

Abstract
Myotonic dystrophy (DM) is the most common adult muscular dystrophy, characterized by autosomal dominant progressive myopathy, myotonia and multiorgan involvement. To date two distinct forms caused by similar mutations have been identified. Myotonic dystrophy type 1 (DM1, Steinert's disease) was described more than 100 years ago and is caused by a (CTG)n expansion in DMPK, while myotonic dystrophy type 2 (DM2) was identified only 18 years ago and is caused by a (CCTG)n expansion in ZNF9/CNBP. When transcribed into CUG/CCUG-containing RNA, mutant transcripts aggregate as nuclear foci that sequester RNA-binding proteins, resulting in spliceopathy of downstream effector genes. Despite clinical and genetic similarities, DM1 and DM2 are distinct disorders requiring different diagnostic and management strategies. DM1 may present in four different forms: congenital, early childhood, adult onset and late-onset oligosymptomatic DM1. Congenital DM1 is the most severe form of DM characterized by extreme muscle weakness and mental retardation. In DM2 the clinical phenotype is extremely variable and there are no distinct clinical subgroups. Congenital and childhood-onset forms are not present in DM2 and, in contrast to DM1, myotonia may be absent even on EMG. Due to the lack of awareness of the disease among clinicians, DM2 remains largely underdiagnosed. The delay in receiving the correct diagnosis after onset of first symptoms is very long in DM: on average more than 5 years for DM1 and more than 14 years for DM2 patients. The long delay in the diagnosis of DM causes unnecessary problems for the patients to manage their lives and anguish with uncertainty of prognosis and treatment.
Key words: Myotonic dystrophy type 1 (Dm1), myotonic dystrophy type 2 (Dm2), management
Introduction
Myotonic dystrophies (DMs) are autosomal dominant, multisystemic diseases with a core pattern of clinical presentation including myotonia, muscular dystrophy, cardiac conduction defects, posterior iridescent cataracts, cerebral involvement and endocrine disorders (1). In 1909 Steinert and colleagues first clearly described the "classic" type of myotonic dystrophy which was called Steinert's disease (OMIM 160900). The gene defect responsible for myotonic dystrophy described by Steinert was discovered in 1992 and found to be caused by expansion of a CTG repeat in the 3' untranslated region of DMPK, a gene encoding a protein kinase (2-4). Subsequently, in 1994, a different multisystemic disorder was described with dominantly inherited myotonia, proximal greater than distal weakness, and cataracts but lacking the gene defect responsible for Steinert's disease (5-7). In Europe, the disease was termed proximal myotonic myopathy (PROMM, OMIM*160900) (6) or proximal myotonic dystrophy (PDM) (7) while in the United States was termed myotonic dystrophy with no CTG repeat expansion or myotonic dystrophy type 2 (DM2) (5). Later studies demonstrated that many of the families identified as having myotonic dystrophy type 2, PROMM or PDM had the same disease, a disorder caused by an unstable tetranucleotide CCTG repeat expansion in intron 1 of Zinc finger protein 9 gene (ZNF9) mapped to 3q21.3 (8, 9). Due to the existence of different types of myotonic dystrophy, the International Myotonic Dystrophy Consortium developed a new nomenclature and guidelines for DNA testing (10). The Steinert's disease that results from an unstable trinucleotide repeat expansion on chromosome 19, is now termed myotonic dystrophy type 1 (DM1). Patients with the clinical picture of myotonic dystrophy type 2/proximal myotonic myopathy, who have positive DNA testing for the unstable tetranucleotide repeat expansion on chromosome 3, are now classified as having myotonic dystrophy type 2 (DM2) (5, 11-12).
Although DM1 and DM2 have similar symptoms, they also present a number of very dissimilar features making them clearly separate diseases (Table 1).
Table 1.
Comparison of clinical manifestations between DM1 and DM2.
| Clinical Features | DM1 | DM2 |
|---|---|---|
|
General features Epidemiology Age of onset (years) Anticipation Congenital form Life expectancy |
Widespread 0 to adult Always present Present Reduced |
European 8-60 Exceptional Absent Normal range |
|
Core features Clinical myotonia EMG myotonia Muscle weakness Cataracts |
Evident in adult- onset Always present Disabling at age 50 Always present |
Present in <50% Absent or variable in many Onset after age 50-70 Present in minority |
|
Muscle symptoms Facial and jaw weakness Bulbar weakness-dysphagia Respiratory muscles weakness Distal limb muscle weakness Proximal limb muscle weakness Sternocleidomastoid weakness Myalgic pain Visible muscle atrophy Calf hypertrophy |
Always present Always later Always later Always prominent May be absent Always prominent Absent or mild Face, temporal, distal hands and legs Absent |
Usually absent Absent Exceptional Only flexor digitorium profundus, rare Main disability in most patients, late Prominent in few Most disabling symptom in many Usually absent Present in ≥50% |
|
Systemic features Tremors Behavioral change Cognitive disorders Hypersomnia Cardiac arrhythmias Male hypogonadism Manifest diabetes |
Absent Early in most Prominent Prominent Always present Manifest Frequent |
Prominent in many Not apparent Not apparent Infrequent From absent to severe Subclinical in most Infrequent |
Myotonic Dystrophy type 1
Clinical features
Myotonic dystrophy type 1 is the most common inherited muscular dystrophy in adults with an estimated prevalence of 1/8000. DM1 is characterized by the phenomenon of anticipation, by which the disease has an earlier onset and more severe course in subsequent generations. Patients with DM1 can be divided into four main categories, each presenting specific clinical features and management problems: congenital, childhood-onset, adult-onset, and late-onset/asymptomatic. Table 2 summarises these subtypes.
Table 2.
Summary of myotonic dystrophy type 1 phenotypes, clinical findings and CTG length.
| Phenotypes | Clinical findings | CTG length | Age of onset |
|---|---|---|---|
| Congenital | Infantile hypotonia Respiratory failure Learning disability Cardiorespiratory complications |
> 1000 | Birth |
| Childhood onset | Facial weakness Myotonia Low IQ Conduction defects |
50-1000 | 1-10 years |
| Adult onset "classic DM1" | Weakness Myotonia Cataracts Conduction defects Insulin resistance Respiratory failure |
50-1000 | 10-30 years |
| Late onset/Asymptomatic | Mild myotonia Cataracts |
50-100 | 20-70 years |
| Pre-mutation | None | 38-49 | N/A |
Congenital DM1
Congenital DM1 (CDM) shows a distinct clinical phenotype with distinct clinical features, therefore it is to be considered a severe early form of 'classical' DM1. CDM often presents before birth as polyhydramnios and reduced fetal movements. After delivery, the main features are severe generalized weakness, hypotonia and respiratory involvement. In up to 50% of CDM, bilateral talipes and other contractures are present at birth. One feature of affected infants is the "fish-shaped" upper lip, an inverted V-shaped upper lip which is characteristic of severe facial weakness and causes weak cry and inability to suck. Mortality from respiratory failure is high. Surviving infants experience gradual improvement in motor function, they can swallow and independently ventilate. Almost all CDM children are able to walk. Cognitive and motor milestones are delayed and all patients with CDM develop learning difficulties and require special needs schooling. Cerebral atrophy and ventricular enlargement are often present endoat birth (13, 14). A progressive myopathy and the other features seen in the classical form of DM1 can develop although this does not start until early adulthood and usually progresses slowly (15). Despite the severe muscular phenotype, clinical myotonia is neither a feature presented in the neonatal period nor can it be disclosed in the electromyogram (EMG). Patients often develop severe problems from cardio-respiratory complications in their third and fourth decades.
Childhood onset DM1
The diagnosis of this form of DM1 is often missed in affected adolescents or children because of uncharacteristic symptoms for a muscular dystrophy and apparently negative family history (16). Cases of DM1 that come to medical attention during childhood typically manifest developmental abnormalities that are less severe than seen in congenital onset cases (17). Unlike the CDM patients, in which maternal transmission is the rule, the sex of the parents does not influence the development of childhood onset DM1. These patients have cognitive deficits and learning abnormalities (18). As in the congenital cases, degenerative features often develop as these children reach adulthood. There is increasing evidence of early conduction abnormalities, and from the age of 10, annual electrocardiograms and consideration of electrophysiological studies should be a part of routine management.
Adult onset DM1
The core features in classic DM1 are distal muscle weakness, leading to difficulty with performing tasks requiring fine dexterity of the hands and foot drop, and facial weakness and wasting, giving rise to ptosis and the typical myopathic or 'hatchet' appearance. The neck flexors and finger/wrist flexors are also commonly involved. Grip and percussion myotonia are regular features; however, myotonia affects other muscle including bulbar, tongue or facial muscles, causing problems with talking, chewing, and swallowing. Elevation of the serum creatine kinase is present. Cardiac involvement is common in DM1 and includes conduction abnormalities with arrhythmias and conduction blocks contributing significantly to the morbidity and mortality of the disease (19-22). In some patients and families, a dilated cardiomyopathy may be observed. Posterior subcapsular cataracts develop in most patients, in some of them at an early age without any other muscle symptoms which will develop later in their disease (23). Minor intellectual deficits are present in many patients in contrast with CDM and childhood onset DM1. Avoidant, obsessive-compulsive and passive-aggressive personality features have also been reported (24, 25). Nocturnal apnoeic episodes and daytime sleepiness are a common manifestation. Gastrointestinal tract involvement covers irritable bowel syndrome, symptomatic gall stones and gamma-glutamyltransferase elevations. Finally, endocrine abnormalities include testicular atrophy, hypotestosteronism, insulin resistance with usually mild type-2 diabetes, thyroid dysfunction.
Late-onset/asymptomatic DM1
In late-onset or asymptomatic patients (with low number of CTG repeats), only limited features are found on clinical and paraclinical assessment. Myotonia, weakness and excessive daytime sleepiness are rarely present. Before DNA tests became available, there were many examples of incorrect ascertainment, even when using markers such as EMG evidence of myotonia and slitlamp examination for the characteristic cataracts (26). In late-onset patients, the search for cataracts is helpful for identifying the transmitting person.
Myotonic Dystrophy type 2
Clinical features
The prevalence of DM2 is not well established, but estimated to be similar to DM1 in European populations (27). In DM2 there are no distinct clinical subgroups although initially different phenotypes of DM2 were described: DM2/PROMM and PDM (5-7). The most important discrepancy between DM1 and DM2 is absence of a congenital or early-onset form in DM2 (12, 28) and the clinical presentation is a more continuum from early adult-onset severe form to very late–onset mild symptoms (paucisymptomatic).
Clinically based ascertainment of DM2 patients is even more difficult because of the large phenotypic variability and a large number of individuals with milder symptoms who remain undiagnosed. Moreover, milder phenotypes with prominent myalgia may easily be misdiagnosed as fibromyalgia (29) and patients with onset of slowly progressive proximal muscle weakness after age 70 years may not be referred for neuromuscular investigations. Further evidence that a large proportion of DM2 patients may be undiagnosed came from a recent study which indicate that co-segregation of heterozygous recessive CLCN1 mutations in DM2 patients is higher than expected (30). In DM2 patients with co-segregating CLCN1 the severity of myotonia appear to be more evident either clinically or on EMG, thus these patients could be more easily identified and diagnosed than DM2 patients without the modifier allele. Consequently the majority of DM2 patients remains undiagnosed even in clinical centers with considerable experience with DM2.
DM2/PROMM typically appears in adult life and has variable manifestations, such as early-onset cataracts (younger than 50 years), varying grip myotonia, thigh muscle stiffness, and muscle pain, as well as weakness (hip flexors, hip extensors, abdominal muscles, or long flexors of the finger muscles) (5, 6, 11, 12, 31-34). These complaints often appear between 20 and 70 years of age, and patients as well as their care providers ascribe them to overuse of muscles, "pinched nerves," "sciatica," arthritis, fibromyalgia, or statin use (35). Early in the presentation of DM2 there is only mild weakness of hip extension, thigh flexion, and finger flexion. Myotonia of grip and thigh muscle stiffness varies from minimal to moderate severity over days to weeks. Myotonia is often less apparent in DM2 compared with patients with DM1. It is more difficult to elicit myotonia on standard EMG testing in DM2 compared to DM1 except for proximal muscles such as the tensor fascia lata and vastus lateralis muscles. In cases of late-onset DM2, myotonia may only appear on electromyographic testing after examination of several muscles (32). Facial weakness is mild in DM2 as is muscle wasting in the face and limbs. The cataracts in DM2 have an appearance identical to that observed in DM1 and develop before 50 years of age as iridescent, posterior capsular opacities on slit-lamp. Cardiac problems appear to be less severe and frequent in patients with DM2 than in patients with DM1 (36, 37). In DM2, cardiac conduction alterations are primarily limited to first-degree atrio-ventricular and bundle branch block. However, sudden death, pacemaker implantation, and severe cardiac arrhythmias have been described in small numbers of patients (33, 38). In DM2, no ventilatory insufficiency has been reported. Central nervous system involvement represents one of the major differences between DM1 and DM2. Although retarded DM2 individuals have been reported, these occurrences may be either accidental or an infrequent disease consequence (12, 31). The type of cognitive impairment that occurs in DM2 is similar to but less severe than that of DM1. Other manifestations, such as hypogonadism, glucose intolerance, excessive sweating, and dysphagia, may also occur and worsen over time in DM2 (5, 11, 12, 34, 39, 40, 41, 42, 43). Pregnancy and menses may also exacerbate muscle pain, myotonia, and muscle cramps (44). PDM patients show many features similar to those found in PROMM, including proximal muscle weakness, cataracts, and electrophysiologically detectable myotonia. Unlike PROMM patients, however, they do not report myalgias, symptomatic myotonia, or muscle stiffness. Instead they present traits not present in PROMM, such as pronounced dystrophicatrophic changes in the proximal muscles and late-onset progressive deafness (7).
Genetics
The DM1 mutation was identified in 1992 as an expansion of an unstable CTG trinucleotide repeat in the 3'untranslated region (UTR) of the myotonic dystrophy protein kinase gene (DMPK; OMIM 605377) which codes for a myosin kinase expressed in skeletal muscle. The gene is located on chromosome 19q13.3 (3, 4). In DM1 patients the repeat size range from 50-4.000 (150-12.000 bp) and is nearly always associated with symptomatic disease although there are patients who have up to 60 repeats who are asymptomatic into old age and similarly patients with repeat sizes up to 500 who are asymptomatic into middle age. Normal individuals have between 5 and 37 CTG repeats. Patients with between 38 and 49 CTG repeats are asymptomatic but are at risk of having children with larger, pathologically expanded repeats (5). This is called a 'pre-mutation' allele. The DM1 mutation length predicts the clinical outcome to some extent: classical DM1 100-1.000 repeats; congenital > 2.000 repeats (10, 45). DM2 results from an unstable tetranucleotide repeat expansion, CCTG, in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9) on chromosome 3q21 (8, 9). The size of the CCTG repeat is below 30 repeats in normal individuals while the range of expansion sizes in DM2 patients is huge. The smallest reported mutation vary between 55-75 CCTG (9, 46) and the largest expansions have been measured to be up about 11.000 repeats (9). Both DM1 and DM2 mutations show instability with variation in different tissue and cell types causing somatic mosaicism (47, 48). The size of the CTG and CCTG repeat appear to increase over time in the same individual, and are dynamic gene defects (12). However DM1 children may inherit repeat lengths considerably longer than those present in the transmitting parent. This phenomenon causes anticipation, which is the occurrence of increasing disease severity and decreasing age of onset in successive generations. A child with congenital DM1 almost always inherits the expanded mutant DMPK allele from their mother. However anticipation may be seen in patients with DM1 who inherit a smaller expanded CTG repeat from their father (49, 50). In DM2 the mutation usually contracts in the next generation, being shorter in children (12). This may explains some distinct features of DM2 such as the missing of a congenital form, the lack of anticipation and the later onset (28). The size of CCTG repeat expansion in leukocyte DNA in DM2 seems to relate in large part to the age of the patient and not necessarily to the severity of symptoms or manifestations. This complicates attempts to correlate the size of the repeat with earlier clinical onset of more severe symptoms as occurs in patients with DM1. However due to somatic mosaicism, CTG repeat size correlates more significantly with age of onset and disease severity below 400 CTG repeats (51). The correlation between CTG repeat size and the severity of the disease can be observed in blood but not in other organs (eg, muscle). In DM1 the repeat lengths in muscle are shown to be larger (52) and there is no correlation between the size of the CTG repeats in muscle and the degree of weakness. It should be noted that in clinical practice, the CTG expansion is measured in blood and there is no additional clinical advantage of measuring repeat size in muscle.
Molecular pathomechanism
As described above the two types of the disease are associated with two different loci: DM1 is caused by the expansion of an unstable CTG trinucleotide repeat in the 3' UTR of the DMPK gene (2, 4) while DM2 mutation consists in the expansion of an unstable CCTG tetranucleotide within the first intron of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9, ZNF9) (9). The fact that two repeat sequences located in entirely different genes can cause such similar disease features implies a common pathogenic mechanism. It is now clear that the gain-of-function RNA mechanism is the predominant cause of pathogenesis of myotonic dystrophies in which the expansion mutation, (CTG)n in DM1 and (CCTG)n in DM2, is transcribed and the mutant RNAs containing the repeat expansions accumulate in the cell nuclei as foci, called ribonuclear inclusions, and are responsible for the pathologic features common to both disorders. The expanded CUG/CCUG-containing transcripts form hairpins, imperfect double-stranded structure which lead to deregulation of two important RNA-binding proteins, muscleblind–like protein 1 (MBNL1) and CUGBP/Elav-like family member 1 (CELF1). In DM1, MBNL1 protein is depleted from the nucleoplasm through recruitment into ribonuclear foci (53, 54, 55) while CELF1 stabilization by PKC phosphorylation results in increased steady-state levels and protein upregulation (56). Recently over-expression of CUGBP1 in skeletal muscle from adult DM1 but not from DM2 has been described (57). A combined effect of decreased MBNL1 and increased CELF1 activity lead to misregulated alternative splicing and other changes of the muscle transcriptome (58, 59). The alteration of pre-mRNA processing strengthens the hypothesis of a spliceopathy which leads to inappropriate expression of embryonic splicing isoforms in adult tissues (60). In DM2, splicing abnormalities are also associated with the sequestration of MBNL1 protein by expanded transcripts (58, 61). However evidence that CUGBP1 upregulation also occurs in DM2 is conflicting (54, 59, 62). However in a recent paper (57) we have shown a normal level of CUGBP1 in a large cohort of Italian DM2 patients. Recent data demonstrate that MBNL1-containing foci in DM2 cells also sequester snRNPs and hnRNPs, splicing factors involved in the early phases of transcript processing, thus strengthening the hypothesis that a general alteration of pre-mRNA post-transcriptional pathway could be at the basis of the multifactorial phenotype of DM2 patients (63, 64).
Misregulation of alternative splicing plays a central role in the development of important DM symptoms (58, 60). For example, among the symptoms of DM, myotonia, insulin resistance and cardiac problems are correlated with the disruption of the alternative splicing of the muscle chloride channel ClC-1, of the insulin receptor (IR) and of the cardiac troponin T (TNNT3), respectively (41, 43, 65, 66, 67). However, spliceopathy may not fully explain the multisystemic disease spectrum. The underlying mechanism responsible for muscle weakness and wasting remains to be established. Recent findings suggest that DM mutations can affect gene expression in multiple ways. Altered activity and/or localization of MBNL1 and CELF1 may alter transcription, translation and cell signaling (68, 69). Moreover it has been demonstrated that in DM1 the highly regulated pathways of miRNA is altered in skeletal muscle and heart tissue potentially contributing to DM1 pathogenetic mechanisms and in DM2 skeletal muscle (70-73). Another open question in the field of DM is to clarify the pathomecanisms underlying the phenotypic differences between DM1 and DM2. Clinical signs in DM1 and DM2 are similar, but there are some distinguishing features: DM2 is generally less severe and lacks a prevalent congenital form. This suggests that other cellular and molecular pathways are involved besides the shared toxic-RNA gain of function hypothesized. Disease-specific manifestations may result from differences in spatial and temporal expression patterns of DMPK and CNBP genes. Similarly, changes in the expression of neighbouring genes may define diseasespecific manifestations. Importantly, the role of CELF1 in DM2 is particularly intriguing with contradictory results being reported (54, 59, 62). Another possible explanation for the clinical differences between the two DM forms is the reduction of DMPK or ZNF9 protein levels in DM1 and DM2 respectively (3, 74-76). Indeed both knockout mouse models for DMPK and ZNF9 show the phenotypic aspects of DM (77, 78). Taken together these observations seem indicate that the emerging pathways of molecular pathogenesis are far more complex than previously appreciated.
Diagnostics
Laboratory tests
As for all genetics diseases with identified mutation, the typical DM1 and DM2 diagnostic method is mutation verification by genetic tests. In the case of DM1, symptoms and family history are often clear and distinctive enough to make a clinical diagnosis, and the mutation can be confirmed by PCR and Southern Blot analysis. PCR analysis is used to detect repeat lengths less than 100 and Southern blot analysis to detect larger expansions. Predictive testing in asymptomatic relatives as well as prenatal and preimplantation diagnosis can also be performed.
On the contrary, the wide clinical spectrum of DM2 phenotype makes the clinical diagnosis more difficult. Moreover conventional PCR and Southern blot analysis are not adequate for a definitive molecular diagnosis in DM2 due to the extremely large size and somatic instability of the expansion mutation (9, 46). The copy number of DM2 CCTG is below 30 in phenotypically normal individuals and up 11.000 in patients (79). A complex genotyping diagnostic procedure is now commonly used consisting of a three step molecular protocol (12, 28): (A) a conventional PCR assay across the mutation locus using probes binding to mutation flanking sequences can be used for mutation exclusion. In all DM2 patients, a single PCR product representing the normal allele can be identified because the DNA polymerase fail to amplify the mutant allele due to length and stable secondary structure. All individuals showing two alleles for the marker are excluded from having the DM2 mutation. However, identical allele size on two normal alleles occurs in 12% of the population; (B) all patients appearing to have one allele need further molecular analysis to determine whether or not they carry a DM2 expansion. Because of the incomplete sensitivity of Southern analysis, a DM2 repeat assay (RP-PCR) was developed; (C) the RP-PCR method involves amplifying the CCTG repeat by PCR, and probing the resultant product with an internal probe to assure specificity. The combined use of these methods allows 99% sensitivity and specificity for known expansions. Several alternative and highly sensitive methods have been developed for DM2 mutation verification including long-range PCR (80) and a tetraplet-primed PCR (81). A modified Southern method using field–inversion electrophoresis (FIGE) is particularly efficient in determining the mutation length (82). However, these methods are still too long and complicated to be part of routine laboratory diagnostics. Nevertheless ribonuclear foci and splicing changes are present before any histological abnormality manifestations (43, 83). This could be important for an early diagnosis before the spectrum of clinical signs of muscle disease appear. So a more practical tool to obtain a definitive DM2 diagnosis in few hours is represented by in situ hybridization (ISH) which is a method that allows the direct visualization of the mutant RNA on muscle biopsy (84, 85). By using specific probes for CCUG expansions, it permits a differential diagnosis between DM2 and DM1. Therefore it may be a simple approach for DM2 diagnosis, which can be performed in a rapid and sensitive manner in any pathology laboratory. ISH with CAGG probe should be considered as a routine laboratory procedure to confirm or refute the clinical suspicion of DM2. It should also be applied routinely to screen patients with myotonic disorders (84, 85). This approach makes muscle biopsy an essential tool for DM2 diagnosis (Fig. 1A). Moreover, since MBNL1 is sequestered by mutant RNA foci, it is possible to visualize the nuclear accumulation of MBNL1 by immunofluorescence on muscle sections (Fig. 1B). However, although MBNL1 represents an histopathological marker of DM, it does not allow to distinguish between DM1 and DM2 (86).
Figure 1.
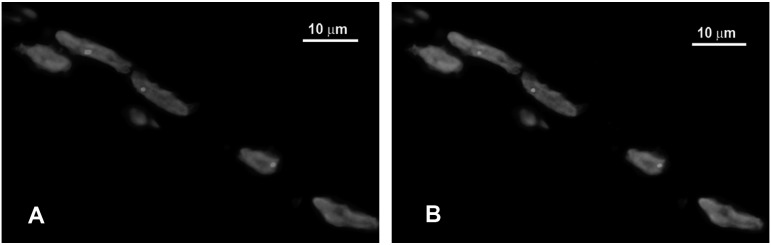
Fluorescence in situ hybridization (FISH) in combination with MBNL1-immunofluorescence on DM2 muscle section. A. Visualization of (CCTG)n expansion on muscle section by FISH using (CAGG)5 specific probe. Red spots within myonuclei (blue, DAPI) represent ribonuclear inclusions containing accumulated mutant RNAs. B. Visualization of nuclear foci of MBNL1 (green spots) colocalizing with ribonuclear inclusions in A.
Muscle biopsy
The histological features of muscle in DM1 and DM2 are very similar (Fig. 2), and sufficiently characteristic so that a diagnosis of DM can be suggested based on muscle biopsy alone (1, 12, 87). In both diseases, affected muscles show a high number of central nuclei and a markedly increased variation in fiber diameter that commonly ranges from less than 10 μm to greater than 100 μm (Fig. 2A, D). Basophilic regenerating fibers, splitting fibers, fibrosis and adipose deposition occur in both diseases to a variable degree depending on the extent of muscle involvement. Ring finger fibers and sarcoplasmic masses are generally more frequent in DM1 muscle biopsy. Recently the comparison of muscle biopsy findings in classic DM1 with those in DM2 has indicated that specific features are present in DM2 muscle biopsy helping the diagnosis of DM2. Severely atrophic fibers with pyknotic nuclear clumps similar in appearance to the severely atrophic fibers in neurogenic atrophy are frequently found in DM2 biopsy also before the occurrence of muscle weakness (Fig. 2D). In DM1, nuclear clumps are present in end-stage muscle biopsy (88). A predominant type 2 fiber atrophy in contrast to the type 1 atrophy observed in DM1, has been described in DM2 (87, 89, 90, 91) (Fig. 2B,C,E,F). Moreover, in DM2 muscle biopsy central nucleation selectively affects type 2 fibers and the atrophic nuclear clumps express fast myosin isoform (type 2 fiber) indicating that DM2 is predominantly a disease of type 2 myofibers (90) (Fig. 2F; Table 3).
Figure 2.

Panel showing muscle histology in DM1 and DM2. A-C. Transversal sections from DM1 muscle biopsies. A. Haematoxylin & Eosin: fiber size variation and central nuclei (arrows) are present. B, C. The population of atrophic fibers (white arrow) are preferentially type 1 fibers as demonstrated in sections stained for ATPase pH 4.3 (B, dark brown) or immunostained for myosin MHCslow (C, brown). Black arrow indicate centrally located nuclei. D-E Transversal sections from DM2 muscle biopsies. D. Haematoxylin & Eosin: as in DM1 muscle, fiber size variation and central nuclei (arrows) are present. Abundant nuclear clumps are also present (arrow heads) despite the muscle shows an early stage pathology. E, F. Type 2 fibers are predominantly affected in DM2 muscle: in routine laboratory muscle staining such as ATPase pH 10.0 (E) or immunostaining for myosin MHCfast (F), type 2 fiber atrophy (white arrows) and type 2 central nucleation (black arrow) are commonly observed.
Table 3.
Muscle histopatology in DM1 and DM2.
| Histopathological findings | DM1 | DM2 |
|---|---|---|
| Fiber size variation | +++ | +++ |
| Internal nuclei | +++ |
+++ more in type 2 fibers |
| Type 1 fiber atrophy | ++ | - |
| Type 2 fiber atrophy | + | ++ |
| Type 2 fiber hypertrophy | - | + |
| Nuclear clump fibers |
+ at advanced stages only |
+++ more in advanced stages |
| Atrophic fibers (diam. ≤ 6μm) |
± type 1 and type 2 fibers at advanced stages only |
+++ type 2 fibers |
| Ring fibers | ++ | + |
| Sarcoplasmic masses | ++ | ± |
| Fibrosis |
+++ at late stages only |
++ at late stages only |
| Fatty replacement |
+++ at late stages only |
++ at late stages only |
+++ present in >75% of biopsies; ++ present in 20-50% of biopsies; + present in 10-24% of biopsies; ± occasionally present; - absent
Management
In general the management of DM2 is similar to that of DM1, but there is less need for supportive care, such as bracing, scooters, or wheelchairs. Cataracts require monitoring. Cardiorespiratory disorders are responsible for 70% of the mortality in DM1 and many of these patients could have been treated by active monitoring and a lower threshold for input. Disturbances in cardiac rhythm are less frequent in DM2, but abnormalities do occur (121, 36-38), and serial monitoring with an electrocardiogram is necessary to check for covert dysrhythmia. Hypogonadism and insulin resistance need monitoring in both diseases. Myotonia tends to be less marked and less troublesome in DM2, but in specific circumstances antimyotonia therapy is helpful, especially if muscle stiffness is frequent and persistent or if pain is prominent (92). Cognitive difficulties also occur in DM2 as in DM1 but become manifest in adult life and appear to be associated with decreased cerebral blood flow to frontal and anterior temporal lobes (39, 93) and decreased brain volume (94, 95). The changes are less severe than in DM1. Their aetiology is unknown but may relate to the toxic effect of intranuclear accumulations of abnormally expanded RNA. Management of these brain symptoms is similar to that for DM1. A frequent and difficult problem in DM2 is the peculiar muscle pain described earlier (35, 29). The exact mechanism underlying the pain is unknown, and there is no well-established, effective treatment. Carbamazepine or mexiletine along with nonsteroidal anti-inflammatory medications or tylenol ameliorate this pain in some patients.
Concluding remarks
The myotonic dystrophies are dominantly inherited multisystemic disorders that include two genetically distinct types. DM1 is the commonest cause of adult onset muscular dystrophy with an estimated prevalence of 1/8000. Due to the lack of awareness of the disease among clinicians, DM2 remains largely underdiagnosed and the prevalence of DM2 is not well established. These diseases have been called 'spliceopathies' and are mediated by a primary disorder of RNA rather than proteins, however, spliceopathy may not fully explain the multisystemic disease spectrum. Although the two forms of myotonic dystrophy share many features, there are definite differences with respect to clinical, muscle biopsy, and genetic findings. In DM2 the core symptoms include proximal muscle weakness, myotonia, cataracts, cardiac conduction defects, insulin resistance and male hypogonadism. In DM1, the muscle weakness and wasting are more severe, preferentially distal and facial with ptosis, and with later evolving dysphagia, generalized weakness, and respiratory failure. A severe congenital form associated with DM1 has not been observed in DM2, and anticipation is the exception in DM2. In contrast to DM1, type 2 fiber are preferentially involved in DM2 with the presence of very atrophic type 2 fibers early in muscle pathogenesis. The basis for the differences between DM1 and DM2 has not been clarified at the molecular level. There is currently no cure but effective management is likely to significantly reduce the morbidity and mortality of patients. The enormous advances in the understanding of the molecular pathogenesis of DM1 and DM2 has revealed pathways of molecular pathogenesis more complex than previously appreciated that could be the right track towards the development of effective therapies.
Acknowledgements
This work was supported by AFM – Association Française contre les Myopathies, CMN – Centro per lo Studio delle Malattie Neuromuscolari and FMM – Fondazione Malattie Miotoniche
References
- 1.Harper PS. Myotonic Dystrophy. In: Karpati G., Hilton-Jones D., Griggs R.C., editors. Disorders of Voluntary Muscle. Cambridge, UK: Cambridge University Press; 2001. pp. 541–559. [Google Scholar]
- 2.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 3.Fu YH, Pizzuti A, Fenwick RG, Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 4.Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 5.Thornton CA, Griggs RC, Moxley RT., 3rd Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol. 1994;35:269–272. doi: 10.1002/ana.410350305. [DOI] [PubMed] [Google Scholar]
- 6.Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology. 1994;44:1448–1452. doi: 10.1212/wnl.44.8.1448. [DOI] [PubMed] [Google Scholar]
- 7.Udd B, Krahe R, Wallgren-Pettersson C, et al. Proximal myotonic dystrophy: a family with autosomal dominant muscular dystrophy, cataracts, hearing loss and hypogonadism: heterogeneity of proximal myotonic syndromes? Neuromuscular Disord. 1997;4:217–228. doi: 10.1016/s0960-8966(97)00041-2. [DOI] [PubMed] [Google Scholar]
- 8.Ranum LP, Rasmussen PF, Benzow KA, et al. Genetic mapping of a second myotonic dystrophy locus. Nature Genetics. 1998;19:196–198. doi: 10.1038/570. [DOI] [PubMed] [Google Scholar]
- 9.Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 10.Ashizawa T, Baiget M. New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1). The International Myotonic Dystrophy Consortium (IDMC) Neurology. 2000;54:1218–1221. doi: 10.1212/wnl.54.6.1218. [DOI] [PubMed] [Google Scholar]
- 11.Day JW, Roelofs R, Leroy B, et al. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2) Neuromuscular Disord. 1999;9:19–27. doi: 10.1016/s0960-8966(98)00094-7. [DOI] [PubMed] [Google Scholar]
- 12.Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657–664. doi: 10.1212/01.wnl.0000054481.84978.f9. [DOI] [PubMed] [Google Scholar]
- 13.Ashizawa T. Myotonic dystrophy as a brain disorder. Arch Neurol. 1998;55:291–293. doi: 10.1001/archneur.55.3.291. [DOI] [PubMed] [Google Scholar]
- 14.Spranger M, Spranger S, Tischendorf M, et al. Myotonic dystrophy. The role of large triplet repeat length in the development of mental retardation. Arch Neurol. 1997;54:251–254. doi: 10.1001/archneur.1997.00550150017009. [DOI] [PubMed] [Google Scholar]
- 15.Joseph JT, Richards CS, Anthony DC, et al. Congenital myotonic dystrophy pathology and somatic mosaicism. Neurology. 1997;49:1457–1460. doi: 10.1212/wnl.49.5.1457. [DOI] [PubMed] [Google Scholar]
- 16.Harper PS, Engelen B, Eymard B, et al. Myotonic dystrophy: present management, future therapy. Neuromuscul Disord. 2002;12:596–599. doi: 10.1016/s0960-8966(02)00020-2. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien TA, Harper PS. Course, prognosis and complications of childhood-onset myotonic dystrophy. Dev Med Child Neurol. 1984;26:62–67. doi: 10.1111/j.1469-8749.1984.tb04407.x. [DOI] [PubMed] [Google Scholar]
- 18.Steyaert J, Die-Smulders C, Fryns JP, et al. Behavioral phenotype in childhood type of dystrophia myotonica. Am J Med Genet. 2000;96:888–889. doi: 10.1002/1096-8628(20001204)96:6<888::aid-ajmg42>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 19.Bassez G, Lazarus A, Desguerre I, et al. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology. 2004;63:1939–1941. doi: 10.1212/01.wnl.0000144343.91136.cf. [DOI] [PubMed] [Google Scholar]
- 20.Chebel S, Ben Hamda K, Boughammoura A, et al. Cardiac involvement in Steinert's myotonic dystrophy. Rev Neurol. 2005;161:932–939. doi: 10.1016/s0035-3787(05)85156-2. [DOI] [PubMed] [Google Scholar]
- 21.Montella L, Caraglia M, Addeo R, et al. Atrial fibrillation following chemotherapy for stage IIIE diffuse large B-cell gastric lymphoma in a patient with myotonic dystrophy (Steinert's disease) Ann Hematol. 2005;84:192–193. doi: 10.1007/s00277-004-0867-6. [DOI] [PubMed] [Google Scholar]
- 22.Dello Russo A, Pelargonio G, Parisi Q, et al. Widespread electroanatomic alterations of right cardiac chambers in patients with myotonic dystrophy type 1. J Cardiovasc Electrophysiol. 2006;17:34–40. doi: 10.1111/j.1540-8167.2005.00277.x. [DOI] [PubMed] [Google Scholar]
- 23.Garrott HM, Walland MJ, O'Day J. Recurrent posterior capsular opacification and capsulorhexis contracture after cataract surgery in myotonic dystrophy. Clin Experiment Ophthalmol. 2004;32:653–655. doi: 10.1111/j.1442-9071.2004.00919.x. [DOI] [PubMed] [Google Scholar]
- 24.Delaporte C. Personality patterns in patients with myotonic dystrophy. Arch Neurol. 1998;55:635–640. doi: 10.1001/archneur.55.5.635. [DOI] [PubMed] [Google Scholar]
- 25.Winblad S, Lindberg C, Hansen S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1) Neuromuscul Disord. 2005;15:287–292. doi: 10.1016/j.nmd.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Barnes PR, Hilton-Jones D, Norbury G, et al. Incorrect diagnosis of myotonic dystrophy and its potential consequences revealed by subsequent direct genetic analysis. J Neurol Neurosurg Psychiatry. 1994;57:662–662. doi: 10.1136/jnnp.57.5.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Udd B, Meola G, Krahe R, et al. 140th ENMC International Workshop: myotonic dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord. 2006;16:403–413. doi: 10.1016/j.nmd.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Udd B, Meola G, Krahe R, et al. Report of the 115th ENMC workshop: myotonic dystrophies. 3rd workshop, 14-16 February 2003, Naarden, the Netherlands. Neuromuscul Disord. 2003;13:589–596. doi: 10.1016/s0960-8966(03)00092-0. [DOI] [PubMed] [Google Scholar]
- 29.Auvinen S, Suominen T, Hannonen P, et al. Myotonic dystrophy type 2 found in two of sixty-three persons diagnosed as having fibromyalgia. Arthritis Rheum. 2008;58:3627–3631. doi: 10.1002/art.24037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suominen T, Schoser B, Raheem O, et al. High frequency of cosegregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and Germany. J Neurol. 2008;255:1731–1736. doi: 10.1007/s00415-008-0010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy. Clinical features of a multisystem disorder similar to myotonic dystrophy. Arch Neurol. 1995;52:25–31. doi: 10.1001/archneur.1995.00540250029009. [DOI] [PubMed] [Google Scholar]
- 32.Meola G. Clinical and genetic heterogeneity in myotonic dystrophies. Muscle Nerve. 2000;12:1789–1799. doi: 10.1002/1097-4598(200012)23:12<1789::aid-mus2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Moxley RT, III, Meola G, Udd B, et al. Report of the 84th ENMC Workshop: PROMM (Proximal Myotonic Myopathy) and Other Myotonic Dystrophy-Like Syndromes: 2nd Workshop. 13-15th October, 2000. Loosdrecht, The Netherlands. Neuromuscular Disord. 2002;12:306–317. doi: 10.1016/s0960-8966(01)00284-x. [DOI] [PubMed] [Google Scholar]
- 34.Schoser BG, Kress W, Walter MC, et al. Homozygosity for CCTG mutation in myotonic dystrophy type 2. Brain. 2004;127:1868–1877. doi: 10.1093/brain/awh210. [DOI] [PubMed] [Google Scholar]
- 35.George A, Schneider-Gold C, Zier S, et al. Musculoskeletal pain in patients with myotonic dystrophy type 2. Arch Neurol. 2004;61:1938–1942. doi: 10.1001/archneur.61.12.1938. [DOI] [PubMed] [Google Scholar]
- 36.Meola G, Sansone V, Marinou K, et al. Proximal myotonic myopathy: a syndrome with a favourable prognosis? J Neurol Sci. 2002;193:89–96. doi: 10.1016/s0022-510x(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 37.Flachenecker P, Schneider C, Cursiefen S, et al. Assessment of cardiovascular autonomic function in myotonic dystrophy type 2 (DM2/PROMM) Neuromuscul Disord. 2003;13:289–293. doi: 10.1016/s0960-8966(02)00277-8. [DOI] [PubMed] [Google Scholar]
- 38.Schoser BG, Ricker K, Schneider-Gold C, et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology. 2004;63:2402–2404. doi: 10.1212/01.wnl.0000147335.10783.e4. [DOI] [PubMed] [Google Scholar]
- 39.Meola G, Sansone V, Perani D, et al. Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology. 1999;5:1042–1050. doi: 10.1212/wnl.53.5.1042. [DOI] [PubMed] [Google Scholar]
- 40.Newman B, Meola G, O'Donovan DG, et al. Proximal myotonic myopathy (PROMM) presenting as myotonia during pregnancy. Neuromuscul Disord. 1999;9:144–149. doi: 10.1016/s0960-8966(98)00118-7. [DOI] [PubMed] [Google Scholar]
- 41.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nature Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 42.Meola G, Sansone V, Perani D, et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM1) and in proximal myotonic myopathy (DM2/PROMM) Neuromuscular Disord. 2003;13:813–821. doi: 10.1016/s0960-8966(03)00137-8. [DOI] [PubMed] [Google Scholar]
- 43.Savkur RS, Philips AV, Cooper TA, et al. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am J Hum Genet. 2004;74:1309–1313. doi: 10.1086/421528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudnik-Schoneborn S, Schneider-Gold C, Raabe U, et al. Outcome and effect of pregnancy in myotonic dystrophy type 2. Neurology. 2006;66:579–580. doi: 10.1212/01.wnl.0000198227.91131.1e. [DOI] [PubMed] [Google Scholar]
- 45.Schoser B, Timchenko L. Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. Curr Genomics. 2010;11:77–90. doi: 10.2174/138920210790886844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bachinsky LL, Czernuszewicz T, Ramagli LS, et al. Premutation allele pool in myotonic dystrophy type 2. Neurology. 2009;72:490–497. doi: 10.1212/01.wnl.0000333665.01888.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lavedan C, Hofmann-Radvanyi H, Shelbourne P, et al. Myotonic dystrophy: size and sex-dependent dynamics of CTG meiotic instability, and somatic mosaicism. Am J Hujm Genet. 1993;52:875–883. [PMC free article] [PubMed] [Google Scholar]
- 48.Monckton DG, Wong LI, Ashizawa T, et al. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. 1995;4:1–8. doi: 10.1093/hmg/4.1.1. [DOI] [PubMed] [Google Scholar]
- 49.Brunner HG, Bruggenwirth HT, Nillesen W, et al. Influence of sex of the transmitting parent as well as of parental allele size on the CTG expansion in myotonic dystrophy (DM) Am J Hum Genet. 1993;53:1016–1023. [PMC free article] [PubMed] [Google Scholar]
- 50.Die-Smulders CE, Smeets HJ, Loots W, et al. Paternal transmission of congenital myotonic dystrophy. J Med Genet. 1997;34:930–933. doi: 10.1136/jmg.34.11.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamshere MG, Harley H, Harper P, et al. Myotonic dystrophy: the correlation of (CTG) repeat length in leukocytes with age at onset is significant only for patients with small expansions. J Med Genet. 1999;36:59–61. [PMC free article] [PubMed] [Google Scholar]
- 52.Thornton C, Johnson K, Moxley RT., 3rd Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol. 1994;35:104–107. doi: 10.1002/ana.410350116. [DOI] [PubMed] [Google Scholar]
- 53.Jiang H, Mankodi A, Swanson MS, et al. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 54.Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 55.Mankodi A, Lin X, Blaxall BC, et al. Nuclear RNA foci in the heart in myotonic dystrophy. Circ Res. 2005;97:1152–1155. doi: 10.1161/01.RES.0000193598.89753.e3. [DOI] [PubMed] [Google Scholar]
- 56.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steadystate levels of CUGBP1 in myotonic dystrophy 1 are due to PKCmediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cardani R, Bugiardini E, Renna LV, et al. Overexpression of CUGBP1 in skeletal muscle from adult classic myotonic dystrophy type 1 but not from myotonic dystrophy type 2. PLOS one. 2013;8(12):e83777–e83777. doi: 10.1371/journal.pone.0083777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 59.Salisbury E, Schoser B, Schneider-Gold C, et al. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol. 2009;175:748–762. doi: 10.2353/ajpath.2009.090047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15:R162–R169. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- 61.Fardaei M, Rogers MT, Thorpe HM, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded- repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 62.Pelletier R, Hamel F, Beaulieu D, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis. 2009;36:181–190. doi: 10.1016/j.nbd.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 63.Fakan S. Perichromatin fibrils are in situ forms of nascent transcriptions. Trend Cell Biol. 1994;4:86–90. doi: 10.1016/0962-8924(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 64.Perdoni F, Malatesta M, Cardani R, et al. RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: an immunocytochemical study. Eur J Histochem. 2009;53:151–158. doi: 10.4081/ejh.2009.e18. [DOI] [PubMed] [Google Scholar]
- 65.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 66.Charlet-B N, Savkur RS, Singh G, et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 67.Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 68.Pascual M, Vicente M, Monferrer L, et al. The muscle blind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- 69.Barreau C, Paillard L, Mereau A, et al. Mammalian CELF/Brunolike RNA-binding proteins: molecular characteristics and biological functions. Biochimie. 2006;88:515–525. doi: 10.1016/j.biochi.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 70.Greco S, Perfetti A, Fasanaro P, et al. Deregulated microRNAs in myotonic dystrophy type 2. PLOS one. 2012;7(6):e39732–e39732. doi: 10.1371/journal.pone.0039732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gambardella S, Rinaldi F, Lepore SM, et al. Overexpression of microRNA- 206 in the skeletal muscle from myotonic dystrophy type 1 patients. J Transl Med. 2010;8:48–54. doi: 10.1186/1479-5876-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perbellini R, Greco S, Sarra-Ferraris G, et al. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord. 2011;21:81–88. doi: 10.1016/j.nmd.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 73.Rau F, Freyermuth F, Fugier C, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol. 2011;18:840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- 74.Maeda M, Taft CS, Bush EW, et al. Identification, tissue-specific expression, and subcellular localization of the 80- and 71- kDa forms of myotonic dystrophy kinase protein. J Biol Chem. 1995;270:20246–20249. doi: 10.1074/jbc.270.35.20246. [DOI] [PubMed] [Google Scholar]
- 75.Huichalaf C, Schoser B, Schneider-Gold C, et al. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J Neurosci. 2009;29:9042–9049. doi: 10.1523/JNEUROSCI.1983-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raheem O, Olufemi SE, Bachinski LL, et al. Mutant (CCTG)n expansion causes abnormal expression of Zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am J Pathol. 2010;177:3025–3036. doi: 10.2353/ajpath.2010.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reddy S, Smith DB, Rich MM, et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet. 1996;13:325–335. doi: 10.1038/ng0796-325. [DOI] [PubMed] [Google Scholar]
- 78.Chen W, Wang Y, Abe Y, et al. Haploinsuffciency for Znf9 in Znf9+/- mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol. 2007;368:8–17. doi: 10.1016/j.jmb.2007.01.088. [DOI] [PubMed] [Google Scholar]
- 79.Day JW, Ranum LP. Genetics and molecular pathogenesis of the myotonic dystrophies. Curr Neurol Neurosci Rep. 2005;5:55–59. doi: 10.1007/s11910-005-0024-1. [DOI] [PubMed] [Google Scholar]
- 80.Bonifazi E, Vallo L, Giardina E, et al. A long PCR-based molecular protocol for detecting normal and expanded ZNF9 alleles in myotonic dystrophy type 2. Diagn Mol Pathol. 2004;13:164–166. [PubMed] [Google Scholar]
- 81.Catalli C, Morgante A, Iraci R, et al. Validation of sensitivity and specificity of tetraplet primed PCR (TP-PCR) in the molecular diagnosis of myotonic dystrophy type 2. J Mol Diagn. 2010;12:601–606. doi: 10.2353/jmoldx.2010.090239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bachinski LL, Udd B, Meola G, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet. 2003;73:835–848. doi: 10.1086/378566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mankodi A, Urbinati CR, Yuan QP, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 84.Cardani R, Mancinelli E, Sansone V, et al. Biomolecular identification of (CCTG)n mutation in myotonic dystrophy type 2 (DM2) by FISH on muscle biopsy. Eur J Histochem. 2004;48:437–442. doi: 10.4081/918. [DOI] [PubMed] [Google Scholar]
- 85.Sallinen R, Vihola A, Bachinski LL, et al. New methods for molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (DM2) Neuromuscul Disord. 2004;14:274–283. doi: 10.1016/j.nmd.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 86.Cardani R, Mancinelli E, Rotondo G, et al. Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2. Eur J Histochem. 2006;50:177–182. [PubMed] [Google Scholar]
- 87.Schoser BG, Schneider-Gold C, Kress W, et al. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve. 2004;29:275–281. doi: 10.1002/mus.10545. [DOI] [PubMed] [Google Scholar]
- 88.Vihola A, Bachinski LL, Sirito M, et al. Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol. 2010;119:465–479. doi: 10.1007/s00401-010-0637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vihola A, Bassez G, Meola G, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology. 2003;60:1854–1857. doi: 10.1212/01.wnl.0000065898.61358.09. [DOI] [PubMed] [Google Scholar]
- 90.Bassez G, Chapoy E, Bastuji-Garin S, et al. Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fiber central nucleation and scattered atrophy. J Neuropathol Exp Neurol. 2008;67:319–325. doi: 10.1097/NEN.0b013e31816b4acc. [DOI] [PubMed] [Google Scholar]
- 91.Pisani V, Panico MB, Terracciano C, et al. Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve. 2008;38:1405–1411. doi: 10.1002/mus.21122. [DOI] [PubMed] [Google Scholar]
- 92.Kwiecinski H, Ryniewicz B, Ostrzycki A. Treatment of myotonia with antiarrhythmic drugs. Acta Neurologica Scandinavica. 1992;86:371–375. doi: 10.1111/j.1600-0404.1992.tb05103.x. [DOI] [PubMed] [Google Scholar]
- 93.Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve. 2007;36:294–306. doi: 10.1002/mus.20800. [DOI] [PubMed] [Google Scholar]
- 94.Chang L, Ernst T, Osborn D, et al. Proton spectroscopy in myotonic dystrophy: correlations with CTG repeats. Arch Neurol. 1998;55:305–311. doi: 10.1001/archneur.55.3.305. [DOI] [PubMed] [Google Scholar]
- 95.Akiguchi I, Nakano S, Shiino A, et al. Brain proton magnetic resonance spectroscopy and brain atrophy in myotonic dystrophy. Arch Neurol. 1999;56:325–330. doi: 10.1001/archneur.56.3.325. [DOI] [PubMed] [Google Scholar]