Proc. R. Soc. Lond. B 241, 132–145. ( 10.1098/rspb.1990.0077) [DOI] [PubMed] [Google Scholar]
- 58.Knight RD, Freeland SJ, Landweber LF. 2001. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2, research 0010 ( 10.1186/gb-2001-2-4-research0010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu YK, Wootton JC, Altschul SF. 2003. The compositional adjustment of amino acid substitution matrices. Proc. Natl Acad. Sci. USA 100, 15 688–15 693. ( 10.1073/pnas.2533904100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biegert A, Soding J. 2009. Sequence context-specific profiles for homology searching. Proc. Natl Acad. Sci. USA 106, 3770–3775. ( 10.1073/pnas.0810767106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boratyn GM, Schaffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL. 2012. Domain enhanced lookup time accelerated BLAST. Biol. Direct 7, 12 ( 10.1186/1745-6150-7-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marchler-Bauer A, et al. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229. ( 10.1093/nar/gkq1189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Becker E, Cotillard A, Meyer V, Madaoui H, Guerois R. 2007. HMM-Kalign: a tool for generating sub-optimal HMM alignments. Bioinformatics 23, 3095–3097. ( 10.1093/bioinformatics/btm492) [DOI] [PubMed] [Google Scholar]
- 64.Panchenko AR. 2003. Finding weak similarities between proteins by sequence profile comparison. Nucleic Acids Res. 31, 683–689. ( 10.1093/nar/gkg154) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson LS, Eddy SR, Portugaly E. 2010. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431 ( 10.1186/1471-2105-11-431) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Remmert M, Biegert A, Hauser A, Soding J. 2012. HHblits: lightning-fast iterative protein sequence searching by HMM–HMM alignment. Nat. Methods 9, 173–175. ( 10.1038/nmeth.1818) [DOI] [PubMed] [Google Scholar]
- 67.Bashton M, Chothia C. 2007. The generation of new protein functions by the combination of domains. Structure 15, 85–99. ( 10.1016/j.str.2006.11.009) [DOI] [PubMed] [Google Scholar]
- 68.Todd AE, Orengo CA, Thornton JM. 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307, 1113–1143. ( 10.1006/jmbi.2001.4513) [DOI] [PubMed] [Google Scholar]
- 69.Vesterstrom J, Taylor WR. 2006. Flexible secondary structure based protein structure comparison applied to the detection of circular permutation. J. Comput. Biol. 13, 43–63. ( 10.1089/cmb.2006.13.43) [DOI] [PubMed] [Google Scholar]
- 70.Bujnicki JM. 2002. Sequence permutations in the molecular evolution of DNA methyltransferases. BMC Evol. Biol. 2, 3 ( 10.1186/1471-2148-2-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeltsch A. 1999. Circular permutations in the molecular evolution of DNA methyltransferases. J. Mol. Evol. 49, 161–164. ( 10.1007/PL00006529) [DOI] [PubMed] [Google Scholar]
- 72.Kelil A, Wang S, Brzezinski R, Fleury A. 2007. CLUSS: clustering of protein sequences based on a new similarity measure. BMC Bioinformatics 8, 286 ( 10.1186/1471-2105-8-286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin J, Anamika K, Srinivasan N. 2010. Classification of protein kinases on the basis of both kinase and non-kinase regions. PLoS ONE 5, e12460 ( 10.1371/journal.pone.0012460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee B, Lee D. 2008. DAhunter: a web-based server that identifies homologous proteins by comparing domain architecture. Nucleic Acids Res. 36, W60–W64. ( 10.1093/nar/gkn172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin K, Zhu L, Zhang DY. 2006. An initial strategy for comparing proteins at the domain architecture level. Bioinformatics 22, 2081–2086. ( 10.1093/bioinformatics/btl366) [DOI] [PubMed] [Google Scholar]
- 76.Terrapon N, Weiner J, Grath S, Moore AD, Bornberg-Bauer E. 2013. Rapid similarity search of proteins using alignments of domain arrangements. Bioinformatics 30, 274–281. ( 10.1093/bioinformatics/btt379) [DOI] [PubMed] [Google Scholar]
- 77.Weiner J, 3rd, Thomas G, Bornberg-Bauer E. 2005. Rapid motif-based prediction of circular permutations in multi-domain proteins. Bioinformatics 21, 932–937. ( 10.1093/bioinformatics/bti085) [DOI] [PubMed] [Google Scholar]
- 78.Edgar RC, Sjolander K. 2004. A comparison of scoring functions for protein sequence profile alignment. Bioinformatics 20, 1301–1308. ( 10.1093/bioinformatics/bth090) [DOI] [PubMed] [Google Scholar]
- 79.Hendlich M, Lackner P, Weitckus S, Floeckner H, Froschauer R, Gottsbacher K, Casari G, Sippl MJ. 1990. Identification of native protein folds amongst a large number of incorrect models. The calculation of low energy conformations from potentials of mean force. J. Mol. Biol. 216, 167–180. ( 10.1016/S0022-2836(05)80068-3) [DOI] [PubMed] [Google Scholar]
- 80.Sippl MJ. 1990. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 213, 859–883. ( 10.1016/S0022-2836(05)80269-4) [DOI] [PubMed] [Google Scholar]
- 81.Godzik A, Kolinski A, Skolnick J. 1992. Topology fingerprint approach to the inverse protein folding problem. J. Mol. Biol. 227, 227–238. ( 10.1016/0022-2836(92)90693-E) [DOI] [PubMed] [Google Scholar]
- 82.Godzik A, Skolnick J. 1992. Sequence-structure matching in globular proteins: application to supersecondary and tertiary structure determination. Proc. Natl Acad. Sci. USA 89, 12 098–12 102. ( 10.1073/pnas.89.24.12098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jones DT, Taylor WR, Thornton JM. 1992. A new approach to protein fold recognition. Nature 358, 86–89. ( 10.1038/358086a0) [DOI] [PubMed] [Google Scholar]
- 84.Sippl MJ, Weitckus S. 1992. Detection of native-like models for amino acid sequences of unknown three-dimensional structure in a data base of known protein conformations. Proteins 13, 258–271. ( 10.1002/prot.340130308) [DOI] [PubMed] [Google Scholar]
- 85.Bryant SH. 1996. Evaluation of threading specificity and accuracy. Proteins 26, 172–185. () [DOI] [PubMed] [Google Scholar]
- 86.Xu Y, Xu D, Uberbacher EC. 1998. An efficient computational method for globally optimal threading. J. Comput. Biol. 5, 597–614. ( 10.1089/cmb.1998.5.597) [DOI] [PubMed] [Google Scholar]
- 87.Xu J, Li M, Kim D, Xu Y. 2003. RAPTOR: optimal protein threading by linear programming. J. Bioinform. Comput. Biol. 1, 95–117. ( 10.1142/S0219720003000186) [DOI] [PubMed] [Google Scholar]
- 88.Ma J, Wang S, Zhao F, Xu J. 2013. Protein threading using context-specific alignment potential. Bioinformatics 29, i257–i265. ( 10.1093/bioinformatics/btt210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fischer D. 2000. Hybrid fold recognition: combining sequence derived properties with evolutionary information. Pac. Symp. Biocomput. 2000, 119–130. [PubMed] [Google Scholar]
- 90.Rice DW, Eisenberg D. 1997. A 3D-1D substitution matrix for protein fold recognition that includes predicted secondary structure of the sequence. J. Mol. Biol. 267, 1026–1038. ( 10.1006/jmbi.1997.0924) [DOI] [PubMed] [Google Scholar]
- 91.Shi J, Blundell TL, Mizuguchi K. 2001. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310, 243–257. ( 10.1006/jmbi.2001.4762) [DOI] [PubMed] [Google Scholar]
- 92.Torda AE, Procter JB, Huber T. 2004. Wurst: a protein threading server with a structural scoring function, sequence profiles and optimized substitution matrices. Nucleic Acids Res. 32, W532–W535. ( 10.1093/nar/gkh357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mooney C, Pollastri G. 2009. Beyond the Twilight Zone: automated prediction of structural properties of proteins by recursive neural networks and remote homology information. Proteins 77, 181–190. ( 10.1002/prot.22429) [DOI] [PubMed] [Google Scholar]
- 94.Rost B, Schneider R, Sander C. 1997. Protein fold recognition by prediction-based threading. J. Mol. Biol. 270, 471–480. ( 10.1006/jmbi.1997.1101) [DOI] [PubMed] [Google Scholar]
- 95.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371. ( 10.1038/nprot.2009.2) [DOI] [PubMed] [Google Scholar]
- 96.Przybylski D, Rost B. 2004. Improving fold recognition without folds. J. Mol. Biol. 341, 255–269. ( 10.1016/j.jmb.2004.05.041) [DOI] [PubMed] [Google Scholar]
- 97.Rost B, Sander C. 1995. Progress of 1D protein structure prediction at last. Proteins 23, 295–300. ( 10.1002/prot.340230304) [DOI] [PubMed] [Google Scholar]
- 98.Wu S, Zhang Y. 2007. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375–3382. ( 10.1093/nar/gkm251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shah PK, Aloy P, Bork P, Russell RB. 2005. Structural similarity to bridge sequence space: finding new families on the bridges. Protein Sci. 14, 1305–1314. ( 10.1110/ps.041187405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sillitoe I, Dibley M, Bray J, Addou S, Orengo C. 2005. Assessing strategies for improved superfamily recognition. Protein Sci. 14, 1800–1810. ( 10.1110/ps.041056105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Skolnick J, Kihara D. 2001. Defrosting the frozen approximation: PROSPECTOR—a new approach to threading. Proteins 42, 319–331. ( 10.1002/1097-0134(20010215)42:3%3C319::AID-PROT30%3E3.0.CO;2-A) [DOI] [PubMed] [Google Scholar]
- 102.Skolnick J, Kihara D, Zhang Y. 2004. Development and large scale benchmark testing of the PROSPECTOR_3 threading algorithm. Proteins 56, 502–518. ( 10.1002/prot.20106) [DOI] [PubMed] [Google Scholar]
- 103.Teichert F, Minning J, Bastolla U, Porto M. 2010. High quality protein sequence alignment by combining structural profile prediction and profile alignment using SABER-TOOTH. BMC Bioinformatics 11, 251 ( 10.1186/1471-2105-11-251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y, Sadreyev RI, Grishin NV. 2009. PROCAIN: protein profile comparison with assisting information. Nucleic Acids Res. 37, 3522–3530. ( 10.1093/nar/gkp212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang W, Liu S, Zhou Y. 2008. SP5: improving protein fold recognition by using torsion angle profiles and profile-based gap penalty model. PLoS ONE 3, e2325 ( 10.1371/journal.pone.0002325) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou H, Zhou Y. 2005. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins 58, 321–328. ( 10.1002/prot.20308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jones DT. 1999. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 287, 797–815. ( 10.1006/jmbi.1999.2583) [DOI] [PubMed] [Google Scholar]
- 108.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 ( 10.1186/1471-2105-9-40) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brylinski M. 2013. The utility of artificially evolved sequences in protein threading and fold recognition. J. Theor. Biol. 328, 77–88. ( 10.1016/j.jtbi.2013.03.018) [DOI] [PubMed] [Google Scholar]
- 110.Pauling L, Corey RB, Branson HR. 1951. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl Acad. Sci. USA 37, 205–211. ( 10.1073/pnas.37.4.205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Geourjon C, Combet C, Blanchet C, Deleage G. 2001. Identification of related proteins with weak sequence identity using secondary structure information. Protein Sci. 10, 788–797. ( 10.1110/ps.30001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Westhead DR, Thornton JM. 1998. Protein structure prediction. Curr. Opin. Biotechnol. 9, 383–389. ( 10.1016/S0958-1669(98)80012-8) [DOI] [PubMed] [Google Scholar]
- 113.Wu S, Zhang Y. 2008. MUSTER: improving protein sequence profile–profile alignments by using multiple sources of structure information. Proteins 72, 547–556. ( 10.1002/prot.21945) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang Y, Faraggi E, Zhao H, Zhou Y. 2011. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics 27, 2076–2082. ( 10.1093/bioinformatics/btr350) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202. ( 10.1006/jmbi.1999.3091) [DOI] [PubMed] [Google Scholar]
- 116.Guo J, Chen H, Sun Z, Lin Y. 2004. A novel method for protein secondary structure prediction using dual-layer SVM and profiles. Proteins 54, 738–743. ( 10.1002/prot.10634) [DOI] [PubMed] [Google Scholar]
- 117.Bondugula R, Xu D. 2007. MUPRED: a tool for bridging the gap between template based methods and sequence profile based methods for protein secondary structure prediction. Proteins 66, 664–670. ( 10.1002/prot.21177) [DOI] [PubMed] [Google Scholar]
- 118.Cheng H, Sen TZ, Jernigan RL, Kloczkowski A. 2007. Consensus Data Mining (CDM) Protein Secondary Structure Prediction Server: combining GOR V and fragment database mining (FDM). Bioinformatics 23, 2628–2630. ( 10.1093/bioinformatics/btm379) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karypis G. 2006. YASSPP: better kernels and coding schemes lead to improvements in protein secondary structure prediction. Proteins 64, 575–586. ( 10.1002/prot.21036) [DOI] [PubMed] [Google Scholar]
- 120.Nguyen MN, Rajapakse JC. 2007. Prediction of protein secondary structure with two-stage multi-class SVMs. Int. J. Data Min. Bioinform. 1, 248–269. ( 10.1504/IJDMB.2007.011612) [DOI] [PubMed] [Google Scholar]
- 121.Pollastri G, McLysaght A. 2005. Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics 21, 1719–1720. ( 10.1093/bioinformatics/bti203) [DOI] [PubMed] [Google Scholar]
- 122.Rost B, Eyrich VA. 2001. EVA: large-scale analysis of secondary structure prediction. Proteins 45, 192–199. ( 10.1002/prot.10051) [DOI] [PubMed] [Google Scholar]
- 123.Rost B. 2001. Review: protein secondary structure prediction continues to rise. J. Struct. Biol. 134, 204–218. ( 10.1006/jsbi.2001.4336) [DOI] [PubMed] [Google Scholar]
- 124.de Brevern AG, Benros C, Hazout S. 2005. Structural alphabet: from a local point of view to a global description of protein 3D structures. In Bioinformatics: new research (ed. Yan PV.), pp. 128–187. New York, NY: Nova Publishers. [Google Scholar]
- 125.Martin J, Letellier G, Marin A, Taly J-F, de Brevern AG, Gibrat J-F. 2005. Protein secondary structure assignment revisited: a detailed analysis of different assignment methods. BMC Struct. Biol. 5, 17 ( 10.1186/1472-6807-5-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tyagi M, Bornot A, Offmann B, de Brevern AG. 2009. Analysis of loop boundaries using different local structure assignment methods. Protein Sci. 18, 1869–1881. ( 10.1002/pro.198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shahsavar A, Erfani Moghaddam M, Antonyuk SV, Khajeh K, Naderi-Manesh H. In preparation. Crystal structures of methylglyoxal synthase from Thermus sp. GH5 in the open and closed conformational states provide insight into the mechanism of allosteric regulation. [Google Scholar]
- 128.Kabsch W, Sander C. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. ( 10.1002/bip.360221211) [DOI] [PubMed] [Google Scholar]
- 129.Rost B, Sander C. 1993. Improved prediction of protein secondary structure by use of sequence profiles and neural networks. Proc. Natl Acad. Sci. USA 90, 7558–7562. ( 10.1073/pnas.90.16.7558) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pollastri G, Przybylski D, Rost B, Baldi P. 2002. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins 47, 228–235. ( 10.1002/prot.10082) [DOI] [PubMed] [Google Scholar]
- 131.Lin K, Simossis VA, Taylor WR, Heringa J. 2005. A simple and fast secondary structure prediction method using hidden neural networks. Bioinformatics 21, 152–159. ( 10.1093/bioinformatics/bth487) [DOI] [PubMed] [Google Scholar]
- 132.Kruus E, Thumfort P, Tang C, Wingreen NS. 2005. Gibbs sampling and helix-cap motifs. Nucleic Acids Res. 33, 5343–5353. ( 10.1093/nar/gki842) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cole C, Barber JD, Barton GJ. 2008. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201. ( 10.1093/nar/gkn238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Duan M, Huang M, Ma C, Li L, Zhou Y. 2008. Position-specific residue preference features around the ends of helices and strands and a novel strategy for the prediction of secondary structures. Protein Sci. 17, 1505–1512. ( 10.1110/ps.035691.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kountouris P, Hirst JD. 2009. Prediction of backbone dihedral angles and protein secondary structure using support vector machines. BMC Bioinformatics 10, 437 ( 10.1186/1471-2105-10-437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Montgomerie S, Sundararaj S, Gallin WJ, Wishart DS. 2006. Improving the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinformatics 7, 301 ( 10.1186/1471-2105-7-301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lin HN, Sung TY, Ho SY, Hsu WL. 2010. Improving protein secondary structure prediction based on short subsequences with local structure similarity. BMC Genomics 11(Suppl. 4), S4 ( 10.1186/1471-2164-11-S4-S4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang H, Zhang T, Chen K, Kedarisetti KD, Mizianty MJ, Bao Q, Stach W, Kurgan L. 2011. Critical assessment of high-throughput standalone methods for secondary structure prediction. Brief. Bioinform. 12, 672–688. ( 10.1093/bib/bbq088) [DOI] [PubMed] [Google Scholar]
- 139.Ginalski K, Grishin NV, Godzik A, Rychlewski L. 2005. Practical lessons from protein structure prediction. Nucleic Acids Res. 33, 1874–1891. ( 10.1093/nar/gki327) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248. ( 10.1093/nar/gki408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shi Y, Zhou J, Arndt D, Wishart DS, Lin G. 2008. Protein contact order prediction from primary sequences. BMC Bioinformatics 9, 255 ( 10.1186/1471-2105-9-255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fontana P, Bindewald E, Toppo S, Velasco R, Valle G, Tosatto SCE. 2005. The SSEA server for protein secondary structure alignment. Bioinformatics 21, 393–395. ( 10.1093/bioinformatics/bti013) [DOI] [PubMed] [Google Scholar]
- 143.Guillet V, Ohta N, Cabantous S, Newton A, Samama J-P. 2002. Crystallographic and biochemical studies of DivK reveal novel features of an essential response regulator in Caulobacter crescentus. J. Biol. Chem. 277, 42 003–42 010. ( 10.1074/jbc.M204789200) [DOI] [PubMed] [Google Scholar]
- 144.De La Cruz X, Thornton JM. 1999. Factors limiting the performance of prediction-based fold recognition methods. Protein Sci. 8, 750–759. ( 10.1110/ps.8.4.750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Venkatachalam CM. 1968. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 6, 1425–1436. ( 10.1002/bip.1968.360061006) [DOI] [PubMed] [Google Scholar]
- 146.Hutchinson EG, Thornton JM. 1994. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 3, 2207–2216. ( 10.1002/pro.5560031206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mansiaux Y, Joseph AP, Gelly JC, de Brevern AG. 2011. Assignment of PolyProline II conformation and analysis of sequence–structure relationship. PLoS ONE 6, e18401 ( 10.1371/journal.pone.0018401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Offmann B, Tyagi M, de Brevern AG. 2007. Local protein structures. Curr. Bioinform. 3, 165–202. ( 10.2174/157489307781662105) [DOI] [Google Scholar]
- 149.Joseph AP, Bornot A, de Brevern AG. 2010. Local structural alphabets. In Introduction to protein structure prediction: methods and algorithms (eds H Rangwala, G Karypis), pp. 75–106. Hoboken, NJ: John Wiley & Sons, Inc. [Google Scholar]
- 150.Bystroff C, Baker D. 1998. Prediction of local structure in proteins using a library of sequence–structure motifs. J. Mol. Biol. 281, 565–577. ( 10.1006/jmbi.1998.1943) [DOI] [PubMed] [Google Scholar]
- 151.Camproux AC, de Brevern AG, Hazout S, Tufféry P. 2001. Exploring the use of a structural alphabet for structural prediction of protein loops. Theor. Chem. Acc. 106, 28–35. ( 10.1007/s002140100261) [DOI] [Google Scholar]
- 152.Chen B, Johnson M. 2009. Protein local 3D structure prediction by Super Granule Support Vector Machines (Super GSVM). BMC Bioinformatics 10(Suppl. 11), S15 ( 10.1186/1471-2105-10-S11-S15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.de Brevern AG, Etchebest C, Hazout S. 2000. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins 41, 271–287. () [DOI] [PubMed] [Google Scholar]
- 154.Fetrow JS, Palumbo MJ, Berg G. 1997. Patterns, structures, and amino acid frequencies in structural building blocks, a protein secondary structure classification scheme. Proteins 27, 249–271. () [DOI] [PubMed] [Google Scholar]
- 155.Hunter CG, Subramaniam S. 2003. Protein fragment clustering and canonical local shapes. Proteins 50, 580–588. ( 10.1002/prot.10309) [DOI] [PubMed] [Google Scholar]
- 156.Rooman MJ, Rodriguez J, Wodak SJ. 1990. Relations between protein sequence and structure and their significance. J. Mol. Biol. 213, 337–350. ( 10.1016/S0022-2836(05)80195-0) [DOI] [PubMed] [Google Scholar]
- 157.Schuchhardt J, Schneider G, Reichelt J, Schomburg D, Wrede P. 1996. Local structural motifs of protein backbones are classified by self-organizing neural networks. Protein Eng. 9, 833–842. ( 10.1093/protein/9.10.833) [DOI] [PubMed] [Google Scholar]
- 158.Hunter CG, Subramaniam S. 2003. Protein local structure prediction from sequence. Proteins 50, 572–579. ( 10.1002/prot.10310) [DOI] [PubMed] [Google Scholar]
- 159.Dong Q, Wang X, Lin L, Wang Y. 2008. Analysis and prediction of protein local structure based on structure alphabets. Proteins 72, 163–172. ( 10.1002/prot.21904) [DOI] [PubMed] [Google Scholar]
- 160.Rangwala H, Kauffman C, Karypis G. 2009. svmPRAT: SVM-based protein residue annotation toolkit. BMC Bioinformatics 10, 439 ( 10.1186/1471-2105-10-439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sander O, Sommer I, Lengauer T. 2006. Local protein structure prediction using discriminative models. BMC Bioinformatics 7, 14 ( 10.1186/1471-2105-7-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yang AS, Wang LY. 2003. Local structure prediction with local structure-based sequence profiles. Bioinformatics 19, 1267–1274. ( 10.1093/bioinformatics/btg151) [DOI] [PubMed] [Google Scholar]
- 163.Bystroff C, Thorsson V, Baker D. 2000. HMMSTR: a hidden Markov model for local sequence-structure correlations in proteins. J. Mol. Biol. 301, 173–190. ( 10.1006/jmbi.2000.3837) [DOI] [PubMed] [Google Scholar]
- 164.Etchebest C, Benros C, Hazout S, de Brevern AG. 2005. A structural alphabet for local protein structures: improved prediction methods. Proteins 59, 810–827. ( 10.1002/prot.20458) [DOI] [PubMed] [Google Scholar]
- 165.de Brevern AG, Benros C, Gautier R, Valadie H, Hazout S, Etchebest C. 2004. Local backbone structure prediction of proteins. In Silico Biol. 4, 381–386. [PMC free article] [PubMed] [Google Scholar]
- 166.de Brevern AG, Etchebest C, Benros C, Hazout S. 2007. ‘Pinning strategy’: a novel approach for predicting the backbone structure in terms of protein blocks from sequence. J. Biosci. 32, 51–72. ( 10.1007/s12038-007-0006-3) [DOI] [PubMed] [Google Scholar]
- 167.Zimmermann O, Hansmann UH. 2008. LOCUSTRA: accurate prediction of local protein structure using a two-layer support vector machine approach. J. Chem. Inf. Model. 48, 1903–1908. ( 10.1021/ci800178a) [DOI] [PubMed] [Google Scholar]
- 168.Li Q, Zhou C, Liu H. 2009. Fragment-based local statistical potentials derived by combining an alphabet of protein local structures with secondary structures and solvent accessibilities. Proteins 74, 820–836. ( 10.1002/prot.22191) [DOI] [PubMed] [Google Scholar]
- 169.de Brevern AG, Hazout S. 2003. ‘Hybrid protein model’ for optimally defining 3D protein structure fragments. Bioinformatics 19, 345–353. ( 10.1093/bioinformatics/btf859) [DOI] [PubMed] [Google Scholar]
- 170.Bornot A, Etchebest C, de Brevern AG. 2009. A new prediction strategy for long local protein structures using an original description. Proteins 76, 570–587. ( 10.1002/prot.22370) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bornot A, Etchebest C, de Brevern AG. 2011. Predicting protein flexibility through the prediction of local structures. Proteins 79, 839–852. ( 10.1002/prot.22922) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ramachandran GN, Ramakrishnan C, Sasisekharan V. 1963. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99. ( 10.1016/S0022-2836(63)80023-6) [DOI] [PubMed] [Google Scholar]
- 173.Kuang R, Leslie CS, Yang AS. 2004. Protein backbone angle prediction with machine learning approaches. Bioinformatics 20, 1612–1621. ( 10.1093/bioinformatics/bth136) [DOI] [PubMed] [Google Scholar]
- 174.Zimmermann O, Hansmann UH. 2006. Support vector machines for prediction of dihedral angle regions. Bioinformatics 22, 3009–3015. ( 10.1093/bioinformatics/btl489) [DOI] [PubMed] [Google Scholar]
- 175.Helles G, Fonseca R. 2009. Predicting dihedral angle probability distributions for protein coil residues from primary sequence using neural networks. BMC Bioinformatics 10, 338 ( 10.1186/1471-2105-10-338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wood MJ, Hirst JD. 2005. Protein secondary structure prediction with dihedral angles. Proteins 59, 476–481. ( 10.1002/prot.20435) [DOI] [PubMed] [Google Scholar]
- 177.Wu S, Zhang Y. 2008. ANGLOR: a composite machine-learning algorithm for protein backbone torsion angle prediction. PLoS ONE 3, e3400 ( 10.1371/journal.pone.0003400) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xue B, Dor O, Faraggi E, Zhou Y. 2008. Real-value prediction of backbone torsion angles. Proteins 72, 427–433. ( 10.1002/prot.21940) [DOI] [PubMed] [Google Scholar]
- 179.Faraggi E, Xue B, Zhou Y. 2009. Improving the prediction accuracy of residue solvent accessibility and real-value backbone torsion angles of proteins by guided-learning through a two-layer neural network. Proteins 74, 847–856. ( 10.1002/prot.22193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Karchin R, Cline M, Mandel-Gutfreund Y, Karplus K. 2003. Hidden Markov models that use predicted local structure for fold recognition: alphabets of backbone geometry. Proteins 51, 504–514. ( 10.1002/prot.10369) [DOI] [PubMed] [Google Scholar]
- 181.Hu Y, Dong X, Wu A, Cao Y, Tian L, Jiang T. 2011. Incorporation of local structural preference potential improves fold recognition. PLoS ONE 6, e17215 ( 10.1371/journal.pone.0017215) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Karplus K, Barrett C, Hughey R. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics 14, 846–856. ( 10.1093/bioinformatics/14.10.846) [DOI] [PubMed] [Google Scholar]
- 183.Zhang Y, Arakaki AK, Skolnick J. 2005. TASSER: an automated method for the prediction of protein tertiary structures in CASP6. Proteins 61(Suppl. 7), 91–98. ( 10.1002/prot.20724) [DOI] [PubMed] [Google Scholar]
- 184.Zhang Y, Kolinski A, Skolnick J. 2003. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145–1164. ( 10.1016/S0006-3495(03)74551-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Grishin NV. 2001. Fold change in evolution of protein structures. J. Struct. Biol. 134, 167–185. ( 10.1006/jsbi.2001.4335) [DOI] [PubMed] [Google Scholar]
- 186.Chivian D, et al. 2003. Automated prediction of CASP-5 structures using the Robetta server. Proteins 53(Suppl. 6), 524–533. ( 10.1002/prot.10529) [DOI] [PubMed] [Google Scholar]
- 187.Rohl CA, Strauss CE, Chivian D, Baker D. 2004. Modeling structurally variable regions in homologous proteins with rosetta. Proteins 55, 656–677. ( 10.1002/prot.10629) [DOI] [PubMed] [Google Scholar]
- 188.Simons KT, Kooperberg C, Huang E, Baker D. 1997. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225. ( 10.1006/jmbi.1997.0959) [DOI] [PubMed] [Google Scholar]
- 189.Gullotto D, Nolassi MS, Bernini A, Spiga O, Niccolai N. 2012. Probing the protein space for extending the detection of weak homology folds. J. Theor. Biol. 320, 152–158. ( 10.1016/j.jtbi.2012.12.005) [DOI] [PubMed] [Google Scholar]
- 190.Sadreyev RI, Tang M, Kim BH, Grishin NV. 2009. COMPASS server for homology detection: improved statistical accuracy, speed and functionality. Nucleic Acids Res. 37, W90–W94. ( 10.1093/nar/gkp360) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Davtyan A, Schafer NP, Zheng W, Clementi C, Wolynes PG, Papoian GA. 2012. AWSEM-MD: protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J. Phys. Chem. B 116, 8494–8503. ( 10.1021/jp212541y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hvidsten TR, Kryshtafovych A, Komorowski J, Fidelis K. 2003. A novel approach to fold recognition using sequence-derived properties from sets of structurally similar local fragments of proteins. Bioinformatics 19(Suppl. 2), ii81–ii91. ( 10.1093/bioinformatics/btg1064) [DOI] [PubMed] [Google Scholar]
- 193.Jones DT, McGuffin LJ. 2003. Assembling novel protein folds from super-secondary structural fragments. Proteins 53(Suppl. 6), 480–485. ( 10.1002/prot.10542) [DOI] [PubMed] [Google Scholar]
- 194.Karplus K, Karchin R, Draper J, Casper J, Mandel-Gutfreund Y, Diekhans M, Hughey R. 2003. Combining local-structure, fold-recognition, and new fold methods for protein structure prediction. Proteins 53(Suppl. 6), 491–496. ( 10.1002/prot.10540) [DOI] [PubMed] [Google Scholar]
- 195.Kim DE, Chivian D, Baker D. 2004. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531. ( 10.1093/nar/gkh468) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lee J, Kim SY. 2005. Protein structure prediction based on fragment assembly and parameter optimization. Biophys. Chem. 115, 209–214. ( 10.1016/j.bpc.2004.12.046) [DOI] [PubMed] [Google Scholar]
- 197.Kosinski J, et al. 2005. FRankenstein becomes a cyborg: the automatic recombination and realignment of fold recognition models in CASP6. Proteins 61(Suppl. 7), 106–113. ( 10.1002/prot.20726) [DOI] [PubMed] [Google Scholar]
- 198.Zhang Y, Skolnick J. 2004. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc. Natl Acad. Sci. USA 101, 7594–7599. ( 10.1073/pnas.0305695101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Handl J, Knowles J, Vernon R, Baker D, Lovell SC. 2011. The dual role of fragments in fragment-assembly methods for de novo protein structure prediction. Proteins 80, 490–504. ( 10.1002/prot.23215) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ghozlane A, Joseph AP, Bornot A, de Brevern AG. 2009. Analysis of protein chameleon sequence characteristics. Bioinformation 3, 367–369. ( 10.6026/97320630003367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Han KF, Baker D. 1996. Global properties of the mapping between local amino acid sequence and local structure in proteins. Proc. Natl Acad. Sci. USA 93, 5814–5818. ( 10.1073/pnas.93.12.5814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kabsch W, Sander C. 1984. On the use of sequence homologies to predict protein structure: identical pentapeptides can have completely different conformations. Proc. Natl Acad. Sci. USA 81, 1075–1078. ( 10.1073/pnas.81.4.1075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kister AE, Potapov V. 2011. Amino acid distribution rules predict protein fold. Biochem. Soc. Trans. 41, 616–619. ( 10.1042/BST20120308) [DOI] [PubMed] [Google Scholar]
- 204.Minor DL, Jr, Kim PS. 1996. Context-dependent secondary structure formation of a designed protein sequence. Nature 380, 730–734. ( 10.1038/380730a0) [DOI] [PubMed] [Google Scholar]
- 205.Adamczak R, Porollo A, Meller J. 2005. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins 59, 467–475. ( 10.1002/prot.20441) [DOI] [PubMed] [Google Scholar]
- 206.Macdonald JR, Johnson WC., Jr 2001. Environmental features are important in determining protein secondary structure. Protein Sci. 10, 1172–1177. ( 10.1110/ps.420101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhu ZY, Blundell TL. 1996. The use of amino acid patterns of classified helices and strands in secondary structure prediction. J. Mol. Biol. 260, 261–276. ( 10.1006/jmbi.1996.0397) [DOI] [PubMed] [Google Scholar]
- 208.Momen-Roknabadi A, Sadeghi M, Pezeshk H, Marashi SA. 2008. Impact of residue accessible surface area on the prediction of protein secondary structures. BMC Bioinformatics 9, 357 ( 10.1186/1471-2105-9-357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gong S, Blundell TL. 2008. Discarding functional residues from the substitution table improves predictions of active sites within three-dimensional structures. PLoS Comput. Biol. 4, e1000179 ( 10.1371/journal.pcbi.1000179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. 2005. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl Acad. Sci. USA 102, 1430–1435. ( 10.1073/pnas.0407146102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Fedyukina DV, Rajagopalan S, Sekhar A, Fulmer EC, Eun YJ, Cavagnero S. 2010. Contribution of long-range interactions to the secondary structure of an unfolded globin. Biophys. J. 99, L37–L39. ( 10.1016/j.bpj.2010.06.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ruvinsky AM, Vakser IA. 2010. Sequence composition and environment effects on residue fluctuations in protein structures. J. Chem. Phys. 133, 155101 ( 10.1063/1.3498743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Crooks GE, Brenner SE. 2004. Protein secondary structure: entropy, correlations and prediction. Bioinformatics 20, 1603–1611. ( 10.1093/bioinformatics/bth132) [DOI] [PubMed] [Google Scholar]
- 214.Kihara D. 2005. The effect of long-range interactions on the secondary structure formation of proteins. Protein Sci. 14, 1955–1963. ( 10.1110/ps.051479505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Gendoo DM, Harrison PM. 2011. Discordant and chameleon sequences: their distribution and implications for amyloidogenicity. Protein Sci. 20, 567–579. ( 10.1002/pro.590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.de Brevern AG, Bornot A, Craveur P, Etchebest C, Gelly JC. 2012. PredyFlexy: flexibility and local structure prediction from sequence. Nucleic Acids Res. 40, W317–W322. ( 10.1093/nar/gks482) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Schlessinger A, Yachdav G, Rost B. 2006. PROFbval: predict flexible and rigid residues in proteins. Bioinformatics 22, 891–893. ( 10.1093/bioinformatics/btl032) [DOI] [PubMed] [Google Scholar]
- 218.Reitzer R, Gruber K, Jogl G, Wagner UG, Bothe H, Buckel W, Kratky C. 1999. Glutamate mutase from Clostridium cochlearium: the structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Structure 7, 891–902. ( 10.1016/S0969-2126(99)80116-6) [DOI] [PubMed] [Google Scholar]
- 219.Rost B, Yachdav G, Liu J. 2004. The PredictProtein server. Nucleic Acids Res. 32(Suppl. 2), W321–W326. ( 10.1093/nar/gkh377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ishida T, Kinoshita K. 2007. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 35(Suppl. 2), W460–W464. ( 10.1093/nar/gkm363) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Paila U, Kondam R, Ranjan A. 2008. Genome bias influences amino acid choices: analysis of amino acid substitution and re-compilation of substitution matrices exclusive to an AT-biased genome. Nucleic Acids Res. 36, 6664–6675. ( 10.1093/nar/gkn635) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Marashi SA, Behrouzi R, Pezeshk H. 2007. Adaptation of proteins to different environments: a comparison of proteome structural properties in Bacillus subtilis and Escherichia coli. J. Theor. Biol. 244, 127–132. ( 10.1016/j.jtbi.2006.07.021) [DOI] [PubMed] [Google Scholar]
- 223.de Brevern AG, Joseph AP. 2011. Species specific amino acid sequence-protein local structure relationships: an analysis in the light of a structural alphabet. J. Theor. Biol. 276, 209–217. ( 10.1016/j.jtbi.2011.01.047) [DOI] [PubMed] [Google Scholar]
- 224.Sadreyev RI, Wang Y, Grishin NV. 2009. Considering scores between unrelated proteins in the search database improves profile comparison. BMC Bioinformatics 10, 399 ( 10.1186/1471-2105-10-399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Costantini S, Colonna G, Facchiano AM. 2006. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem. Biophys. Res. Commun. 342, 441–451. ( 10.1016/j.bbrc.2006.01.159) [DOI] [PubMed] [Google Scholar]
- 226.Bhardwaj N, Langlois RE, Zhao G, Lu H. 2005. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 33, 6486–6493. ( 10.1093/nar/gki949) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Fariselli P, Finelli M, Rossi I, Amico M, Zauli A, Martelli PL, Casadio R. 2005. TRAMPLE: the transmembrane protein labelling environment. Nucleic Acids Res. 33, W198–W201. ( 10.1093/nar/gki440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Battey JN, Kopp J, Bordoli L, Read RJ, Clarke ND, Schwede T. 2007. Automated server predictions in CASP7. Proteins 69(Suppl. 8), 68–82. ( 10.1002/prot.21761) [DOI] [PubMed] [Google Scholar]
- 229.Xu D, Zhang Y. 2012. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735. ( 10.1002/prot.24065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wallner B, Larsson P, Elofsson A. 2007. Pcons.net: protein structure prediction meta server. Nucleic Acids Res. 35, W369–W374. ( 10.1093/nar/gkm319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zhou H, Skolnick J. 2009. Protein structure prediction by pro-Sp3-TASSER. Biophys. J. 96, 2119–2127. ( 10.1016/j.bpj.2008.12.3898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Li SC, Bu D, Xu J, Li M. 2008. Fragment-HMM: a new approach to protein structure prediction. Protein Sci. 17, 1925–1934. ( 10.1110/ps.036442.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Pei J, Grishin NV. 2004. Combining evolutionary and structural information for local protein structure prediction. Proteins 56, 782–794. ( 10.1002/prot.20158) [DOI] [PubMed] [Google Scholar]
- 234.Peng J, Xu J. 2009. Boosting protein threading accuracy. In Research in computational molecular biology (ed. S Batzoglou). Lecture Notes in Computer Science, vol. 5541, pp. 31–45. Berlin, Germany: Springer. ( 10.1007/978-3-642-02008-7_3) [DOI] [PMC free article] [PubMed]
- 235.Mirny LA, Shakhnovich EI. 1998. Protein structure prediction by threading. Why it works and why it does not. J. Mol. Biol. 283, 507–526. ( 10.1006/jmbi.1998.2092) [DOI] [PubMed] [Google Scholar]
- 236.Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. 2008. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 70, 611–625. ( 10.1002/prot.21688) [DOI] [PubMed] [Google Scholar]
- 237.Douguet D, Labesse G. 2001. Easier threading through web-based comparisons and cross-validations. Bioinformatics 17, 752–753. ( 10.1093/bioinformatics/17.8.752) [DOI] [PubMed] [Google Scholar]
- 238.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 2003. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics 19, 1015–1018. ( 10.1093/bioinformatics/btg124) [DOI] [PubMed] [Google Scholar]
- 239.Kurowski MA, Bujnicki JM. 2003. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31, 3305–3307. ( 10.1093/nar/gkg557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wallner B, Elofsson A. 2003. Can correct protein models be identified? Protein Sci. 12, 1073–1086. ( 10.1110/ps.0236803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wang Z, Eickholt J, Cheng J. 2010. MULTICOM: a multi-level combination approach to protein structure prediction and its assessments in CASP8. Bioinformatics 26, 882–888. ( 10.1093/bioinformatics/btq058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bujnicki JM. 2006. Protein-structure prediction by recombination of fragments. Chembiochem 7, 19–27. ( 10.1002/cbic.200500235) [DOI] [PubMed] [Google Scholar]
- 243.Contreras-Moreira B, Fitzjohn PW, Bates PA. 2003. In silico protein recombination: enhancing template and sequence alignment selection for comparative protein modelling. J. Mol. Biol. 328, 593–608. ( 10.1016/S0022-2836(03)00309-7) [DOI] [PubMed] [Google Scholar]
- 244.Siew N, Elofsson A, Rychlewski L, Fischer D. 2000. MaxSub: an automated measure for the assessment of protein structure prediction quality. Bioinformatics 16, 776–785. ( 10.1093/bioinformatics/16.9.776) [DOI] [PubMed] [Google Scholar]
- 245.Zhang Y, Skolnick J. 2004. Tertiary structure predictions on a comprehensive benchmark of medium to large size proteins. Biophys. J. 87, 2647–2655. ( 10.1529/biophysj.104.045385) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.John B, Sali A. 2003. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 31, 3982–3992. ( 10.1093/nar/gkg460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Liu T, Guerquin M, Samudrala R. 2008. Improving the accuracy of template-based predictions by mixing and matching between initial models. BMC Struct. Biol. 8, 24 ( 10.1186/1472-6807-8-24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Wallner B, Fang H, Elofsson A. 2003. Automatic consensus-based fold recognition using Pcons, ProQ, and Pmodeller. Proteins 53(Suppl. 6), 534–541. ( 10.1002/prot.10536) [DOI] [PubMed] [Google Scholar]
- 249.Wallner B, Elofsson A. 2005. Pcons5: combining consensus, structural evaluation and fold recognition scores. Bioinformatics 21, 4248–4254. ( 10.1093/bioinformatics/bti702) [DOI] [PubMed] [Google Scholar]
- 250.Roche DB, Buenavista MT, Tetchner SJ, McGuffin LJ. 2011. The IntFOLD server: an integrated web resource for protein fold recognition, 3D model quality assessment, intrinsic disorder prediction, domain prediction and ligand binding site prediction. Nucleic Acids Res. 39, W171–W176. ( 10.1093/nar/gkr184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Margelevicius M, Venclovas C. 2010. Detection of distant evolutionary relationships between protein families using theory of sequence profile–profile comparison. BMC Bioinformatics 11, 89 ( 10.1186/1471-2105-11-89) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Brylinski M, Lingam D. 2012. eThread: a highly optimized machine learning-based approach to meta-threading and the modeling of protein tertiary structures. PLoS ONE 7, e50200 ( 10.1371/journal.pone.0050200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhang Y, Skolnick J. 2005. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309. ( 10.1093/nar/gki524) [DOI] [PMC free article] [PubMed] [Google Scholar]