

Abstract
Sonoporation is based upon an ultrasound–microbubble cavitation routine that physically punctures the plasma membrane on a transient basis. During such a process, the actin cytoskeleton may be disrupted in tandem because this network of subcellular filaments is physically interconnected with the plasma membrane. Here, by performing confocal fluorescence imaging of single-site sonoporation episodes induced by ultrasound-triggered collapse of a single targeted microbubble, we directly observed immediate rupturing of filamentary actin (F-actin) at the sonoporation site (cell type: ZR-75-30; ultrasound frequency: 1 MHz; peak negative pressure: 0.45 MPa; pulse duration: 30 cycles; bubble diameter: 2–4 µm). Also, through conducting a structure tensor analysis, we observed further disassembly of the F-actin network over the next 60 min after the onset of sonoporation. The extent of F-actin disruption was found to be more substantial in cells with higher uptake of sonoporation tracer. Commensurate with this process, cytoplasmic accumulation of globular actin (G-actin) was evident in sonoporated cells, and in turn the G-actin : F-actin ratio was increased in a trend similar to drug-induced (cytochalasin D) actin depolymerization. These results demonstrate that sonoporation is not solely a membrane-level phenomenon: organization of the actin cytoskeleton is concomitantly perturbed.
Keywords: single-site sonoporation, actin cytoskeleton, F-actin disassembly, G-actin accumulation, structure tensor analysis, characteristic time
1. Introduction
As the physical interface of mammalian cells, the plasma membrane effectively serves as a dynamic barrier that maintains cellular homoeostasis through active regulation of transmembranous transport. Pulsed disruption of membrane integrity would intrinsically give rise to a temporary permeability increase that favours enhanced passage of substances into and out of the cell [1]. Such an over-arching principle has been the norm for the design of physically oriented drug/gene delivery paradigms [2,3]. Among various physical perforation approaches, sonoporation has been well considered as an emerging method in view of its strong potential in achieving high spatio-temporal specificity [4–6]. It is now established that this perforation strategy, which is founded upon acoustic cavitation principles [7,8], can readily enhance transmembranous transport [9]. Also, it has been demonstrated that sonoporation can be controllably instigated through the use of an ultrasound–microbubble-mediated routine in which synthetic microbubbles are leveraged as cavitation sources nearby cells, while pulsed ultrasound is used to initiate acoustic cavitation [10,11].
Although sonoporation has shown significant potential as a controllable way of transiently breaching plasma membrane integrity, its crude impact on cellular structure has remained relatively unexplored in a biophysical context [12]. Often, membrane puncturing is considered as the sole impact of sonoporation on living cells; as long as post-sonoporation cellular viability is retained, the perforation process is considered to be sustainable [13–16]. This way of conceiving the cytostructural impact of sonoporation is perhaps over-simplified because post hoc changes in cellular morphology have in fact been observed downstream from the onset of sonoporation [17,18]; also, physical stress symptoms, for example cell shrinkage, have been shown to emerge in sonoporated cells on a time-lapsed basis [19]. For such reason, it is essential to elucidate the potential relationship between sonoporation and constituent organelles involved in regulating cytomechanical dynamics. The resulting insights acquired would fundamentally benefit ongoing efforts that aim to pursue rational use of sonoporation in biomedical applications.
In this investigation, we seek to unravel how sonoporation as a physical phenomenon would affect the actin cytoskeleton when a cell undergoes localized sonoporation. In particular, it is our intent to characterize how the actin cytoskeleton, as a network of subcellular filamentary polymers directly connected with the plasma membrane [20,21], would actively participate in constituting a cell's functional reaction to the physical impact brought about by sonoporation. Analysing the cytoskeletal response to sonoporation is of particular importance because this subcellular scaffold is well recognized to play a central role in modulating a cell's mechanical properties [22,23]. In turn, the knowledge gained from our present study can help establish an overall understanding of the impact of sonoporation on the physical dynamics of living cells.
The specific aim of this investigation is to acquire a new body of in situ direct observations and quantitative measures on the spatio-temporal response of the actin network induced by an episode of sonoporation. Our underlying hypothesis is that the actin cytoskeleton would undergo dramatic rearrangement in sonoporated cells to accommodate the temporary loss of membrane integrity triggered by acoustic cavitation. To test this hypothesis, we have designed a detailed experimental protocol that makes use of: (i) single-pulse ultrasound exposure, (ii) targeted microbubbles that can bind to plasma membrane surface, and (iii) live confocal imaging to monitor actin dynamics in sonoporated cells. Note that our work is readily distinguished from a few previous reports on ultrasound-induced cytoskeletal changes. In those studies, albeit the cells were subjected to low-intensity ultrasound [24,25] or ultrasound pulsing in the presence of microbubbles [26], the occurrence of sonoporation was not tracked and confirmed in situ at a single-cell level.
2. Material and methods
2.1. Acoustic exposure set-up
2.1.1. Overall description
An overview of the ultrasound apparatus used in our investigation is shown in figure 1. This apparatus was an in-house design that supports live in situ imaging of cellular response through a confocal fluorescence microscopy system (LSM 710, Carl Zeiss, Jena, Germany). It transmitted ultrasound through a single-element piston transducer that operated at 1 MHz centre frequency (25.4 mm effective diameter; Wuxi Beisheng Technology Ltd, Wuxi, Jiangsu, China). For the transducer driving waveform, its parameters were defined using an arbitrary waveform generator (33120A, Agilent Technologies, Santa Clara, CA, USA) that was serially connected to a 50 dB gain broadband amplifier (2100L, Electronics & Innovation Ltd, Rochester, NY, USA). The modulating frequency of the arbitrary waveform generator was set to 1 MHz (i.e. same as the transducer's centre frequency). The pulse-cycle length was set to 30 cycles, and this value in turn yielded a pulse duration of 30 µs. The amplitude was adjusted to yield an in situ peak negative pressure of 0.45 MPa (see the electronic supplementary material for field profile and calibration details). In line with a recent biophysical study on sonoporation [10], a single-shot ultrasound-pulsing strategy was adopted to ensure that sonochemical effects would not be elicited owing to sustained induction of cavitation activities. It was triggered manually on the arbitrary waveform generator.
Figure 1.

A schematic of the acoustic exposure apparatus used to investigate actin cytoskeleton dynamics induced by sonoporation. The main diagram shows the key hardware components in the apparatus. Right inset shows the composition of the cassette-shaped cell chamber. Lower-left inset provides an enlarged illustration of the cell monolayer and the location of targeted microbubbles.
2.1.2. Waveguide design
The ultrasound apparatus was coupled to the confocal microscope's scan stage through a leg-shaped waveguide and a cassette-shaped cell chamber, both of which were custom-designed components. For the waveguide, its hollow cylindrical shaft (with 1.5 mm thick acrylic wall) was 70 mm in length and 35 mm in inner diameter, and it was angled at 40° with respect to the waveguide's rectangular cuboid foot with 55 × 40 mm base dimensions. The distal closed end of the waveguide foot was padded with acoustic absorbing rubber, while the basal surface was carved out and was replaced with a 0.13 mm thick glass coverslip to improve acoustic field transmission into the cell chamber. During operation, the cavity of the waveguide was filled with deionized water, and the transducer was mounted onto the upper end of the shaft (a rubber toric joint was used to seal the spacing between the transducer and the shaft wall).
2.1.3. Cell chamber design
The cell chamber was of a cassette structure whose size was carefully defined based on the physical dimensions of the waveguide's cuboid foot and the confocal microscope's stage-top 37°C incubation device (Chamlide TC; Live Cell Instrument, Seoul, Korea). As shown in figure 1, this device consisted of three acrylic layers: (i) a 2 mm thick outer frame with a 55 × 40 mm window for coherent fitting with the waveguide's foot, (ii) a 1 mm thick inner frame that served as a structural adapter, and (iii) a 5 mm thick observation deck with an 24 × 18 mm window and a 0.13 mm thick glass coverslip sealed onto the bottom surface. In each experiment, the cell chamber was mounted onto the microscope's scan stage by simply slotting the observation deck into the stage-top incubator that was positioned immediately above the objective lens. Cell samples were seeded onto the basal coverslip of the observation deck (protocol described in §2.2). To facilitate registration of their in situ location, a numerated line grid with 1 × 1 mm box resolution was impressed onto the bottom surface of the coverslip using heat transfer printing principles. This grid allowed us to precisely trace cells residing in different box coordinates by simply adjusting the microscope's scan stage position.
2.2. Cell sample preparations
2.2.1. Cell line and culturing protocol
In line with our previous study [19], ZR-75-30 human breast carcinoma cells (CCL-1504; ATCC, Manassas, VA, USA) were used for experimentation. They were grown in a culture flask inside a 37°C incubator environment with 5% carbon dioxide; culturing medium was based on Roswell Park Memorial Institute 1640 medium (R8758; Sigma-Aldrich, St Louis, MO, USA) with 10% fetal bovine serum supplements (30-2020; ATCC). The cells were cultured to their exponential growth phase before they were trypsinized and transferred to the cell chamber. They were seeded onto the coverslip of the cell chamber at a working density of 10 000 cells per slip. Subsequently, they were fostered to reattach to the coverslip by filling the observation deck with fresh culture medium and placing the cell chamber in the 37°C incubator for 24 h.
Note that, as compared to normal cells, breast carcinoma cells exhibit higher membrane deformability [27], and this cytomechanical property is known to protect cells from physical injury and favour recovery after a wounding event [28]. Such propensity against membrane wounding represents a suitable baseline condition for our investigation that involves pulsed disruption of membrane integrity via sonoporation. Cancerous cells are also known to express vascular endothelial growth factor (VEGF) receptors on their membrane [29]. In our experiments, these receptors are leveraged as binding sites for VEGF-receptor-targeted microbubbles that are introduced as agents for site-specific induction of sonoporation (methods described in §2.2.3).
2.2.2. Pre-exposure labelling of actin
For live microscopy experiments, pre-exposure labelling of the actin cytoskeleton of the ZR-75-30 cells in the coverslip was carried out with a commercial viral transfection stain. In particular, 2 μl of CellLight actin green fluorescent protein (actin-GFP) (C10582, Invitrogen, Carlsbad, CA, USA; excitation/emission maxima: 488/510 nm) was added to the observation deck, and the transfection was allowed to take place in the dark for 24 h inside the 37°C incubator. Afterward, the observation deck was washed with phosphate buffered saline (PBS).
2.2.3. Supplementation with targeted microbubbles
To facilitate induction of sonoporation on a site-specific basis, targeted microbubbles with binding preference to VEGF receptors were added to the observation deck of the cell chamber. These lipid-shelled targeted microbubbles were 1–4 µm in diameter; their fabrication procedure and cell-binding efficacy considerations are described in the electronic supplementary material. Before applying ultrasound exposure, 20 µl of targeted microbubbles in mixture form (with 108 bubbles ml−1 concentration) was added to the cell chamber, and a 5 min idle period was allocated to foster their attachment to the cell surface. Unbound microbubbles were subsequently washed away using PBS, and the cell chamber was refilled with fresh culturing medium.
2.2.4. Addition of sonoporation tracer
Before ultrasound exposure was applied, the culturing medium in the cell chamber was supplemented with a fluorescent tracer to enable tracing of cells that experienced sonoporation. For live imaging studies, propidium iodide (PI) (P4170, Sigma-Aldrich; excitation/emission maxima: 493/630 nm) was used as the tracer (applied at 100 µM concentration). It is well known that PI is membrane impermeant and would only be taken up by a cell if membrane permeability is increased, and its intracellular fluorescence would become plateaued once membrane permeability returns to homoeostatic level. Hence, it can be suitably leveraged as an indicator for sonoporation that is marked by a transitory increase in membrane permeability [10,11]. For fixed-cell imaging studies, 2.5 µM Sytox-Orange (S34861, Invitrogen; excitation/emission maxima: 547/570 nm) was used in lieu of PI to avoid cross-talk with other fluorescent labels. The cellular uptake kinetics of this alternative sonoporation tracer is largely similar to PI. Indeed, both tracers would exhibit significantly increased fluorescence upon binding with nucleic acids inside the cell.
2.3. Live microscopy of sonoporation-induced actin dynamics
2.3.1. Pre-exposure imaging
Bright-field images were acquired before ultrasound exposure to locate cells and to determine the microbubble attachment position on the cell surface. Also, three-dimensional scanning of actin-GFP fluorescence was carried out over the entire cell. Both imaging processes were performed using our platform's confocal microscope and a 40× oil-immersion lens (420461–9910, Carl Zeiss; numerical aperture: 1.3; refractive index: 1.518; working distance: 0.21 mm). The fluorescence of actin-GFP was elicited using a laser wavelength of 488 nm, and a detection band of 500–540 nm was used. Pinhole size and pixel dwell time were set to 91 µm and 6.3 µs, respectively. The scan plane was swept from the cell's basal layer to the apical surface (the sectional step size was set to 0.436 µm; top slice was at 3.05 µm from the base).
2.3.2. Monitoring of actin response after sonoporation
Following initial scanning, a single-shot ultrasound pulse was applied to trigger microbubble collapse (confirmed by bright-field imaging). The resulting actin-GFP dynamics was monitored live over the scan plane that was 1.744 µm from the cell base. Note that, with a 91 µm pinhole size, the scan plane's sectional thickness was 3 µm as required to cover the entire cell height. To trace sonoporated cells, PI fluorescence was elicited using 488 nm laser wavelength and 610–650 nm detection band. Using these parameters, one frame was acquired every 10 s in the first 3 min after ultrasound pulsing; after that, the frame acquisition period was lengthened to 1 min per frame. The imaging process lasted until 1 h following the onset of sonoporation, after which a three-dimensional sweep was performed similar to pre-exposure scanning.
2.3.3. Tensor analysis of actin structure
To quantify the extent of actin cytoskeleton changes in response to sonoporation, structure tensor analysis was carried out on the acquired actin fluorescence images. This technique has recently emerged as a quantitative way of analysing the structure of actin cytoskeleton [30]. In conducting the analysis, image frames were transferred to the ImageJ software (v. 1.47n; National Institutes of Health, Bethesda, MD, USA) that was installed with a structure tensor analysis plugin called OrientationJ. For each image, the cell boundary was first marked. Subsequently, using OrientationJ, a coherency index for each pixel position within the cell was derived by calculating the local tensor orientation with respect to its spatial neighbourhood. As described elsewhere [31], coherency would approach unity for pixels with a dominant orientation (e.g. those that belong to a filamentary structure), and it would tend to zero for pixels without significant orientation. To summatively characterize the tensor coherency distribution over the entire cell, we defined a global measure called total weighted coherency, and it was calculated by summing intracellular coherency values which were not less than 90% of the intracellular maximum (i.e. pixels with relatively low coherency were zero-weighted). For each cell being analysed, this global measure was calculated before exposure and at various post-sonoporation time points (10 s, 1 min, 5 min, 60 min).
2.3.4. Temporal kinetics estimation
With the temporal estimates of total weighted coherency, an exponential decay regression was performed using GraphPad (v. 5; GraphPad Software Inc., La Jolla, USA) to determine the characteristic time for actin cytoskeleton changes to take place. The rate of change was in turn derived. Similarly, a growth saturation analysis was conducted on the intracellular PI level to quantify the uptake kinetics of sonoporation. For this analysis, intracellular PI fluorescence was measured as a function of post-sonoporation time using ImageJ's built-in functions, and they were fitted onto the growth saturation model to estimate the characteristic time and rate of PI influx.
The relationship between actin changes and PI influx was evaluated in three ways. First, we compared the temporal evolution of actin's total weighted coherency between sonoporated cells with mild and high PI uptake; the threshold between these two categories was nominally defined as 10-fold increase in PI fluorescence. Each cell subgroup comprised eight cells, and in total 16 cells were analysed; inter-group differences were statistically analysed using Student's t-test. Second, the characteristic time constants for total weighted coherency were compared between the mild and high PI uptake groups. Third, a Pearson correlation analysis was performed on the estimated rates of change (total weighted coherency versus PI uptake).
2.4. Post-exposure microscopy of actin contents
2.4.1. Initial procedure
To further analyse the distribution of actin contents in sonoporated cells, a set of experiments was conducted via a post-exposure fixed-cell staining approach. We particularly focused on characterizing the post-exposure level of two physical forms of actin [32]: (i) filamentary actin (F-actin; the polymeric form) and (ii) globular actin (G-actin; the monomer form). For these experiments, cell samples were prepared similar to what has been described earlier except that pre-exposure actin-GFP labelling was not performed and Sytox-Orange was used as the sonoporation tracer. After acquiring pre-exposure bright-field images to register the in situ position of cells and microbubbles, single-shot pulsed ultrasound was applied, and the cell chamber was placed in the incubator for 1 h (i.e. the same timeframe for live imaging). Non-internalized Sytox-Orange was then washed away using PBS. For comparative analysis, a subset of experiments was conducted under sham exposure conditions (i.e. without ultrasound pulsing). Another subset of positive control experiments was also performed by chemically inducing actin depolymerization through treating the cells with 100 nM cytochalasin D (Cyto D) (C8273; Sigma-Aldrich) for 1 h in a 37°C incubator environment.
2.4.2. Live–dead pre-staining and imaging
To distinguish between observations on the actin distribution of viable sonoporated cells and those of non-viable ones, a live–dead bicolour staining cocktail was added to the cell chamber. In this work, the live and dead stains were, respectively, 5 µM Calcein-Blue-AM (C1429, Invitrogen; excitation/emission maxima: 360/449 nm) and 5 nM Sytox-Red (S34859, Invitrogen; excitation/emission maxima: 640/658 nm). The live–dead stain was washed away 30 min later, and confocal images of cell viability and sonoporation tracer were acquired using the following laser excitation wavelengths and detection bands: (i) Calcein-Blue-AM (live cells): 360/485–510 nm, (ii) Sytox-Red (dead cells): 633/645–670 nm, and (iii) Sytox-Orange (sonoporation tracer): 543/555–580 nm. Other scan parameters were the same as before.
2.4.3. Actin staining and imaging
Staining of cellular F-actin and G-actin contents was carried out after determining the post-exposure viability of individual sonoporated cells. Cells were first fixed by washing the cell chamber twice with PBS and then adding 4% paraformaldehyde for 15 min at room temperature. Note that, given the time required to conduct live–dead staining (about 1 h including imaging), the cell fixation time point corresponded to 2 h after sonoporation; this time point was synchronized with our previous study that showed fixed-cell images of repressive morphological features in sonoporated cells [19]. To permanently permeabilize the fixed cells, 0.1% Triton X-100 was added after three rounds of PBS washing, and it was followed by another round of PBS washing afterward. Subsequently, a PBS cocktail supplemented with 1% bovine serum albumin (A7030, Sigma-Aldrich) was added to reduce non-specific binding of actin stains.
F-actin and G-actin were, respectively, labelled using 0.165 µM fluorescein-isothiocyanate-conjugated phalloidin (phalloidin-FITC) (P5282, Sigma-Aldrich; excitation/emission maxima: 495/520 nm) and 0.3 µM Alexa Fluor 594 deoxyribonuclease I (DNase I) (D12372, Invitrogen; excitation/emission maxima: 590/617 nm). Reaction was allowed to take place for 20 min. After that, the actin stains were washed away with PBS and the cells were imaged by the confocal microscope. Laser wavelengths of 488 and 543 nm were used, respectively, for phalloidin-FITC and DNase I, while the detection bands were set to 505–530 nm and 605–630 nm, respectively (other scan settings remained constant as before).
2.4.4. Ratiometric analysis of actin contents
Using ImageJ, the fluorescence levels of phalloidin-FITC and DNase I were measured. Their ratio (G : F-actin ratio) was then calculated to estimate the post-exposure balance between F-actin and G-actin. Results were compared between viable and non-viable sonoporated cells, sham control group and positive control group (N = 50 in each group). The significance of trends was statistically confirmed via an analysis of variance test.
3. Results
3.1. Actin network disassembly owing to single-microbubble-mediated sonoporation
Disruption of the actin cytoskeleton was generally observed upon the onset of microbubble-mediated sonoporation at a single site. Figure 2a shows a time-series rendering of this observation based on live confocal images acquired from our platform (green and red fluorescence, respectively, depict actin contents and PI which serves as sonoporation tracer). As can be observed, prior to exposure, one targeted microbubble was attached to the cell membrane (indicated by bright-field greyscale contrast), and the actin cytoskeleton resembled an apparent network structure with significant presence of filamentary fibres. Upon single-shot ultrasound pulsing, the microbubble had collapsed, and it in turn induced membrane perforation as indicated by rapid, localized PI influx into the intracellular space through the pre-exposure microbubble site. Note that PI had filled the entire nucleus within 1 min.
Figure 2.

Sonoporation induces progressive disorientation of actin cytoskeleton network over a 60 min period. (a) Each frame shows a live confocal fluorescence image of actin contents (green; actin-GFP) and sonoporation indicator (red; PI). Sonoporation was induced by the collapse of a 2.5 µm microbubble, whose position is indicated in the pre-exposure frame. Rapid cytoplasmic filling of PI took place as indicated by the yellow arrow in the short-term frames (10 s, 20 s, 30 s, 1 min). Subsequently, as shown in the long-term frames (10–60 min), there was gradual reduction of actin fluorescence, and its appearance was increasingly disorganized. (b) Enlarged view of the dashed window marked in (a). Each new episode of filamentary actin loss, deduced with respect to the previous time point shown, is marked by a yellow cross. These episodes took place on an acute basis immediately after sonoporation (see frames at 10, 20 and 30 s) and on a time-delayed basis (see frames at 40 and 60 min).
Synchronous with the physical action of sonoporation, an immediate rupture of F-actin fibres can be observed at the position corresponding to the microbubble's pre-collapse location. This finding is highlighted in figure 2b that gives an enlarged rendering of the temporal dynamics at the sonoporation site (field of view corresponds to the dashed window demarcated in figure 2a; only actin fluorescence is depicted here). As illustrated, within the first 10 s after sonoporation, rapid disappearance of F-actin fibres (with high green fluorescence) was evident in the vicinity of the sonoporation site (these events were marked in situ by yellow crosses). Indeed, this local loss of F-actin fibres subsequently propagated to other parts of the actin cytoskeleton. As shown in the other short-term image frames (20 s, 30 s, 1 min), F-actin fibres in the outskirts of this cell had largely vanished within 1 min after the onset of sonoporation. As a result, there was a significantly different appearance in the actin cytoskeleton morphology before exposure and at 1 min after sonoporation.
In addition to the initial F-actin rupturing in response to sonoporation, further disassembly of the actin cytoskeleton was apparent over a 60 min timeframe. As depicted in figure 2b(ii), the F-actin fibres that remained visible at 10 min after sonoporation (distal from the perforation site) were progressively disintegrated in the next 50 min. Consequently, at 60 min after sonoporation, the actin contents simply exhibited a granular texture without directional orientation. It should be emphasized that such observation occurred on a whole-cell level and was not localized at the perforation site. According to images in figure 2a(ii), the actin cytoskeleton's network-like structure had essentially faded away by this time.
3.2. Structure tensor coherency of actin network: progressively decreased in sonoporated cells
As a quantitative insight into the actin structure disassembly induced by sonoporation, figure 3 plots, for the sonoporated cell shown in figure 2, a temporal set of coherency maps derived from structure tensor analysis of actin fluorescence. Note that coherency was defined in this context as a normalized measure of whether a dominant directional orientation was exhibited at a certain part of the actin cytoskeleton. It can be observed that, before exposure, the cell had distinct bands of high tensor coherency values (blue–magenta colour codes) as indicative of the presence of F-actin cables at those positions. This organized pattern was progressively lost owing to sonoporation. Within 60 min after the onset of sonoporation, the tensor coherency became low (red–yellow colour codes) over the entire cell.
Figure 3.

Tensor coherency maps of local actin structure for the sonoporated cell shown in figure 2 (cell boundary indicated by dashed line). Comparing to the pre-exposure form (a), post-sonoporation actin structure exhibited progressive loss of tensor coherency (b) for 10 s, 1 min, and 60 min after sonoporation. For pixels with coherency values near unity (blue–magenta colour codes), their actin structure exhibits a dominant orientation; pixels without significant actin structure orientation have coherency values near zero (red–yellow colour codes).
Upon analysing different cells with a single sonoporation site, a more substantial impact on actin disruption can be observed in those with stronger intracellular fluorescence of PI that served as sonoporation tracer. Evidence of this observation is shown in figure 4 that plots a set of box–whisker plots of the total weighted coherency of actin fluorescence at four different post-sonoporation time points: 10 s, 1 min, 5 min and 60 min. Results are shown for two equally sized cell subgroups (N = 8) that, respectively, comprised cells with less than or more than 10-fold increase in total PI fluorescence (referred to as mild and high uptake groups accordingly). As can be noted, 10 s after sonoporation, a decrease in total weighted coherency can already be observed (on average about 10% and 40%, respectively, for the mild and high uptake groups). In addition, cells in the high PI uptake group exhibited a more dramatic reduction in total weighted coherency. As time elapsed, sonoporated cells with high PI uptake showed on average 95% reduction in total weighted coherency, indicating that the actin cytoskeletons of these cells were almost completely disassembled from their fibrous network form.
Figure 4.

Total weighted coherency statistics indicate disorientation of actin structure in sonoporated cells over time. Normalized actin tensor coherency values (relative to each cell's pre-exposure level) are shown for four post-sonoporation time points (10 s, 1 min, 5 min and 60 min) and two categories of sonoporated cells: mild and high PI uptake (classification criterion: less than 10× or more than 10×). Each box plots the minimum–maximum range observed over multiple cells (N = 8). ***p < 0.001.
3.3. Actin network disassembly time: correlated with propidium iodide uptake level
By fitting the temporal decrease of total weighted coherency onto an exponential decay model, we observed that the characteristic time of actin cytoskeleton disassembly was different for sonoporated cells with mild and high PI uptake. Supporting data for this observation are shown in figure 5a. As can be observed, for cells with mild PI uptake, their actin disassembly time constants were generally of the order of tens of seconds. These estimated values were significantly shorter than those deduced for sonoporated cells with high PI uptake, of which the duration was generally over 100 s.
Figure 5.

Temporal kinetics analysis showing the dependence of actin network disassembly on the extent of sonoporation (quantified by PI uptake level). (a) Actin disassembly time constants derived from exponential decay regression of total weighted coherency values shown in figure 4 for sonoporated cells with mild and high PI uptake; each box shows minimum–maximum range (N = 8; ***p < 0.001). (b) PI uptake time constants obtained from growth saturation regression of intracellular PI measurements taken from the same cells. (c) Scatter plot of actin disassembly rate versus PI uptake rate; data for all 16 cells in the two groups are shown.
As an adjunct observation of the above trend, the characteristic time constants of PI uptake were also found to be statistically different between the mild and high PI internalization cell groups. The corresponding statistical results are summarized in the box–whisker plots shown in figure 5b. Note that the PI uptake time constant represents an indicator for the duration in which transmembrane permeability was enhanced owing to microbubble-mediated sonoporation. For cells sonoporated with our protocol, this constant was generally higher for those with high total PI internalization (on average 27 s versus 253 s between the mild and high uptake groups).
Building upon our results on characteristic time constants, we found that the time rate of decrease in total weighted coherency of actin was positively correlated with the time rate of PI uptake. As illustrated in the scatter plot of figure 5c, for sonoporated cells that exhibited a high PI uptake rate, their decrease rate in total weighted coherency would also be large (R = 0.9532). This shows that, for sonoporation to facilitate rapid internalization of exogenous substance, the rate of impact on actin network disruption is concomitantly high.
3.4. Accumulation of globular actin in sonoporated cells
For the observed loss of F-actin in sonoporated cells, we found that there was a commensurate gain in the amount of G-actin within the cytoplasm. As an illustration of this finding, figure 6 shows a series of post-exposure confocal images (2 h after exposure) for cells in different categories: sham exposure, positive control and sonoporation (viable and non-viable). The primary information shown in these images is fluorescence levels of F-actin (phalloidin-FITC) and G-actin (DNase I); data are also provided on the cell's morphology, live–dead status and sonoporation tracer fluorescence. It can be observed that, relative to the sham exposure cell (leftmost column), the viable sonoporated cell (third column) showed a significantly stronger G-actin fluorescence; also, it did not show the presence of F-actin cables, consistent with our live-cell imaging results (figure 2). A similar trend can be observed for the non-viable sonoporated cell (fourth column), indicating that the actin disruption impact of sonoporation may be persistent as a cell progressed towards death. It is also in line with the positive control cell that experienced actin depolymerization owing to Cyto D treatment (second column).
Figure 6.

Monomeric G-actin accumulates in sonoporated cells, and response is similar to cells treated with Cyto D actin depolymerization drug (positive control). Each column shows a set of post-exposure (2 h) fixed-cell confocal images for a cell with: sham exposure, Cyto D drug treatment, viable sonoporation and non-viable sonoporation. For each cell, four forms of images are shown: bright-field image merged with sonoporation tracer fluorescence (Sytox-Orange), live–dead fluorescence (Calcein-Blue-AM for live, Sytox-Red for dead), F-actin expression (phalloidin-FITC) and G-actin expression (DNase I 594).
Another observation to be noted from figure 6 is that, similar to the positive control cell, the viable sonoporated cell was significantly more rounded in shape. This morphology is rather different from that of the sham exposure cell, which exhibited a stretched appearance. It is also not the same as the morphology of the non-viable sonoporated cell (rightmost column) that showed irregular budding along the cell boundary, characteristic of the apoptosis programme.
Statistical analysis across multiple cells (N = 50) confirmed that a significant increase in G : F-actin ratio was evident downstream from the onset of sonoporation. This trend is illustrated in the series of box–whisker plots shown in figure 7. In line with the image-level observations in figure 6, sonoporated cells exhibited a statistically significant reduction in F-actin fluorescence (figure 7a; mean level was 32.2% lower than that for sham control) and concomitant increase in G-actin fluorescence (figure 7b; mean level was 2.6 times higher than control). These in turn resulted in an average increase in G : F-actin ratio by 3.7-fold comparing with sham exposure group (figure 7c). Note that the results are consistent with that observed in cells in the positive control group (middle column of boxes in figure 7).
Figure 7.
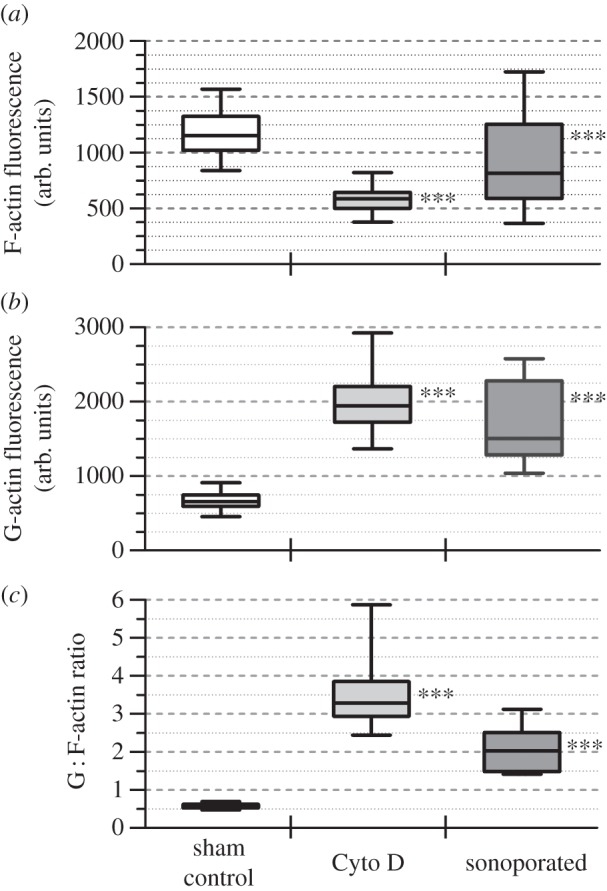
Altered F-actin and G-actin expression in sonoporated cells resulted in a net increase in G : F-actin ratio. Results are shown for sham control cells, positive control cells (Cyto D drug treatment) and viable sonoporated cells. Box–whisker plots of (a) F-actin fluorescence (phalloidin-FITC), (b) G-actin fluorescence (DNase I 594) and (c) G : F-actin ratio are given. Each box plots the minimum–maximum range (N = 50). ***p < 0.001.
4. Discussion and conclusion
4.1. Immediate impact of sonoporation on actin network
As we have demonstrated recently in another study, sonoporation is in essence an act of traumatizing the plasma membrane on an acute but transitory basis [33]. Its course of action may well disrupt the integrity of the actin cytoskeleton because this network of subcellular filaments is physically interconnected with the plasma membrane. In this study, we have demonstrated how each episode of sonoporation, as realized via ultrasound-triggered collapse of a single targeted microbubble attached on the cell membrane, would lead to rapid rupture of the F-actin network adjacent to the perforation site (figure 2). This finding effectively serves as new evidence to illustrate that sonoporation is not solely a membrane-level phenomenon. Furthermore, using a structure tensor analysis approach (figures 3 and 4) coupled with exponential decay regression, we have estimated that the characteristic time for actin network disassembly was of the order of seconds, and its rate of change was directly correlated with the rate of sonoporation tracer uptake into the cell (figure 5). Although this time scale of actin network disruption is not as short as the microsecond-range microbubble dynamics that take place during ultrasound pulsing [34], it can still be considered as acute in that it matches well with the perforation phase of a sonoporation episode which is known to happen of the order of seconds [33].
Note that, in a sonoporation episode, immediate disruption of the actin network is after all not an undesirable event. As suggested in the cell biology literature [35], this event would favour wound repair because the resulting sparser cytoskeleton lattice would structurally make it easier for intracellular vesicles to translocate to the perforation site to undergo homotypic patch fusion (an important course of action for living cells to reseal membrane pores). Therefore, acute remodelling of actin near the site of sonoporation can by and large be regarded as a cytoprotective process that would facilitate pore closure [20,21]. In fact, a similar actin disruption phenomenon has been reported in cells punctured by a micro-tip [36] or pulsed by short-duration electrical fields [37,38].
4.2. Downstream reorganization of actin contents in sonoporated cells
It is worth highlighting the fact that the impact of sonoporation on the actin cytoskeleton was not limited to the initial disruption. In particular, further disassembly of the actin network was observed in sonoporated cells as time elapsed (figures 2–4). Post-exposure microscopy has revealed that such a disassembly event was due to enhanced disintegration of F-actin into its globular monomer form (G-actin), and during this process cell viability remained unaffected (figure 6). In turn, the G : F-actin ratio was significantly increased in sonoporated cells: a trend that was characteristically similar to that for cells which underwent biochemically induced actin disruption via Cyto D treatment (figure 7).
As F-actin is the primary cytoskeletal component that provides mechanical support to the cell [39,40], its net reduction in sonoporated cells (and the resulting increase in G : F-actin ratio) can be interpreted as an act of cell softening or fluidization [41]. However, temporal persistence of this phenomenon, which was indeed observed in sonoporated cells, would have implications on the long-term functional activeness of the actin machinery. It would not only affect cell locomotion and motility [42], but also trigger the activation of pro-apoptotic stress signals [43,44]. The latter implication matches well with our team's previous flow cytometry findings that showed: (i) time-lapsed loss of viability in sonoporated cells starting at 2 h after the onset of sonoporation [45] and (ii) retardation in cell-cycle progression over a multi-hour timeframe [19,46]. It is also seemingly linked to other reports on sonoporation-mediated bioeffects that showed upregulation of various stress messengers like reactive oxygen species and calcium ions [47,48]. As such, the range of biological consequences that can be instigated by the physical action of sonoporation (in addition to its biophysical impact on actin) should not be underplayed. Indeed, recent statistics have raised concerns over the efficiency of sonoporation in facilitating drug uptake without fatally injuring the cells [49].
4.3. Perspectives for further investigations
From a biophysical context, this investigation is essentially poised between our team's recent reports on the spatio-temporal dynamics of sonoporation sites [33] and the downstream stress response elicited in sonoporated cells [19,45,46]. In the future, it would be worthwhile to strengthen the connections between these bodies of findings by performing a correlational study between sonoporation-induced actin disruption kinetics and other sonoporation-related cellular phenomena. For instance, a three-way correlation analysis should be conducted to explicitly establish the link between the size of sonoporation sites, actin structure tensor coherency and sonoporation tracer uptake.
As another avenue for extension beyond our current body of findings, direct measurements of the mechanical properties of sonoporated cells can be pursued using established rheology techniques [23]. Various cytomechanical parameters would be of interest, such as fluidity, stiffness, surface viscosity and membrane tension. The acquired data would represent important information on the biophysical details related to sonoporation. Coupled with other research efforts that seek to establish the key acoustic cavitation conditions for realizing sonoporation [50,51], it should eventually be possible to formulate an analytical model for sonoporation to associate between cavitation physics, membrane perforation dynamics and biophysical impact. This model would be important to substantiate the merit of sonoporation as an emerging membrane perforation technique that is different from other established methods like electroporation, whose biophysical details have already been well characterized and modelled [52,53].
Funding statement
This work has been supported in part by the Hong Kong Innovation and Technology Fund (ITS/292/11) and the University of Hong Kong (201109176229).
References
- 1.Stephens DJ, Pepperkok R. 2001. The many ways to cross the plasma membrane. Proc. Natl Acad. Sci. USA 98, 4295–4298. ( 10.1073/pnas.081065198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villemejane J, Mir LM. 2009. Physical methods of nucleic acid transfer: general concepts and applications. Br. J. Pharmacol. 157, 207–219. ( 10.1111/j.1476-5381.2009.00032.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escoffre JM, Teissie J, Rols MP. 2010. Gene transfer: how can the biological barriers be overcome? J. Membr. Biol. 236, 61–74. ( 10.1007/s00232-010-9275-0) [DOI] [PubMed] [Google Scholar]
- 4.Geers B, Dewitte S, de Smedt C, Lentacker I. 2012. Crucial factors and emerging concepts in ultrasound-triggered drug delivery. J. Control. Release 164, 248–255. ( 10.1016/j.jconrel.2012.08.014) [DOI] [PubMed] [Google Scholar]
- 5.Mehier-Humbert S, Guy RH. 2005. Physical methods for gene transfer: improving the kinetics of gene delivery into cells. Adv. Drug Deliv. Rev. 57, 733–753. ( 10.1016/j.addr.2004.12.007) [DOI] [PubMed] [Google Scholar]
- 6.Frenkel V. 2008. Ultrasound mediated delivery of drugs and genes to solid tumors. Adv. Drug Deliv. Rev. 60, 1193–1208. ( 10.1016/j.addr.2008.03.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohl CD, Arora M, Ikink R, de Jong N, Versluis M, Delius M, Lohse D. 2006. Sonoporation from jetting cavitation bubbles. Biophys. J. 91, 4285–4286. ( 10.1529/biophysj.105.075366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Y, Cui J, Deng CX. 2008. Dynamics of sonoporation correlated with acoustic cavitation activities. Biophys. J. 94, L51–L53. ( 10.1529/biophysj.107.125617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zarnitsyn V, Rostad CA, Prausnitz MR. 2008. Modeling transmembrane transport through cell membrane wounds created by acoustic cavitation. Biophys. J. 95, 4124–4138. ( 10.1529/biophysj.108.131664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kudo N, Okada K, Yamamoto K. 2009. Sonoporation by single-shot pulsed ultrasound with microbubbles adjacent to cells. Biophys. J. 96, 4866–4876. ( 10.1016/j.bpj.2009.02.072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Z, Liu H, Mayer M, Deng CX. 2012. Spatiotemporally controlled single cell sonoporation. Proc. Natl Acad. Sci. USA 109, 16 486–16 491. ( 10.1073/pnas.1208198109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell P, Prausnitz MR. 2007. Future directions of therapeutic ultrasound. Ultrasound Med. Biol. 33, 657 ( 10.1016/j.ultrasmedbio.2006.11.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzman HR, Nguyen DX, Khan S, Prausnitz MR. 2001. Ultrasound-mediated disruption of cell membranes. I. Quantification of molecular uptake and cell viability. J. Acoust. Soc. Am. 110, 588–596. ( 10.1121/1.1376131) [DOI] [PubMed] [Google Scholar]
- 14.Sundaram J, Mellein BR, Mitragotri S. 2003. An experimental and theoretical analysis of ultrasound-induced permeabilization of cell membranes. Biophys. J. 84, 3087–3101. ( 10.1016/S0006-3495(03)70034-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hallow DM, Mahajan AD, McCutchen TE, Prausnitz MR. 2006. Measurement and correlation of acoustic cavitation with cellular bioeffects. Ultrasound Med. Biol. 32, 1111–1122. ( 10.1016/j.ultrasmedbio.2006.03.008) [DOI] [PubMed] [Google Scholar]
- 16.Lai CY, Wu CH, Chen CC, Li PC. 2006. Quantitative relations of acoustic inertial cavitation with sonoporation and cell viability. Ultrasound Med. Biol. 32, 1931–1941. ( 10.1016/j.ultrasmedbio.2006.06.020) [DOI] [PubMed] [Google Scholar]
- 17.Schlicher RK, Radhakrishna H, Tolentino TP, Apkarian RP, Zarnitsyn V, Prausnitz MR. 2006. Mechanism of intracellular delivery by acoustic cavitation. Ultrasound Med. Biol. 32, 915–924. ( 10.1016/j.ultrasmedbio.2006.02.1416) [DOI] [PubMed] [Google Scholar]
- 18.Schlicher RK, Hutcheson JD, Radhakrishna H, Apkarian RP, Prausnitz MR. 2010. Changes in cell morphology due to plasma membrane wounding by acoustic cavitation. Ultrasound Med. Biol. 36, 677–692. ( 10.1016/j.ultrasmedbio.2010.01.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Wan JMF, Yu ACH. 2013. Sonoporation as a cellular stress: induction of morphological repression and developmental delays. Ultrasound Med. Biol. 39, 1075–1086. ( 10.1016/j.ultrasmedbio.2013.01.008) [DOI] [PubMed] [Google Scholar]
- 20.Ko KS, McCulloch CAG. 2000. Partners in protection: interdependence of cytoskeleton and plasma membrane in adaptions to applied forces. J. Membr. Biol. 174, 85–95. ( 10.1007/s002320001034) [DOI] [PubMed] [Google Scholar]
- 21.Doherty GJ, McMahon HT. 2008. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Annu. Rev. Biophys. 37, 65–95. ( 10.1146/annurev.biophys.37.032807.125912) [DOI] [PubMed] [Google Scholar]
- 22.Trepat X, Lenormand G, Fredburg JJ. 2008. Universality in cell mechanics. Soft Matter 4, 1750–1759. ( 10.1039/b804866e) [DOI] [Google Scholar]
- 23.Mofrad MRK. 2009. Rheology of the cytoskeleton. Annu. Rev. Fluid Mech. 41, 433–453. ( 10.1146/annurev.fluid.010908.165236) [DOI] [Google Scholar]
- 24.Mizrahi N, Zhou EH, Lenormand G, Krishnan R, Weihs D, Butler JP, Weitz DA, Fredberg JJ, Kimmel E. 2012. Low intensity ultrasound perturbs cytoskeleton dynamics. Soft Matter 8, 2438–2443. ( 10.1039/c2sm07246g) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Cheng J, Qin YX. 2012. Mechanobiological modulation of cytoskeleton and calcium influx in osteoblastic cells by short-term focused acoustic radiation force. PLoS ONE 7, e38343 ( 10.1371/journal.pone.0038343) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juffermans LJM, van Dijk A, Jongenelen CAM, Drukarch B, Reijerkerk A, de Vries HE, Kamp O, Musters RJP. 2009. Ultrasound and microbubble-induced intra- and intercellular bioeffects in primary endothelial cells. Ultrasound Med. Biol. 35, 1917–1927. ( 10.1016/j.ultrasmedbio.2009.06.1091) [DOI] [PubMed] [Google Scholar]
- 27.Suresh S. 2007. Biomechanics and biophysics of cancer cells. Acta Biomater. 3, 413–438. ( 10.1016/j.actbio.2007.04.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yalcin HC, Hallow KM, Wang J, Wei MT, Ou-Yang HD, Ghadiali SN. 2009. Influence of cytoskeletal structure and mechanics on epithelial cell injury during cycle airway reopening. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L881–L891. ( 10.1152/ajplung.90562.2008) [DOI] [PubMed] [Google Scholar]
- 29.Carmeliet P. 2005. VEGF as a key mediator of angiogenesis in cancer. Oncology 69(Suppl. 3), 4–10. ( 10.1159/000088478) [DOI] [PubMed] [Google Scholar]
- 30.Weichsel J, Herold N, Lehmann MJ, Kräusslich H-G, Schwarz US. 2010. A quantitative measure for alterations in the actin cytoskeleton investigated with automated high-throughput microscopy. Cytometry A 77A, 52–63. ( 10.1002/cyto.a.20818) [DOI] [PubMed] [Google Scholar]
- 31.Rezakhaniha R, Agianniotis A, Schrauwen JTC, Griffa A, Sage D, Bouten CVC, van de Vosse FN, Unser M, Stergiopulos N. 2011. Experimental investigation of collagen waviness and orientation in the arterial adventitia using confocal laser scanning microscopy. Biomech. Model. Mechanobiol. 11, 461–473. ( 10.1007/s10237-011-0325-z) [DOI] [PubMed] [Google Scholar]
- 32.Dominguez R, Holmes KC. 2011. Actin structure and function. Annu. Rev. Biophys. 40, 169–186. ( 10.1146/annurev-biophys-042910-155359) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Wan JMF, Yu ACH. 2013. Membrane perforation and recovery dynamics in microbubble-mediated sonoporation. Ultrasound Med. Biol. 39, 2393–2405. ( 10.1016/j.ultrasmedbio.2013.08.003) [DOI] [PubMed] [Google Scholar]
- 34.van Wamel A, Kooiman K, Hartzveld M, Emmer M, ten Cate FJ, Versluis M, de Jong N. 2006. Vibrating microbubbles poking indvidual cells: drug transfer into cells via sonoporation. J. Control. Release 112, 149–155. ( 10.1016/j.jconrel.2006.02.007) [DOI] [PubMed] [Google Scholar]
- 35.Miyake K, McNeil PL, Suzuki K, Tsunoda R, Sugai N. 2001. An actin barrier to resealing. J. Cell Sci. 114, 3487–3494. [DOI] [PubMed] [Google Scholar]
- 36.Godin LM, Vergen J, Prakash YS, Pagano RE, Hubmayr RD. 2011. Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L615–L623. ( 10.1152/ajplung.00265.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berghofer T, Eing C, Flickinger B, Hohenberger P, Wegner LH, Frey W, Nick P. 2009. Nanosecond electric pusles trigger actin responses in plant cells. Biochem. Biophys. Res. Commun. 387, 590–595. ( 10.1016/j.bbrc.2009.07.072) [DOI] [PubMed] [Google Scholar]
- 38.Stacey M, Fox P, Buescher S, Kolb J. 2011. Nanosecond pulsed electric field induced cytoskeleton, nuclear membrane and telomere damage adversely impact cell survival. Bioelectrochemistry 82, 131–134. ( 10.1016/j.bioelechem.2011.06.002) [DOI] [PubMed] [Google Scholar]
- 39.Pollard TD, Cooper JA. 2009. Actin, a central player in cell shape and movement. Science 326, 1208–1212. ( 10.1126/science.1175862) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tojkander S, Gateva G, Lappalainen P. 2012. Actin stress fibers: assembly, dynamics and biological roles. J. Cell Sci. 125, 1855–1864. ( 10.1242/jcs.098087) [DOI] [PubMed] [Google Scholar]
- 41.Trepat X, Deng L, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, Butler JP, Fredburg JJ. 2007. Universal physical responses to stretch in the living cell. Nature 447, 592–595. ( 10.1038/nature05824) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Revenu C, Athman R, Robine S, Louvard D. 2004. The co-workers of actin filaments: from cell structures to signals. Nat. Rev. Mol. Cell Biol. 5, 1–12. ( 10.1038/nrm1437) [DOI] [PubMed] [Google Scholar]
- 43.Gourlay CW, Ayscough KR. 2005. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat. Rev. Mol. Cell Biol. 6, 583–589. ( 10.1038/nrm1682) [DOI] [PubMed] [Google Scholar]
- 44.Desouza M, Gunning PW, Stehn JR. 2012. The actin cytoskeleton as a sensor and mediator of apoptosis. BioArchitecture 2, 75–87. ( 10.4161/bioa.20975) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong W, Chen X, Jiang P, Wan JMF, Qin P, Yu ACH. 2013. Induction of endoplasmic reticulum stress by sonoporation: linkage to mitochondria-mediated apoptosis initiation. Ultrasound Med. Biol. 39, 2382–2392. ( 10.1016/j.ultrasmedbio.2013.08.005) [DOI] [PubMed] [Google Scholar]
- 46.Zhong W, Sit WH, Wan JMF, Yu ACH. 2011. Sonoporation induces apoptosis and cell cycle arrest in human promyelocytic leukemia cells. Ultrasound Med. Biol. 37, 2149–2159. ( 10.1016/j.ultrasmedbio.2011.09.012) [DOI] [PubMed] [Google Scholar]
- 47.Honda H, Kondo T, Zhao QL, Feril LB, Jr, Kitagawa H. 2004. Role of intracellular calcium ions and reactive oxygen species in apoptosis induced by ultrasound. Ultrasound Med. Biol. 30, 683–692. ( 10.1016/j.ultrasmedbio.2004.02.008) [DOI] [PubMed] [Google Scholar]
- 48.Hassan MA, Campbell P, Kondo T. 2010. The role of Ca2+ in ultrasound-elicited bioeffects: progress, perspectives and prospects. Drug Discov. Today 15, 892–906. ( 10.1016/j.drudis.2010.08.005) [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Yan J, Prausnitz MR. 2012. Can ultrasound enable efficient intracellular uptake of molecules? A retrospective literature review and analysis. Ultrasound Med. Biol. 38, 876–888. ( 10.1016/j.ultrasmedbio.2012.01.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zinin PV, Allen JS., III 2009. Deformation of biological cells in the acoustic field of an oscillating bubble. Phys. Rev. E 79, 021910 ( 10.1103/PhysRevE.79.021910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forbes MM, O'Brien WD., Jr 2012. Development of a theoretical model describing sonoporation activity of cells exposed to ultrasound in the presence of contrast agents. J. Acoust. Soc. Am. 131, 2723–2729. ( 10.1121/1.3687535) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teissie J, Golzio M, Rols MP. 2005. Mechanisms of cell membrane electropermeabilization: a minireview of our present (lack of?) knowledge. Biochim. Biophys. Acta 1724, 270–280. ( 10.1016/j.bbagen.2005.05.006) [DOI] [PubMed] [Google Scholar]
- 53.Krassowska W, Filev PD. 2007. Modeling electroporation in a single cell. Biophys. J. 92, 404–417. ( 10.1529/biophysj.106.094235) [DOI] [PMC free article] [PubMed] [Google Scholar]