

Abstract
Nonalcoholic steatohepatitis (NASH) is a major risk factor for hepatic fibrogenesis. NASH is often found in diabetic patients with hyperglycemia. Hyperglycemia induces non-enzymatic glycation of proteins, yielding advanced glycation end-products (AGEs). Effects of AGEs are mainly mediated by two categories of cytoplasmic membrane receptors. Receptor for AGEs (RAGE) is associated with increased oxidative stress and inflammation, whereas AGE receptor-1 (AGE-R1) is involved in detoxification and clearance of AGEs. Activation of hepatic stellate cells (HSC) is crucial to the development of hepatic fibrosis. We recently reported that AGEs stimulated HSC activation likely by inhibiting gene expression of AGE-R1 and inducing gene expression of RAGE in HSC, which were eliminated by the antioxidant curcumin. This study is to test our hypothesis that curcumin eliminates the effects of AGEs on the divergent regulation of the two receptors of AGEs in HSC by interrupting the AGEs-caused activation of leptin signaling, leading to the inhibition of HSC activation. We observed herein that AGEs activated leptin signaling by inducing gene expression of leptin and its receptor in HSC. Like AGEs, leptin differentially regulated gene expression of RAGE and AGE-R1. Curcumin eliminated the effects of AGEs in HSC by interrupting leptin signaling and activating transcription factor Nrf2, leading to the elevation of cellular glutathione and the attenuation of oxidative stress. In conclusions, curcumin eliminated the effects of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC by interrupting the AGEs-caused activation of leptin signaling, leading to the inhibition of HSC activation.
Keywords: advanced glycation end-products, hepatic fibrosis, hepatic stellate cells, glutathione, oxidant stress
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is a major risk factor for the development of hepatic fibrosis (1). Major liver-related clinical consequences are minimized in NASH patients if fibrogenesis can be halted (2, 3). NASH is often found in obese and diabetic patients with hyperglycemia (4). Hyperglycemia induces non-enzymatic glycation of proteins, yielding advanced glycation end-products (AGEs) (5), which are accelerated by oxidative stress (6). The liver is not only a site for clearing circulating AGEs, but also a target organ (7). Elevated levels of serum AGEs are observed in patients with NASH (8). Effects of AGEs are mediated by two categories of cytoplasmic membrane receptors. Receptor for AGEs (RAGE) is associated with increased oxidative stress, cell growth, and inflammation (9), The axis of AGE-RAGE-oxidative stress is involved in diabetic and cardiovascular complications (10). AGE receptor-1 (AGE-R1) is involved in detoxification and clearance of AGEs (11). In contrast to a dramatic increase in expression of RAGE in diabetic patients with high levels of plasma AGEs (12, 13), the abundance of AGE-R1 is significantly reduced in diabetic patients (14).
NASH is featured with fat accumulation and inflammation in the liver and accompanied with abnormally elevated levels of plasma leptin, i.e. hyperleptinemia, (15, 16). Leptin, a 16 kD adipose-derived protein, is the product of an obese gene (ob) and is mainly responsible for control of food uptake and regulation of energy balance (17). Binding of leptin induces the activation of its receptors and downstream signaling cascades, including JAK2/STAT3 and PI3K (17). Leptin-deficient mice (ob/ob) fail to develop hepatic fibrosis during steatohepatitis or in response to chronic toxic liver injury (18). However, the administration of physiological levels of circulating leptin in ob/ob mice by injecting exogenous leptin resumes liver fibrosis caused by dietary manipulations (18). On the other hand, thioacetamide-induced liver fibrosis is almost completely diminished in leptin receptor-deficient Zucker rats (19). These observations strongly imply that leptin might play a permissive role in the development of hepatic fibrosis.
Hepatic stellate cells (HSC) are the primary source of extracellular matrix (ECM) during hepatic fibrogenesis (20). Activation of quiescent HSC undergo profound phenotypic changes, including enhanced cell proliferation, de novo expression of α-smooth muscle actin (α-SMA), over-production of ECM and depletion of cellular lipids (20). Enormous evidence has demonstrated that leptin induces HSC activation in vitro and in vivo (21–24). Although NASH-related hepatic fibrosis is currently the target of significant scientific and clinical interest in the world, very few breakthroughs have occurred in therapeutic intervention of this disease (1). Research identifying novel, safe and effective anti-fibrotic agents is, thus, of the highest priority (25). Most evolving anti-fibrotic therapies are aimed at inhibiting the activation of HSC. No effective medicine is currently available for treatment of obesity- & NASH-related hepatic fibrosis (26, 27). The antioxidant curcumin, an ingredient derived from turmeric, is a promising dietary component for protection of the liver against fibrogenic insults (28, 29). We and others have shown that curcumin inhibits the activation of HSC in vitro (30–34) and protects the liver from fibrogenesis in vivo (28, 35–37). Curcumin suppresses gene expression of leptin and its receptor and interrupts leptin signaling in HSC (24).
We recently reported that AGEs inhibited gene expression of AGE-R1 and induced gene expression of RAGE in HSC, and stimulated HSC activation (38, 39). Curcumin eliminated the effects of AGEs on the divergent regulation of gene expression of the receptors (38, 39). Additional experiments are necessary to elucidate the underlying mechanisms. The aim of this study was to test our hypothesis that curcumin eliminated the effects of AGEs on the induction of gene expression of RAGE and the suppression of gene expression of AGE-R1 in HSC by interrupting AGEs-caused activation of leptin signaling, leading to the inhibition of HSC activation. Culturing quiescent HSC on plastic plates causes spontaneous activation, mimicking the process seen in vivo, which provides a good model for elucidating underlying mechanisms of HSC activation and studying the potential therapeutic intervention of the process.
MATERIALS AND METHODS
Materials and chemicals
AGEs was generated as we previously described (38, 39). The AGE concentration was determined by measuring AGE-specific fluorescence with excitation at 360nm and emissions at 440 nm. The fluorescence of the BSA control was used as a base line, which was at least 70 fold less than that of the AGE-BSA sample. No contamination with insulin-like growth factor-1 and/or endotoxin was detected. Curcumin (purity > 94%), recombinant leptin, the JAK2 inhibitor AG490, the PI3K inhibitor LY294002, N-acetyl-cysteine (NAC), L-buthioninesulfoximine (BSO) were purchased from Sigma, Inc. (Saint Louis, MO).
HSC isolation and cell culture
Male Sprague Dawley rats (200–250 g), C57BL/6J mice (20–40 g) and leptin deficient mice (ob/ob) with C57BL/6J background (20–40 g) were purchased from the Harlan Laboratories, Inc. (Indianapolis, IN). They were housed in a temperature-controlled animal facility with a 12-h light, 12-h dark cycle and allowed free access to regular chew and water ad libitum. HSC were isolated by the pronase-collagenase perfusion in situ before density gradient centrifugation, as we previously described (24). The animal protocol for the use of rats or mice was approved by Institutional Animal Care and Use Committee of Saint Louis University. [It is noteworthy that all of experiments in this report used HSC from rats, except experiments using ob/ob, in which HSC were respectively isolated from leptin deficient (Lept −/−) mice and from wild-type (Lept +/+) mice.] Primary HSC were cultured in Dulbecco’s modification of Eagle’s medium (DMEM) supplemented with 20% of fetal bovine serum (FBS). HSC were passaged in DMEM with 10% of FBS. Semi-confluent HSC with 4 to 9 passages were used for experiments in this report. In some experiments, cells were cultured in serum-depleted media for 24 h before treatment, which rendered HSC more sensitive to exogenous leptin or AGEs. Cells were subsequently treated and cultured in serum-depleted media, which excluded the interference from other factors in FBS.
Cell growth assays
Cell growth was determined by using the CellTiter 96 aqueous nonradioactive cell proliferation assay kit (MTS assays) (Promega, Madison, WI), following the protocol provided by the manufacturer. Each group was carried out in triplicates and repeated for at least three times. Results were expressed as fold changes in the density of viable cells.
Immuno-precipitation (IP) and Western blotting analyses
Whole cell extracts were prepared from semi-confluent passaged HSC after treatment. Protein concentrations were determined by using the BCA Protein Assay kit according to the protocol provided by the manufacturer (Pierce, Rockford, IL). IP, electrophoresis, transblotting, and immuno-detection were conducted as previously described (24). Primary antibodies against leptin (sc-843), Ob-R (sc-8325), RAGE (sc-5563), AGE-R1 (sc-25558), phospho-JAK2 (sc-16566-R), JAK2 (sc-294), phospho-STAT3 (sc-8059), STAT3 (sc-482), phospho-PI3K (sc-12929-R), PI3K (sc-423), Nrf2 (sc-13032), and horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibody against β-actin was purchased from Sigma-Aldrich, Inc. (Saint Louis, MO). β-actin was used as an internal control for equal loading. Representatives were presented from three independent experiments. The commercial antibodies for detecting RAGE and AGE-R1 recognized the proteins from both rat and mouse. [Results from Western blotting analyses were representatives from at least three independent experiments.]
RNA isolation and real-time PCR
Preparation of total RNA and real-time PCR assays using SYBR green were carried out as we previously described (24). Total RNA was treated with deoxyribonuclease I before the synthesis of the first strand of cDNA. mRNA levels were expressed as fold changes after normalization with endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH), as suggested by Schmittgen et al (40). Real-time PCR primers for leptin, Ob-Rb, and GAPDH were previously described (24). [The primer sequences for measuring RAGE and AGE-R1 mRNA were shared by the genes from both rats and mice:]
| RAGE: | 5′-GAA TCC TCC CCA ATG GTT CA-3′ (F) 5′-GCC CGA CAC CGG AAA GT-3′ (R). |
| AGE-R1: | 5′-GCT CTG ATA TCG GTG ACC CT-3′ (F) 5′-TCG TAG TTG TGG TGGTCG AT-3′ (R). |
Plasmids and transient transfection assays
The Nrf2 trans-activity luciferase reporter plasmid p8xARE-luc, which contained 8 copies of antioxidant response elements (ARE), was a gift from Dr. Roland Wolf (41). The luciferase reporter plasmid pRAGE-luc, containing a fragment (−2168 bp) of the mouse RAGE gene promoter, was previously described (39). The luciferase reporter plasmid pAGE-R1-luc with a fragment (−3838 bp) of the mouse AGE-R1 gene promoter was previously described (38). The cDNA expression plasmid pNrf2, or pdn-Nrf2, respectively contained a full length of wild-type Nrf2 cDNA, or dominant negative (dn) Nrf2 cDNA, subcloned in a cDNA expression plasmid (42). Both of the plasmids were generously provided by Dr. Raekil Park (42). The cDNA expression plasmids pwt-STAT3 and pdn-STAT3 were kindly provided by Dr. Gerhard Müller-Newen (43). pwt-STAT3 contains a full length of wild-type STAT3 cDNA subcloned in the expression plasmid pcDNA 5FRT. However, in pdn-STAT3, a nucleotide “A” was mutated to “T” in STAT3 cDNA, leading to the substitution of tyrosine 705 to phenylalanine and to the generation of dominant negative (dn) STAT3 (43). Semi-confluent HSC in six-well cell culture plates were transiently transfected using the Lipofectamine reagent (Invitrogen Corp. Carlsbad, CA), as we previously described (24). In experiments of transient transfection, 2 μg of luciferase reporter plasmid DNA and 0.5 μg of the β-galactosidase activity reporter plasmid pSV-β-gal (Promega, Madison, WI, USA) were used in each well of a 6-well plate. Each sample was in triplicate in every experiment. Transfection efficiency was normalized by co-transfection of pSV-β-gal (Promega, Madison, WI). β-galactosidase activities were measured by using a chemiluminescence assay kit (Tropix, Bedford, MA). Luciferase activities were measured by the luciferase assay system from Promega (Madison, WI, USA), using an automated luminometer (Turner Designs, Inc., Sunnyvale, CA, USA). Luciferase activities were presented in arbitrary units after normalization with β-galactosidase activities based on per microgram of proteins.
Immuno-fluorescent staining
HSC were seeded in the slide-flasks. After treatment, cells were fixed with 4% paraformaldahyde for 10 minutes and washed in 1xPBS for 5 minutes, then blocked in a solution containing 5% normal goat serum, 1% FBS, and 0.05% TX100 in PBS, for 30 minutes. Cells on slides were incubated in primary rabbit polyclonal antibodies against Nrf2 (1:50) at 4° C for overnight. After wash with PBS for 15 minutes for 3 times, cells were incubated with goat anti-rabbit secondary antibodies (1:500) conjugated with green-fluorescent Alexa Fluor® 488 dye (A11008, Invitrogen, CA, USA) at room temperature for 1 h. After wash in PBS, the slides were finally mounted with mounting solution containing 4′-6-Diamidino-2-phenylindole (DAPI) in PBS for nuclei staining and evaluated under a fluorescent microscope (Leico DM4000B microscope, North Central Instruments, Plymouth, MN). An oil lens (100x) was used when photos were taken. Images were merged using Image J software. [Views from immuno-staining were representatives from at least three independent experiments.]
Analyses of the activity of glutamate-cysteine ligase (GCL)
GCL activities were determined at 25°C spectrophotometrically using a coupled assay with pyruvate kinase and lactate dehydrogenase as we described previously (34).
Determination of cellular glutathione (GSH)
The levels of cellular GSH and GSSG in HSC were, respectively, determined by using the enzyme immune assay kit GSH-400 (Cayman, Ann Arbor, MI), following the protocol provided by the manufacturer, as we described previously (34).
Statistical analysis
Differences between means were evaluated using an unpaired two-sided Student’s t test (P≤0.05 was considered as significant). Where appropriate, comparisons of multiple treatment conditions with controls were analyzed by ANOVA with the Dunnett’s test for post hoc analysis. [It is noteworthy that results from real-time PCR were combined from three repeats and statistically analyzed (n=3). Results from luciferase activity assays were from at least six repeats (n≥6).]
RESULTS
AGEs dose-dependently induced gene expression of leptin and leptin receptor in activated HSC in vitro
We recently reported that AGEs stimulated HSC activation, likely by inhibiting gene expression of AGE-R1 and inducing gene expression of RAGE in HSC (38, 39). In addition, the activation of HSC induced gene expression of leptin and activated its signaling, which, in turn, accelerated the activation of HSC (21, 24, 44, 45). We, therefore, assumed that the effects of AGEs on the divergent regulation of the expression of the two receptor genes in HSC might be mediated by inducing gene expression of leptin and activating its signal transduction pathways, leading to the activation of HSC. To test the assumption, we started by evaluating the effect of AGEs on gene expression of leptin and its receptor Ob-Rb. Serum-starved HSC were treated with AGEs at indicated doses (0–200 μg/ml) in serum-depleted media for 24 h. The subsequent culture in serum-depleted media excluded the interference from factors in FBS. Total RNA or whole cell extracts were prepared for real-time PCR or Western blotting analyses. As shown in Fig. 1A, AGEs dose-dependently enhanced the mRNA level of leptin and Ob-Rb in passaged HSC. Further experiments by IP and Western blotting analyses revealed that AGEs elevated the protein abundance of leptin and Ob-R in the cells. The results collectively demonstrated that AGEs dose-dependently stimulated gene expression of leptin and its receptor Ob-R in HSC in vitro.
Figure 1. AGEs dose-dependently induced gene expression of leptin and its receptor Ob-R in activated HSC in vitro.
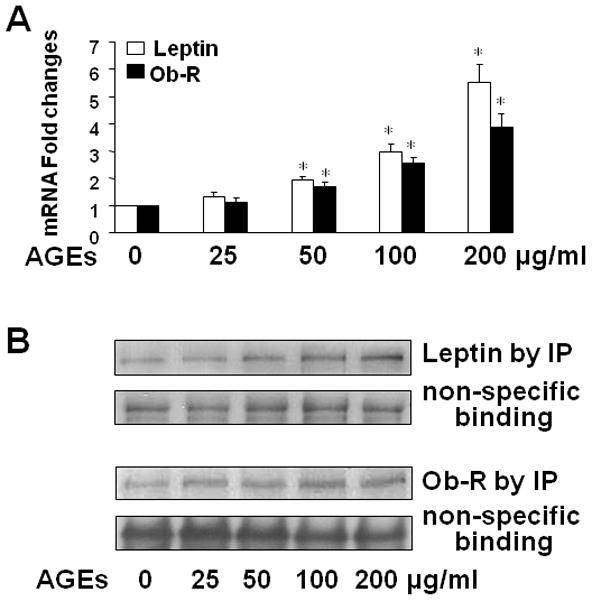
Serum-starved HSC were treated with AGEs as indicated. Total RNA or whole cell extracts were prepared. A. Real-time PCR analyses of leptin and Ob-Rb mRNA. Values were expressed as mRNA fold changes (means ± SD) (n= 3). *P<0.05 vs. the untreated control (the corresponding 1st column). B. Western blotting analyses of immune-precipitated leptin and Ob-R. Nonspecifically recognized heavy chain was used as an invariant control for equal loading. Representatives were presented from three independent experiments.
Leptin dose-dependently and differentially regulated gene expression of RAGE and AGE-R1 in activated HSC in vitro
To determine the role of leptin in regulating gene expression of RAGE and AGE-R1 in HSC, serum-starved cells were treated with leptin at different doses (0–150 ng/ml) in serum-depleted media for 24 h. Total RNA or whole cell extracts were prepared. As shown in Fig. 2A, compared with untreated control (the corresponding 1st column), leptin dose-dependently and differentially altered the mRNA levels of RAGE and AGE-R1 in HSC by elevating the mRNA level of RAGE and reducing the mRNA content of AGE-R1 (the corresponding 2nd to 6th columns). Additional Western blotting analyses assured the observation and indicated that leptin increased the protein abundance of RAGE and reduced the content of AGE-R1 in HSC in a dose dependent manner (Fig. 2B). Taken together, these data demonstrated that leptin dose-dependently and differentially regulated gene expression of RAGE and AGE-R1 in activated HSC in vitro.
Figure 2. Leptin dose-dependently and differentially regulated gene expression of RAGE and AGE-R1 in activated HSC in vitro.
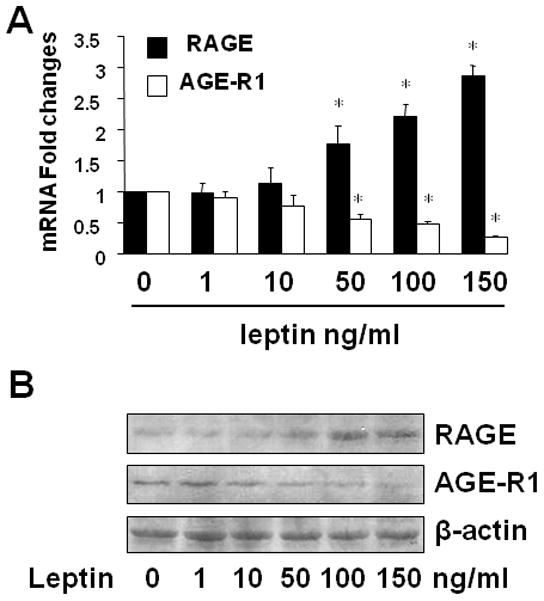
Serum-starved HSC were treated with leptin (100 ng/ml) as indicated. Total RNA or whole cell extracts were prepared. A. Real-time PCR analyses of RAGE and AGE-R1 mRNA. Values were expressed as mRNA fold changes (means ± SD) (n= 3). *P<0.05 vs. the untreated control (the corresponding 1st column). B. Western blotting analyses of RAGE and AGE-R1. β-actin was used as an invariant control for equal loading. Representatives were presented from three independent experiments.
Leptin deficient HSC were irresponsive to AGEs in cell growth and in the regulation of gene expression of RAGE and AGE-R1, which were dose-dependently restored by exogenous leptin
We recently showed that AGEs dose-dependently induced the activation of HSC by stimulating cell growth and the expression of pro-fibrogenic genes (38). We assumed that leptin might mediate the stimulatory effects of AGEs. To test the assumption, HSC were isolated from wild-type mice or from leptin deficient (ob/ob) mice. After serum-starvation in serum depleted media for 24 h, both genotypes of passaged mouse HSC were respectively pretreated with AGEs at indicated concentrations (0–100 μg/ml) for 1 h with or without the addition of exogenous recombinant leptin at indicated concentrations for additional 24 h. Cell growth was determined by using MTS assays. As shown in Figure 3A, AGEs showed no effect on cell growth in ob/ob HSC (the 1st to 4th columns). In great contrast, in the presence of exogenous leptin (20 ng/ml), AGEs caused a dose-dependent increase in cell growth of the ob/ob HSC (the 5th to 8th columns). Further experiments in Fig. 3B showed that AGEs caused, as expected, an increase in cell growth of HSC from wild-type mice (the 1st to 4th of the white columns), but not HSC from ob/ob mice (the 1st to 4th of the black columns). However, exogenous leptin facilitated the stimulatory role of AGEs at 100 μg/ml and caused a dose-dependent increase in both genotypes of HSC (the 5th to 8th of the columns). Nevertheless, the stimulatory effect of leptin on cell growth in ob/ob HSC was significantly weaker (black columns), compared with that in wild-type HSC (white columns). Taken together, the results indicated that leptin was a necessary factor and played a critical role in the process of the AGEs-caused cell growth of HSC in vitro.
Figure 3. Leptin-deficient HSC were irresponsive to AGEs in cell growth and in the regulation of gene expression of RAGE and AGE-R1, which were dose-dependently restored by exogenous leptin.
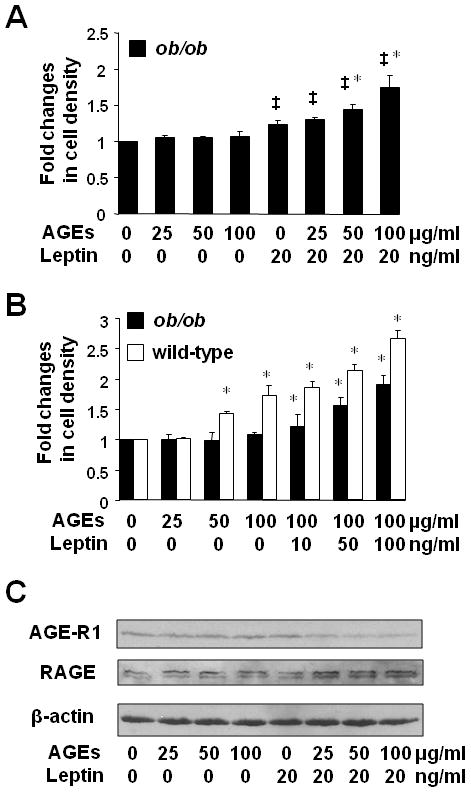
[HSC were isolated from wild-type mice or from leptin deficient (ob/ob) mice.] After serum-starvation in serum depleted media for 24 h, passaged mouse HSC were pretreated with AGEs at indicated concentrations for 1 h with or without the addition of exogenous recombinant leptin at indicated concentrations for additional 24 h. A & B. Cell growth was determined by colorimetric MTS assays. Results were expressed as fold changes in cell density. Values were expressed as means ± SD (n=3). *P<0.05 vs. the untreated control (the corresponding 1st column). C. Western blotting analyses of RAGE and AGE-R1. β-actin was used as an invariant control for equal loading. Representatives were presented from three independent experiments.
To explore the underlying mechanisms, we presumed that leptin critically mediated the effect of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC. To test the presumption, serum-starved leptin deficient HSC were treated with AGEs at indicated concentrations (0–100 μg/ml) with or without exogenous leptin (20 ng/ml) for 24 h. Whole cell extracts were prepared. As shown in Fig. 3C by Western blotting analyses, AGEs had no apparent impact on the abundance of RAGE or AGE-R1 in ob/ob HSC (the 1st to 4th wells). However, in the present of exogenous leptin at 20 ng/ml, AGEs significantly reduced the level of AGE-R1 and increased the abundance of RAGE in a dose dependent manner in ob/ob HSC (the 5th to 8th wells), suggesting the necessity of leptin in the process. It is noteworthy that unlike rat RAGE, mouse RAGE always shows two bands detected by antibodies used in this report. It has been reported that RAGE has variant splicing products in mammals (46). Taken together, these results indicated that leptin was necessary for AGEs to stimulate cell growth and mediated the impact of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC.
Inhibition of the activation of JAK2/STAT3 or PI3K/AKT abrogated the impacts of AGEs or leptin on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC
Prior studies have shown that JAK2/STAT3 and PI3K/AKT are the major intermediators in signal transduction cascades activated by leptin (47, 48). To explore the role of leptin signaling pathways in the impact of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1, serum-starved HSC were pretreated with AG490 (20 μM), a specific JAK2 inhibitor, or LY294002 (20 μM), a selective inhibitor of PI3K/AKT, for 1 h prior to the addition of leptin (100 ng/ml) or AGEs (100 μg/ml) for additional 24 h. Total RNA or whole cell extracts were prepared for real-time PCR or Western blotting analyses. As shown in Fig. 4A and 4B, compared with the untreated control (the corresponding 1st column or well), leptin as well as AGEs, as expected, elevated the levels of RAGE transcript and protein and reduced the contents of AGE-R1 mRNA and protein (the corresponding 2nd or 4th columns or wells). It was noteworthy that the differential effects of leptin or AGEs were abrogated by the JAK2 inhibitor AG490 (the corresponding 3rd or 5th columns or wells). Similarly, the inhibition of PI3K/AKT by LY294002 (LY) also eliminated the effects of leptin or AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 (Fig. 4C and D).
Figure 4. Inhibition of the activation of JAK2/STAT3 or PI3K/AKT abrogated the impacts of AGEs or leptin on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC.
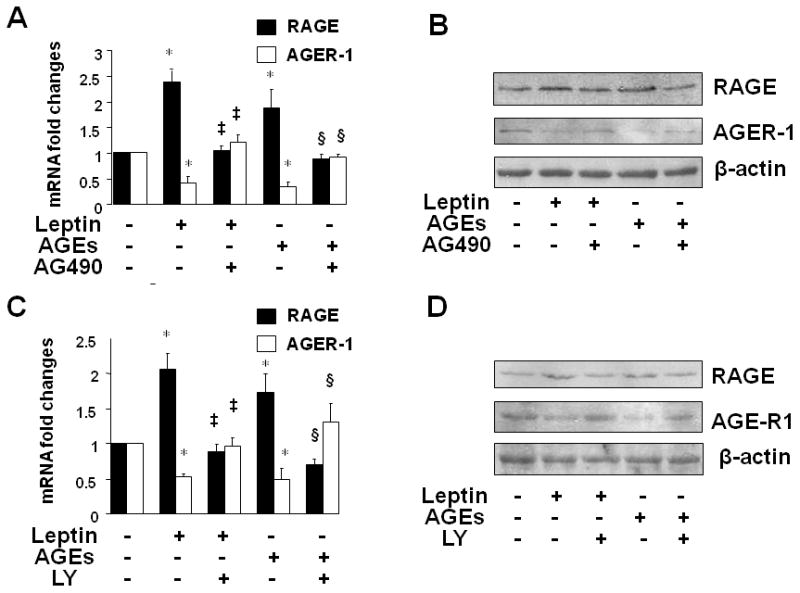
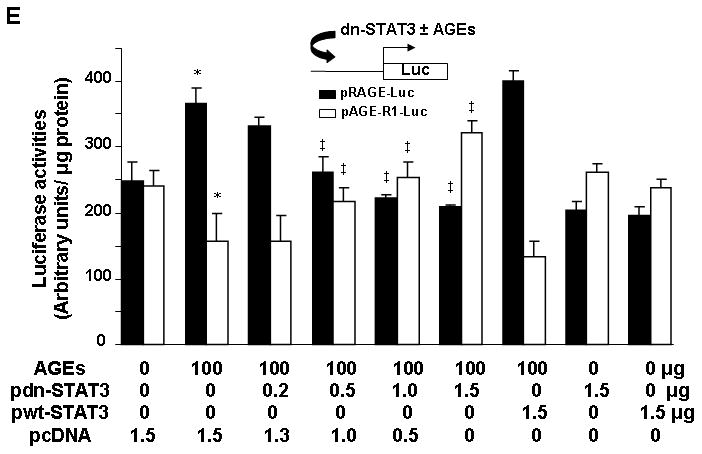
Serum-starved HSC were pretreated with AG490 (20 μM), a specific JAK2 inhibitor, or LY294002 (20 μM), a selective inhibitor of PI3K/AKT, for 1 h prior to the addition of leptin (100 ng/ml) or AGEs (100 μg/ml) for additional 24 h. Total RNA or whole cell extracts were prepared for real-time PCR or Western blotting analyses of RAGE or AGE-R1. A & B. Cells were treated with AG490 and analyzed by real-time PCR (A), or Western blotting analyses (B); C & D. Cells were treated with LY294002 (LY) and analyzed by real-time PCR (C), or Western blotting analyses (D). Values in A & C were expressed as mRNA fold changes (means ± SD) (n=3). *P<0.05 vs. the untreated control (the corresponding 1st column); ‡P<0.05 vs. the cells treated with leptin only (the corresponding 2nd column); §P<0.05 vs. the cells treated with AGES only (the corresponding 5th column). β-actin was used as an invariant control in Western blotting analyses for equal loading. Representatives were presented from three independent experiments. [E. Luciferase activity assays of HSC co-transfected with the rage or age-r1 promoter luciferase reporter plasmid pRAGE-luc or pAGE-R1-luc and a cDNA expression plasmid pdn-STAT3 or pwt-STAT3, followed by the treatment with or without AGEs (100 μg/ml) for 24 h. The plasmids pdn-STAT3 and pwt-STAT3, respectively, encode dominant negative STAT3 and wild-type STAT3. Luciferase activities were expressed as relative units after normalization with β-galactosidase activities (means ± SD, n=6). *P<0.05 vs. the untreated control (the corresponding 1st column); ‡P<0.05 vs. the cells only transfected with pRAGE-luc or pAGE-R1-luc, without pdn-STAT3 or pwt-STAT3 (the corresponding 2nd column).]
[To eliminate potential false effects from the chemical inhibitors used in the experiments, additional experiments were conducted. HSC were co-transfected with the rage or age-r1 promoter luciferase reporter plasmid pRAGE-luc or pAGE-R1-luc (38, 39) and a cDNA expression plasmid pdn-STAT3 or pwt-STAT3 (43). The plasmids pdn-STAT3 and pwt-STAT3, respectively, encode dominant negative STAT3 and wild-type STAT3 (43). A total of 4.0 μg of a DNA mixture was added to each well of cells in 6-well culture plates, including 2 μg of pRAGE-luc or pAGE-R1-luc, 0.5 μg of pSV-β-gal and 1.5 μg of pdn-STAT3, or pwt-STAT3, at various doses plus the empty vector pcDNA. The latter was used to ensure an equal amount of total DNA in transfection assays. After recovery, cells were serum-starved for 4 hr prior to the treatment with or without AGEs (100 μg/ml) in serum-depleted media for additional 24 hr. As shown in Fig. 4E by luciferase activity assays, forced expression of dominant negative STAT3 dose-dependently attenuated the effects of AGEs on divergently regulating the promoter activity of RAGE and AGE-R1 genes in HSC (the 2nd to 6th columns, compared with the corresponding 1st column). In contrast, forced expression of wild-type STAT3 slightly, but not significantly, enhanced the effects of AGEs on the divergent regulation of the promoter activity of RAGE and AGE-R1 genes (the 7th column, compared with the corresponding 2nd column). Forced expression of dominant negative STAT3 or wild-type STAT3, without AGEs, had no apparent impact on the promoter activity of the two genes in the cells (the 8th or 9th column, compared with the corresponding 1st column).] These results collectively indicated that the activation of JAK2/STAT3 and PI3K/AKT played a vital role in the effects of AGEs or leptin on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC.
AGEs activated JAK2/STAT3 and PI3K/AKT in HSC, leading to the divergent regulation of RAGE and AGE-R1, which was interrupted by curcumin
We recently reported that the antioxidant curcumin eliminated the effect of AGEs on stimulating HSC activation by inducing gene expression of AGE-R1 and inhibiting gene expression of RAGE in HSC (38, 39). The underlying mechanisms remain largely to be defined. We presumed that AGEs activated leptin signaling and the activation of JAK2/STAT3 and PI3K/AKT in HSC, leading to the divergent regulation of RAGE and AGE-R1, and that curcumin eliminated these effects of AGEs by interrupting the activation of JAK2/STAT3 and PI3K/AKT. To begin to test the presumption, passaged HSC were treated with the JAK2 inhibitor AG490 at different doses (0–20 μM) for 24 h. Whole cell extracts were prepared for Western blotting analyses. As shown in Fig. 5A, the inhibition of JAK2 by AG490 reduced the abundance of RAGE, meanwhile increased the content of AGE-R1 in a dose-dependent manner. The results confirmed and highlighted the vital role of the activation of JAK2/STAT3, PI3K/AKT in the divergent regulation of gene expression of RAGE and AGE-R1 in HSC. To test our presumption, serum-starved HSC were pretreated with curcumin at different concentrations (0–30 μM) for 1 h before the treatment with or without AGEs (100 μg/ml) for additional 30 minutes. Pilot experiments indicated that AGEs rapidly activated its signaling in HSC and reached its peak within 20 to 30 min (unpublished observations). Whole cell extracts were prepared for Western blotting analyses. As shown in Fig. 5B, C & D, compared with the untreated control (the corresponding 1st wells), AGEs elevated the levels of phosphorylation of JAK2 (5B), STAT3 (5C) and PI3K (5D) (the corresponding 2nd wells), which were eliminated by curcumin in a dose-dependent manner (the corresponding 3rd to 6th wells). These results supported our presumption and demonstrated that curcumin abolished the role of AGEs in the activation of JAK2/STAT3 and PI3K/AKT, which might lead to the elimination of the effect of AGEs on the divergent regulation of RAGE and AGE-R1 in HSC.
Figure 5. AGEs activated JAK2/STAT3 and PI3K/AKT in HSC, likely leading to the divergent regulation of RAGE and AGE-R1, which was interrupted by curcumin.
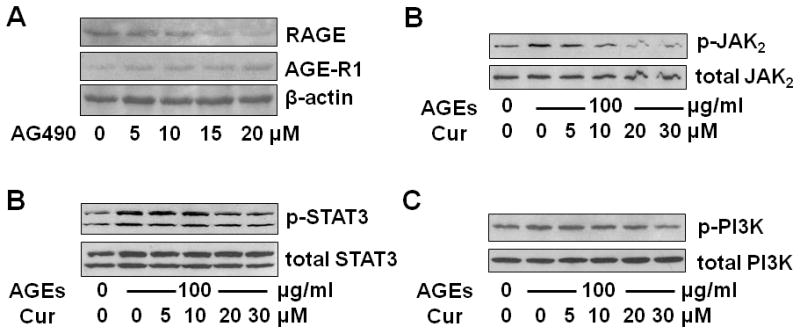
A. Western blotting analyses of RAGE and AGE-R1 in passaged HSC treated with AG490 at indicated concentrations for 24 h. β-actin was used as an invariant control for equal loading. Representatives were presented from three independent experiments. B, C & D. Serum-starved HSC were pretreated with curcumin (Cur) at indicated concentrations (0–30 μM) for 1 h before the treatment with or without AGEs (100 μg/ml) for additional 30 minutes. Whole cell extracts were prepared for Western blotting analyses of phosphorylated JAK2 (p-JAK2) (B), phosphorylated STAT3 (p-STAT3) (C) and phosphorylated PI3K (p-PI3K) (D). Representatives were presented from three independent experiments. Corresponding total proteins were used as internal invariant controls for equal loading.
Curcumin eliminated the impacts of leptin on differentially regulating gene expression of RAGE and AGE-R1
To explore the role of curcumin in the interruption of the AGEs-activated leptin signaling and the elimination of the AGEs-caused divergent regulation of RAGE and AGE-R1, HSC were transfected with the rage or age-r1 promoter luciferase reporter plasmid pRAGE-luc or pAGE-R1-luc. Each of the plasmid contained a fragment of the gene promoter of RAGE or AGE-R1 subcloned in a luciferase reporter plasmid (38, 39). After overnight recovery, cells were serum-starved for 4 h and subsequently pretreated with curcumin (0–30 μM) for 1 h prior to the treatment with or without leptin (100 ng/ml) in serum-depleted media for additional 24 h. Luciferase activity assays were conducted. As shown in Fig. 6A, compared with the untreated control (the corresponding 1st column), leptin significantly increased the luciferase activity in cells transfected with pRAGE-luc (the 2nd black column) and reduced the luciferase activity in cells transfected with pAGE-R1-luc (the 2nd white column). Curcumin dose-dependently eliminated the divergent effects of leptin (the corresponding 3rd to 6th columns). To further explore the observations, serum-starved HSC were similarly pretreated with curcumin (0–30 μM) for 1 h prior to the treatment with or without leptin (100 ng/ml) in serum-depleted media for additional 24 h. Total RNA or whole cell extracts were prepared. Results from real-time PCR (Fig. 6B) and Western blotting analyses (Fig. 6C) substantiated the above observation at the levels of transcript and protein. These results indicated that curcumin eliminated the impacts of leptin on differentially regulating gene expression of RAGE and AGE-R1 in HSC. In summary, results in Fig. 4, 5 and 6 collectively supported our presumptions and revealed that the activation of leptin signaling and the JAK2/STAT3, PI3K/AKT signal pathway played a critical role in the AGEs-caused divergent regulation of RAGE and AGE-R1 in HSC. Curcumin inhibited the activation of JAK2/STAT3 and PI3K/AKT and eliminated the effects of AGEs, or leptin, on the divergent regulation of the expression of the genes in HSC.
Figure 6. Curcumin eliminated the impacts of leptin on differentially regulating gene expression of RAGE and AGE-R1.
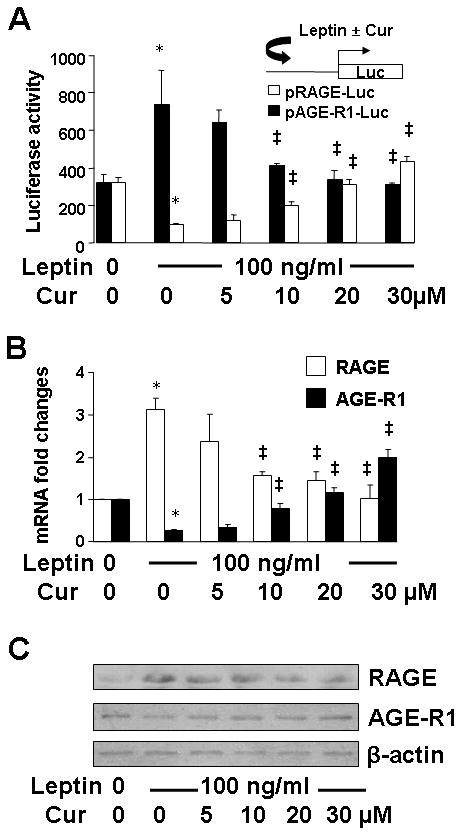
Serum-starved HSC were treated with or without leptin (100 ng/ml) in the presence of curcumin (Cur) at indicated concentrations in serum-depleted DMEM for 24 h. *P<0.05 vs. the untreated control (the corresponding 1st column); ‡P<0.05 vs. the cells treated with leptin only (the corresponding 2nd column). A. Luciferase activity assays of cells transiently transfected with the plasmid pRAGE-luc or pAGE-R1-luc. Luciferase activities were expressed as relative units after normalization with β-galactosidase activities (means ± SD, n≥6). The inset denoted the luciferase reporter plasmids in use and the application of leptin with or without Cur to the system; B. Real-time PCR analyses of RAGE and AGE-R1 mRNA. Values were expressed as mRNA fold changes (means ± SD) (n= 3); C. Western blotting analyses of RAGE and AGE-R1. β-actin was used as an invariant control for equal loading. Representatives were presented from three independent experiments.
Like leptin, AGEs inhibited the activation of Nrf2 in HSC, which was abolished by curcumin
We previously reported that leptin induced oxidative stress in HSC, which was dose-dependently attenuated by curcumin (24). However, the underlying mechanisms remain largely elusive. Activation of the transcription factor NF-E2 p45-related factor 2 (Nrf2) is initiated by phosphorylation of Nrf2, leading to its translocation to the nucleus where it binds to antioxidant/electrophile responsive elements (ARE) in the promoter of target genes (49, 50). We postulated that the AGEs-caused activation of leptin signaling stimulated oxidative stress, and that curcumin attenuated the stimulatory effect by inducing the activation of Nrf2, leading to the elimination of the effect of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1. To test the postulation, first of all, we needed to determine if AGEs or leptin could inhibit the activation of Nrf2 in HSC, and whether curcumin could eliminate the inhibitory effect. Serum-starved HSC were pretreated with curcumin (20 μM) for 1 h prior to the addition of leptin (100 ng/ml) or AGEs (100 μg/ml) for additional 24 h. Immuno-fluorescent stain was used to determine the location of Nrf2 in the cells. As demonstrated in Fig. 7A, Nrf2 was evenly distributed in both of the cytoplasm and nuclei in the untreated control HSC. Like leptin, the treatment with AGEs resulted in the accumulation of Nrf2 in the cytoplasm of HSC, indicating the inhibition of Nrf2 activation. It was of interest to observe that the pretreatment with curcumin eliminated the impact of leptin or AGEs and induced the translocation of Nrf2 into nuclei. These observations suggested that like leptin, AGEs inhibited Nrf2 activation in HSC, which was eliminated by curcumin.
Figure 7. AGEs, like leptin, inhibited the activation of Nrf2 in HSC, which was abolished by curcumin.
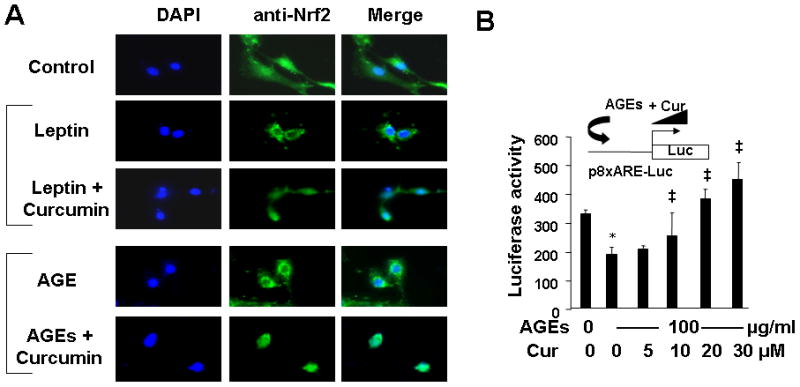
A. Serum-starved HSC were pretreated with curcumin (Cur) (20 μM) for 1 h prior to the addition of leptin (100 ng/ml) or AGEs (100 μg/ml) for additional 24 h. Control cells received no treatment. Cells were fixed with 4% paraformaldahyde for the subsequent immuno-stain of Nrf2. An oil lens (100x) was used. [Views from immuno-staining were representatives from at least three independent experiments.] B. HSC were transiently transfected with the plasmid p8xARE-luc. After recovery, cells were treated with or without AGEs (100 μg/ml) in the presence of curcumin at indicated concentrations in serum-depleted DMEM for 24 h. Luciferase activity assays were conducted. Luciferase activities were expressed as relative units after normalization with β-galactosidase activities (means ± SD, n≥6). *P<0.05 vs. the untreated control cells (the corresponding 1st column). ‡P<0.05 vs. the cells treated with AGEs only (the 2nd column). The inset denoted the plasmid p8xARE-luc in use and the application of AGEs plus curcumin (Cur) to the system.
To further evaluate the impact of AGEs or leptin with or without curcumin on the activation of Nrf2 in HSC, HSC were transfected with the Nrf2 trans-activity luciferase reporter plasmid p8XARE-luc, which contained 8 copies of ARE subcloned in a luciferase reporter plasmid (41). After overnight recovery, cells were serum-starved for 4 h and subsequently pretreated with curcumin at different doses (0–30 μM) for 1 h prior to the addition of AGEs (100 μg/ml), or leptin (100 ng/ml), for additional 24 h. As shown in Fig. 7B and in Supplementary 1 by luciferase activity assays, AGEs, like leptin, significantly reduced luciferase activities in the cells (the corresponding 2nd columns). However, the pretreatment with curcumin dose-dependently abrogated the inhibitory effect of AGEs (or leptin), and increased luciferase activities (the corresponding 3rd to 5th columns). [(Results for leptin treatment were presented in Supplementary 1.)] Taken together, these results indicated that like leptin, AGEs inhibited the activation of Nrf2 in HSC, which was abrogated by curcumin.
Forced expression of Nrf2, or dn-Nrf2, cDNA showed opposite impacts on the regulation of the promoter activities of RAGE and AGE-R1 genes in HSC
To further test our aforesaid postulation and to elucidate the role of Nrf2 in the AGEs-caused divergent regulation of gene expression of RAGE and AGE-R1, passaged HSC were co-transfected with the plasmid pRAGE-luc, or pAGE-R1-luc, plus the cDNA expression plasmid pNrf2 or pdn-Nrf2. The plasmids pNrf2 and pdn-Nrf2, respectively, contained a full length of wild-type Nrf2 cDNA, or dominant negative (dn) Nrf2 cDNA, subcloned in a cDNA expression plasmid (42). After overnight recovery, cells were serum-starved for 4 h and subsequently treated with or without AGEs (100 μg/ml), or curcumin (20 μM) for additional 24 h. Luciferase activity assays were conducted in the cells. As shown in Fig. 8A, AGEs, as expected, significantly activated the rage promoter demonstrated by elevating the luciferase activity in cells with pRAGE-luc, and inhibited the age-r1 promoter shown by reducing the luciferase activity in cells with pAGE-R1-luc (the corresponding 2nd columns). It was of interest to observe that forced expression of Nrf2 cDNA dose-dependently diminished the effect of AGEs on the divergent regulation of the promoter activity of RAGE and AGE-R1 genes (the corresponding 3rd to 7th columns).
Figure 8. Forced expression of Nrf2, or dn-Nrf2, cDNA showed opposite impacts on the regulation of the promoter activities of RAGE and AGE-R1 genes in HSC.
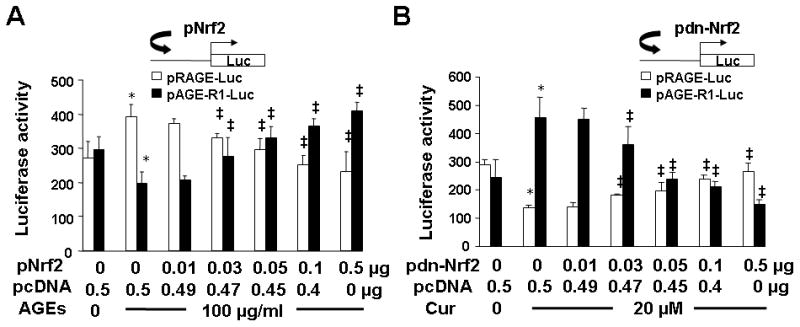
Passaged cells were co-transfected with the luciferase report plasmid pRAGE-luc (A), or pAGE-R1-luc (B), and the cDNA expression plasmid pNrf2 (A), or pdn-Nrf2 cDNA (B), at various doses plus the empty vector pcDNA. The latter was used to ensure equal amount of total DNA in the co-transfection. After recovery, cells were serum-starved for 4 h and treated with or without AGEs (100 μg/ml) (A), or curcumin (Cur) (20 μM) (B), in serum-depleted DMEM for 24 h. Luciferase activities were analyzed and expressed as relative units after normalization with β-galactosidase activities (means ± SD, n=6). *P<0.05 vs. the untreated control cells (the corresponding 1st column). ‡P<0.05 vs. the cells treated with AGEs (A), or curcumin only, (the corresponding 2nd column) (B). The insets denoted the co-transfection of HSC with a luciferase reporter plasmid plus a cDNA expression plasmid in the system.
On the other hand in Fig. 8B, in contrast to AGEs, curcumin, as expected, significantly reduced the luciferase activity in cells with pRAGE-luc and increased the luciferase activity in cells with pAGE-R1-luc (the corresponding 2nd columns). Forced expression of dn-Nrf2 cDNA, which prevented Nrf2 from activation, dose-dependently diminished the differential effects of curcumin on the promoter activity of RAGE and AGE-R1 genes (the corresponding 3rd to 7th columns). Taken together, these results demonstrated that forced expression of Nrf2 or dn-Nrf2 cDNA showed opposite impacts and differentially regulated the promoter activities of RAGE and AGE-R1 genes in HSC. In summary, the results in Fig. 7 and 8 collectively indicated that the activation of Nrf2 had differential impacts on the promoter activity of RAGE and AGE-R1 genes. AGEs, as well as leptin, inhibited the activation of Nrf2 and ARE, leading to the divergent regulation of the promoter activity of RAGE and AGE-R1 genes in HSC, which were eliminated by curcumin.
AGEs decreased the abundance of GCL and its activity in HSC, leading to the reduction in the content of cellular GSH, which was eliminated by curcumin
Glutamate-cysteine ligase (GCL) is a key rate-limiting enzyme in de novo synthesis of glutathione (GSH), a main non-protein thiol (51). GSH reacts with reactive oxygen species (ROS) or functions as a cofactor of antioxidant enzymes, leading to the attenuation of oxidative stress. During that process, GSH is converted to its oxidized form (GSSG). The ratio of GSH/GSSG is regarded as a sensitive indicator of oxidant stress (52, 53). A higher ratio of GSH/GSSH indicates a lower level of oxidant stress. The enzyme of GCL is a heterodimer with a large catalytic subunit (GCLc, ~73 kDa) and a small modifier subunit (GCLm, ~30 kDa), which are encoded by different genes and dissociated under reducing conditions (54, 55). ARE were found in the gene promoters of both GCL subunits (56, 57). The activation of Nrf2 plays a critical role in regulation of gene expression of GCL subunits (58).
Since our earlier results suggested that AGEs inhibited the activation of Nrf2 in HSC, it was, therefore, plausible to explore the impact of AGEs on the expression of GCL and its activity, and further determine the level of GSH and the ratio of GSH/GSSG in HSC. Passaged HSC were treated with AGEs at indicated concentrations for 24 h. Whole cell extracts were prepared respectively for Western blotting analyses of GCL subunits and for GCL activity assays. As shown in Fig. 9A and B, AGEs dose-dependently reduced the abundance of GCLc and GCLm, as well as the activity of GCL in HSC. To further explore the impact of AGEs on oxidative stress in HSC, and elucidate the role of curcumin in eliminating the effect of AGEs and in attenuating the AGEs-caused oxidative stress, HSC were treated with or without AGEs (100 μg/ml) in the presence of curcumin at indicated doses for 24 h. As shown in Fig. 9C and D, compared with the untreated control (the corresponding 1st column), AGEs significantly increased cellular oxidative stress demonstrated by reducing the level of cellular GSH (9C) and the ratio of GSH/GSSG in HSC (9D) (the corresponding 2nd columns). Curcumin dose-dependently eliminated the inhibitory effect of AGEs. Taken together, these results indicated that AGEs elevated oxidative stress by reducing the abundance of GCL subunits and its enzymatic activity in HSC. The latter was attenuated by curcumin.
Figure 9. AGEs decreased the abundance of GCL and its activity in HSC, leading to the reduction in the content of cellular GSH, which was eliminated by curcumin.
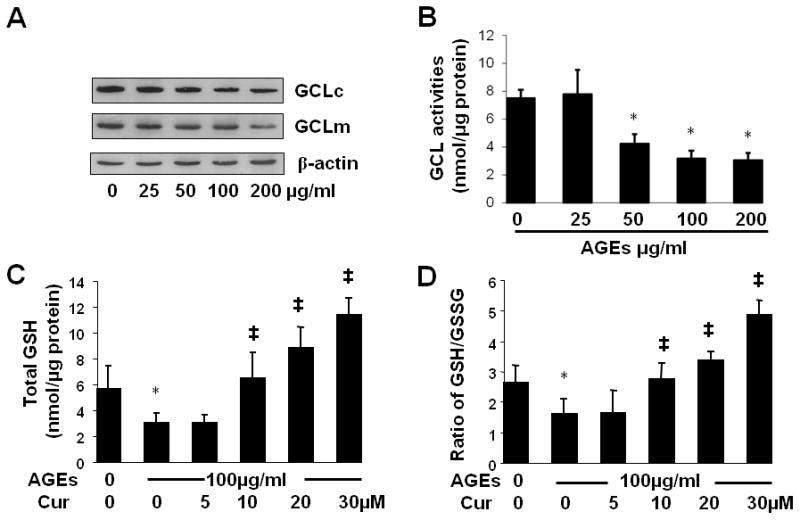
A & B. Passaged HSC were treated with AGEs at indicated concentrations for 24 h. Whole cell extracts were prepared respectively for Western blotting analyses of GCL subunits (A), or for GCL activity assays (B). β-actin was used in Western blotting analyses as an invariant control for equal loading. Representatives were presented from three independent experiments. GCL activities were shown as nmol/μg protein (mean ± SD, n=3). *P<0.05 vs. the untreated control cells (the 1st column). C & D. HSC were treated with or without AGEs (100 μg/ml) in the presence of curcumin (Cur) at indicated doses for 24 h. Cell extracts were prepared for assays. *P<0.05 vs. the untreated control (the 1st column); ‡P<0.05 vs. cells treated with AGEs only (the 2nd column). (C). Determination of cellular GSH contents. GSH contents were shown as nmol/μg protein (mean ± SD, n=3). (D). Determination of the ratio of GSH to GSSG (mean ± SD, n=3).
The inhibition of the activity of GCL abolished the role of NAC and curcumin, and mimicked the effects of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC
Further experiments were necessary to determine if GSH played a critical role in the AGEs-caused divergent regulation of RAGE and AGE-R1 in HSC. Serum-starved HSC were divided into two groups. In one group, cells were stimulated with AGEs (100 μg/ml), or AGEs plus curcumin (Cur) (20 μM), or NAC (5 mM), in serum-depleted media for 24 h. Control cells received no treatment. In the other group, cells were pretreated with BSO (0.25 mM) for 1 h before the addition of AGEs (100 μg/ml), or AGEs plus curcumin (20 μM), or NAC (5 mM), in serum-free media for additional 24 h. Total RNA or whole cells extracts were respectively prepared for real-time PCR or Western blotting analyses. As shown in Fig. 10, compared to the untreated control (the corresponding 1st column or well), AGEs, as expected, significantly induced gene expression of RAGE and reduced gene expression of AGE-R1 (the corresponding 2nd columns and wells). Mimicking curcumin (the corresponding 3rd columns and wells), NAC apparently eliminated the effect of AGEs on the divergent regulation of the expression of the two genes (the corresponding 4th columns and wells). It was of interest to observe that the depletion of cellular GSH by the GCL inhibitor BSO abolished the roles of curcumin and NAC in the elimination of the effects of AGEs (the corresponding 6th and 7th columns and wells). Taken together, these results indicated that the inhibition of the activity of GCL and depletion of cellular GSH by BSO abolished the role of NAC and curcumin, and mimicked the effects of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC, suggesting the critical role of GSH in the process.
Figure 10. The inhibition of the activity of GCL by BSO abolished the role of NAC and curcumin, and mimicked the effects of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC.
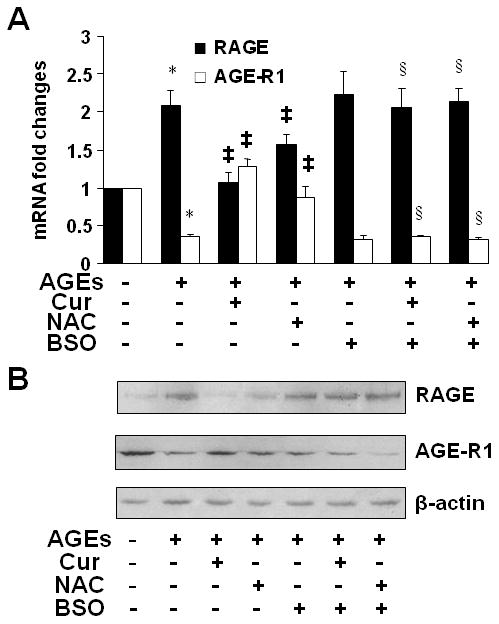
Serum-starved HSC were divided into two groups. In one group, cells were stimulated with AGEs (100 μg/ml), or AGEs plus curcumin (Cur) (20 μM), or NAC (5 mM), in serum-depleted media for 24 h. Control cells received no treatment. In the other group, cells were pretreated with BSO (0.25 mM) for 1 h before the addition of AGEs (100 μg/ml), or AGEs plus curcumin (20 μM), or NAC (5 mM), in serum-free media for additional 24 h. Total RNA or whole cell extracts were prepared. A, Real-time PCR analyses of RAGE and AGE-R1 mRNA. Values were expressed as mRNA fold changes (means ± SD) (n= 3). *P<0.05 vs. the untreated control (the corresponding 1st column); ‡P<0.05 vs. cells treated with AGEs only (the corresponding 2nd column); §P<0.05, vs. cells treated with AGEs plus curcumin, or NAC, (the corresponding 3rd or 4th column, respectively). B, Western blotting analyses of RAGE and AGE-R1. β-actin was used as an invariant control for equal loading. Representatives were presented from three independent experiments.
DISCUSSION
We recently reported that AGEs stimulated HSC activation likely by inducing gene expression of RAGE and suppressing gene expression of AGE-R1, which were abrogated by curcumin (38, 39). However, the underlying mechanisms remained to be defined. In this report, we demonstrated that AGEs induced gene expression of leptin, activated its signaling and elevated cellular oxidative stress in HSC, leading to the divergent regulation of gene expression of RAGE and AGE-R1. Curcumin eliminated the effects of AGEs by interrupting leptin signaling, activating Nrf2 and elevating the level of cellular GSH.
AGEs are non-enzymatic adducts of proteins, lipids, and nucleic acids which form in a time-dependent manner in a pro-oxidant environment (5). A great deal of AGEs is formed endogenously due to hyperglycemia, hyperlipidemia and oxidative stress in diabetic patients (59). AGEs are accumulated in tissues and circulation during liver fibrogenesis (59). In addition, high-fat diets are another major resource of blood AGEs (59). It bears indication that AGEs used in this report were generated in vitro system. It remains unknown to what extent these AGEs could represent “native” AGEs created in vivo system. Although AGEs generated in vivo could be detected in AGEs formed in vitro, the extent of AGE modifications formed in vitro might be unlikely as high as those generated in vivo (60).
There is an inverse correlation between the dietary AGE content and the ratio of AGE-R1 to RAGE, leading to the increase in oxidative stress, organ damage and the short of life span (61). AGEs interact with cells through specific receptors (62). Among them, AGE-R1 is a scavenger receptor (11). RAGE down-regulates cellular defense mechanisms. Studies have implicated that AGEs act as causal factors in the vascular complications of diabetes (61), and that RAGE contributes, at least in part, to the development of diabetic complications (5). The role of AGEs and RAGE has being implied in the development of NASH-associated hepatic fibrosis (63). On the other hand, AGE-R1 acts as a negative regulator of the inflammatory response to AGEs (11). Blockade of AGEs-RAGE signaling attenuates acetaminophen-induced hepatotoxicity in mice (64). It has been suggested that inhibition of AGE formation, blockade of the AGE-RAGE interaction, and suppression of RAGE expression or its downstream pathways may be novel therapeutic strategies for the treatment of AGEs-caused complications (65). Our results revealed that curcumin showed a novel mechanism to diminish the deleterious effects of AGEs and protect HSC from activation by suppressing gene expression of RAGE and inducing gene expression of AGE-R1.
Our results suggested that the activation of key intermediators JAK2/STAT3 and PI3K induced by leptin or AGEs played a critical role in the AGEs-caused divergent regulation of gene expression of RAGE and AGE-R1 (Fig. 4). However, the rapid activation of JAK2/STAT3 and PI3K within 30 min by AGEs in Fig. 5B-D was unlikely induced by leptin produced by HSC. It has been reported that binding AGEs to RAGE induces sustained post-receptor signaling, including activation of p21ras, MAP kinases, and the NF-κB pathway (5). Whether AGE-R1 could transduce signals from AGEs remains elusive. It is plausible to assume that the interaction of AGEs and RAGE could rapidly activate JAK2/STAT3 and PI3K, which initiates an instant action for the induction of gene expression of leptin and its receptor, and the subsequent activation of leptin signaling, ultimately leading to the divergent regulation of gene expression of RAGE and AGE-R1. [This assumption is supported by our observation. The knockdown of RAGE by RAGE shRNA diminished the stimulant effect of AGEs on the activation of HSC in vitro, suggesting a critical role for the induction of rage expression in the process (39). Additional experiments using RAGE shRNA are necessary to further test the assumption, which are being conducted in our lab.]
[The leptin receptor Ob-R is a member of the class I cytokine receptor family. Alternative splicing of Ob-R gene results in at least six transcripts designated Ob-Ra through Ob-Rf (66). Among them, Ob-Rb contains the longest intracellular domain, which is crucial for leptin signaling. Ob-Ra, Ob-Rc and Ob-Rd contain only short cytoplasmic domains. Ob-Rd is only found in mice, while Ob-Rf only exists in rats. In human, expression of Ob-Ra, Ob-Rb and Ob-Rc mRNA has been reported (67). We have shown that curcumin dose-dependently reduces the steady-state levels of mRNA of leptin and different isoforms of Ob-Rs, including Ob-Ra, Ob-Rb, and Ob-Re, in rat HSC in vitro (24). Similar results were also observed in mice (data not published). Ob-Rb was the target in this report (Fig. 1A) by using primers which only hybridized with the region of DNA encoding the intracellular domain. However, we could not exclude the possibility in our report that different receptor isoforms may play different roles in the biology of leptin.]
Although underlying mechanisms remain incompletely understood, accumulating evidence has indicated that oxidative stress plays critical roles in the activation of HSC (20, 68). Oxidative stress is a deleterious imbalance between the production and removal of free radicals, including reactive oxygen species. They are generated from aerobic cells by inflammation and aerobic metabolism (52, 53). Reducing oxidative stress in mammalian cells is through several antioxidant systems, including enzymes and non-enzymatic molecules. Among them, GSH is the main non-protein thiol. The GSH/GSSG ratio is regarded as a sensitive indicator of oxidant stress (52, 53). The interaction between AGEs and RAGE generates oxidative stress and subsequently evokes vascular inflammation, thereby playing a central role in diabetic complications (69). In this study, we demonstrated that, similar to curcumin, the elevation of the level of cellular GSH by NAC eliminated the effect of AGEs on the divergent regulation of RAGE and AGE-R1. These results also highlighted the critical role of elevated oxidative stress in the AGEs-caused divergent regulation of the expression of RAGE and AGE-R1 genes, which, in turn, further induced oxidative stress in a manner of positive feedback and accelerated the activation of HSC.
[We previously demonstrated that curcumin dose-dependently increased the activity of GCL in passaged HSC by inducing gene expression of both subunits of GCL (34). However, the underlying mechanisms were elusive. In this report, we observed that curcumin stimulated the nuclear translocation of Nrf2 and elevated its transcriptional activity in HSC (Fig. 7).] The transcription factor Nrf2 normally forms a complex with the cytoskeleton-associated protein Keap1 in cytoplasm (49, 70). Keap1 and Nrf2 constitute a crucial cellular sensor for oxidative stress (49, 70). ARE is often found in the promoter of many genes and confers a general transcriptional sensitivity to oxidative stress (50). Nrf2 is essential for the ARE-mediated induction of gene expression of anti-oxidative stress proteins/enzymes. Our results in this report suggested that in responding to AGEs, the inactivation of Nrf2 resulted in the suppression of gene expression of GCL subunits in HSC. The underlying mechanisms are under investigation. [Studies have shown that leptin activates several downstream molecules involved in key pathways related to cell survival such as JAK2, STAT3, PI3K, MAP kinases, AMP-activated protein kinase (AMPK), CDK5 and GSK3β (71). Binding AGEs to RAGE induces sustained post-receptor signaling, including activation of p21ras, MAP kinases, and the NF-κB pathway (5).] Our preliminary data suggested that AMPK might be a key regulator in the regulation of Nrf2 activation (data not presented here). Our observations are consistent with prior reports. Phosphorylation of AMPK induces the activation of AMPK (72), and in turn, promotes the phosphorylation of Nrf2, which triggers the translocation of Nrf2 from the cytoplasm into the nucleus, leading to its binding to ARE and the induction of expression of target genes (50). After computer-aided analyses, no consensus ARE was found in the promoters of rodent RAGE or AGE-R1 genes (data not shown here). It suggested that Nrf2 might be indirectly involved in the divergent regulation of the expression of RAGE and AGE-R1. Although beyond the scope of the current project, additional experiments are ongoing in our lab.
Based on our observations, a simplified model is proposed to explain the mechanism of AGEs in the divergent regulation of gene expression of RAGE and AGE-R1 in HSC, which is eliminated by curcumin (Fig. 11). The interaction of AGEs with RAGE induces gene expressions of leptin and its receptor. The activation of leptin signaling suppresses the activity of Nrf2 and gene expression of GCL and its activity, leading to the reduction in cellular GSH and the elevation of oxidative stress. The latter facilitates the divergent regulation of gene expression of RAGE and AGE-R1. The elevated abundance of RAGE shows a positive feedback mechanism and, in turn, enhances the interaction with AGEs and the subsequent events, collectively leading to the activation of HSC. Curcumin interrupts leptin signaling and blocks the occurrences of the subsequent events, resulting in the elimination of the effects of AGEs and the inhibition of HSC activation. It bears emphasis that this model does not exclude any other mechanisms involved in the effects of AGEs. In addition, we should not simply conclude that the protective roles of curcumin solely result from these mechanisms studied in this report. No single unequivocal mechanism accounts for all of versatile roles of curcumin against HSC activation. Our results provide novel mechanisms by which curcumin eliminates the effects of AGEs on the divergent regulation of gene expression of RAGE and AGE-R1 in HSC in vitro. Additional experiments are necessary to explore its role in protection of the liver from hepatic fibrogenesis facilitated by hyperglycemia-associated AGEs in vivo.
Figure 11. A simplified model of the mechanism of AGEs in the divergent regulation of gene expression of RAGE and AGE-R1 in HSC, which is eliminated by curcumin.
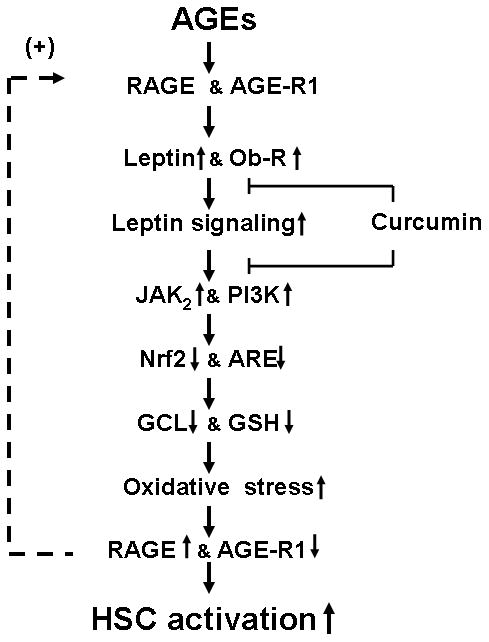
The interaction of AGEs with RAGE induces gene expressions of leptin and its receptor, leading to the activation of leptin signaling, including JAK2 and PI3K. The latter suppresses the activity of Nrf2 and gene expression of GCL and its activity, leading to the reduction in cellular GSH and the elevation of oxidative stress. The latter facilitates the divergent regulation of gene expression of RAGE and AGE-R1. The elevated abundance of RAGE shows a positive feedback mechanism and, in turn, enhances its interaction with AGEs and the subsequent events, collectively leading to the activation of HSC. Curcumin interrupts leptin signaling and blocks the occurrences of the subsequent events, resulting in the elimination of the effects of AGEs and the inhibition of HSC activation. “↑” or “↓” indicates the effects of AGEs. “⊢” represents the inhibitory role of curcumin in the process.
Supplementary Material
HSC were transiently transfected with the plasmid p8xARE-luc. After recovery, cells were treated with or without leptin (100 ng/ml) in the presence of curcumin at indicated concentrations in serum-depleted DMEM for 24 h. Luciferase activity assays were conducted. Luciferase activities were expressed as relative units after normalization with β-galactosidase activities (means ± SD, n≥6). *P<0.05 vs. the untreated control cells (the corresponding 1st column). ‡P<0.05 vs. the cells treated with leptin only (the 2nd column). The inset denoted the plasmid p8xARE-luc in use and the application of leptin plus curcumin (Cur) to the system.
Acknowledgments
The work was supported by RO1 DK 047995 from NIH/NIDDK, the awards from the President Research Fund of Saint Louis University, and from the Liver Center of Saint Louis University to A. Chen
ABBREVIATIONS
- AGEs
advanced glycation end-products
- AGE-R1
AGE receptor-1
- α-SMA
alpha-smooth muscle actin
- ARE
antioxidant responsive elements
- BSO
L-buthioninesulfoximine
- DAPI
4′-6-Diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle’s medium
- ECM
extracellular matrix
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GCL
glutamate-cysteine ligase
- GSH
glutathione
- HSC
hepatic stellate cells
- NAC
N-acetyl-cysteine
- NASH
non-alcoholic steatohepatitis
- Nrf2
NF-E2 p45-related factor 2
- RAGE
Receptor for AGEs
- ROS
reactive oxygen species
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Pinzani M. Pathophysiology of non-alcoholic steatohepatitis and basis for treatment. Dig Dis. 2011;29(2):243–248. doi: 10.1159/000323928. [DOI] [PubMed] [Google Scholar]
- 2.Ribeireiro T, Swain J, Sarr M, et al. NAFLD and insulin resistance do not increase the risk of postoperative complications among patients undergoing bariatric surgery--a prospective analysis. Obes Surg. 2011;21(3):310–315. doi: 10.1007/s11695-010-0228-6. [DOI] [PubMed] [Google Scholar]
- 3.Charlton M, Krishnan A, Viker K, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301(5):G825–834. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefanovic A, Kotur-Stevuljevic J, Spasic S, Bogavac-Stanojevic N, Bujisic N. The influence of obesity on the oxidative stress status and the concentration of leptin in type 2 diabetes mellitus patients. Diabetes Res Clin Pract. 2008;79(1):156–163. doi: 10.1016/j.diabres.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 5.Bierhaus A, Humpert PM, Morcos M, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83(11):876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 6.Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40(4):405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- 7.Smedsrod B, Melkko J, Araki N, Sano H, Horiuchi S. Advanced glycation end products are eliminated by scavenger-receptor-mediated endocytosis in hepatic sinusoidal Kupffer and endothelial cells. Biochem J. 1997;322 (Pt 2):567–573. doi: 10.1042/bj3220567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyogo H, Yamagishi S, Iwamoto K, et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22(7):1112–1119. doi: 10.1111/j.1440-1746.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498(2–3):99–111. doi: 10.1016/s0167-4889(00)00087-2. [DOI] [PubMed] [Google Scholar]
- 10.Ramasamy R, Yan SF, Schmidt AM. The diverse ligand repertoire of the receptor for advanced glycation endproducts and pathways to the complications of diabetes. Vascul Pharmacol. 2012;57(5–6):160–167. doi: 10.1016/j.vph.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu C, He JC, Cai W, Liu H, Zhu L, Vlassara H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc Natl Acad Sci U S A. 2004;101(32):11767–11772. doi: 10.1073/pnas.0401588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brett J, Schmidt AM, Yan SD, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143(6):1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 13.Tanji N, Markowitz GS, Fu C, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11(9):1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- 14.Li YM, Mitsuhashi T, Wojciechowicz D, et al. Molecular identity and cellular distribution of advanced glycation endproduct receptors: relationship of p60 to OST-48 and p90 to 80K-H membrane proteins. Proc Natl Acad Sci U S A. 1996;93(20):11047–11052. doi: 10.1073/pnas.93.20.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman JM. Modern science versus the stigma of obesity. Nat Med. 2004;10(6):563–569. doi: 10.1038/nm0604-563. [DOI] [PubMed] [Google Scholar]
- 16.Yokaichiya DK, Galembeck E, Torres BB, Da Silva JA, de Araujo DR. Insulin and leptin relations in obesity: a multimedia approach. Adv Physiol Educ. 2008;32(3):231–236. doi: 10.1152/advan.00014.2007. [DOI] [PubMed] [Google Scholar]
- 17.Lee GH, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379(6566):632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 18.Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37(2):206–213. doi: 10.1016/s0168-8278(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 19.Sakaida I, Jinhua S, Uchida K, Terai S, Okita K. Leptin receptor-deficient Zucker (fa/fa) rat retards the development of pig serum-induced liver fibrosis with Kupffer cell dysfunction. Life Sci. 2003;73(19):2491–2501. doi: 10.1016/s0024-3205(03)00653-2. [DOI] [PubMed] [Google Scholar]
- 20.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35(4):762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saxena NK, Saliba G, Floyd JJ, Anania FA. Leptin induces increased alpha2(I) collagen gene expression in cultured rat hepatic stellate cells. J Cell Biochem. 2003;89(2):311–320. doi: 10.1002/jcb.10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aleffi S, Petrai I, Bertolani C, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42(6):1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- 24.Tang Y, Zheng S, Chen A. Curcumin eliminates leptin’s effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology. 2009;150(7):3011–3020. doi: 10.1210/en.2008-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wells RG. Liver fibrosis: challenges of the new era. Gastroenterology. 2009;136(2):387–388. doi: 10.1053/j.gastro.2008.12.028. [DOI] [PubMed] [Google Scholar]
- 26.Calamita G, Portincasa P. Present and future therapeutic strategies in non-alcoholic fatty liver disease. Expert Opin Ther Targets. 2007;11(9):1231–1249. doi: 10.1517/14728222.11.9.1231. [DOI] [PubMed] [Google Scholar]
- 27.Federico A, Niosi M, Vecchio Blanco CD, Loguercio C. Emerging drugs for non-alcoholic fatty liver disease. Expert Opin Emerg Drugs. 2008;13(1):145–158. doi: 10.1517/14728214.13.1.145. [DOI] [PubMed] [Google Scholar]
- 28.O’Connell MA, Rushworth SA. Curcumin: potential for hepatic fibrosis therapy? Br J Pharmacol. 2008;153(3):403–405. doi: 10.1038/sj.bjp.0707580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aggarwal BB, Sundaram C, Malani N, Ichikawa H. Curcumin: the Indian solid gold. Adv Exp Med Biol. 2007;595:1–75. doi: 10.1007/978-0-387-46401-5_1. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Fu Y, Chen A. Activation of peroxisome proliferator-activated receptor-gamma contributes to the inhibitory effects of curcumin on rat hepatic stellate cell growth. Am J Physiol Gastrointest Liver Physiol. 2003;285(1):G20–30. doi: 10.1152/ajpgi.00474.2002. [DOI] [PubMed] [Google Scholar]
- 31.Zheng S, Chen A. Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J. 2004;384(Pt 1):149–157. doi: 10.1042/BJ20040928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng S, Chen A. Curcumin suppresses the expression of extracellular matrix genes in activated hepatic stellate cells by inhibiting gene expression of connective tissue growth factor. Am J Physiol Gastrointest Liver Physiol. 2006;290(5):G883–893. doi: 10.1152/ajpgi.00450.2005. [DOI] [PubMed] [Google Scholar]
- 33.Zheng S, Chen A. Disruption of transforming growth factor-beta signaling by curcumin induces gene expression of peroxisome proliferator-activated receptor-gamma in rat hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2007;292(1):G113–123. doi: 10.1152/ajpgi.00200.2006. [DOI] [PubMed] [Google Scholar]
- 34.Zheng S, Yumei F, Chen A. De novo synthesis of glutathione is a prerequisite for curcumin to inhibit hepatic stellate cell (HSC) activation. Free Radic Biol Med. 2007;43(3):444–453. doi: 10.1016/j.freeradbiomed.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu Y, Zheng S, Lin J, Ryerse J, Chen A. Curcumin protects the rat liver from CCl4-caused injury and fibrogenesis by attenuating oxidative stress and suppressing inflammation. Mol Pharmacol. 2008;73(2):399–409. doi: 10.1124/mol.107.039818. [DOI] [PubMed] [Google Scholar]
- 36.Nanji AA, Jokelainen K, Tipoe GL, Rahemtulla A, Thomas P, Dannenberg AJ. Curcumin prevents alcohol-induced liver disease in rats by inhibiting the expression of NF-kappa B-dependent genes. Am J Physiol Gastrointest Liver Physiol. 2003;284(2):G321–327. doi: 10.1152/ajpgi.00230.2002. [DOI] [PubMed] [Google Scholar]
- 37.Park EJ, Jeon CH, Ko G, Kim J, Sohn DH. Protective effect of curcumin in rat liver injury induced by carbon tetrachloride. J Pharm Pharmacol. 2000;52(4):437–440. doi: 10.1211/0022357001774048. [DOI] [PubMed] [Google Scholar]
- 38.Lin J, Tang Y, Kang Q, Chen A. Curcumin eliminates the inhibitory effect of advanced glycation end-products (AGEs) on gene expression of AGE receptor-1 in hepatic stellate cells in vitro. Lab Invest. 2012;92(6):827–841. doi: 10.1038/labinvest.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin J, Tang Y, Kang Q, Feng Y, Chen A. Curcumin inhibits gene expression of receptor for advanced glycation end-products (RAGE) in hepatic stellate cells in vitro by elevating PPARgamma activity and attenuating oxidative stress. Br J Pharmacol. 2012;166(8):2212–2227. doi: 10.1111/j.1476-5381.2012.01910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285(2):194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 41.Wang XJ, Hayes JD, Wolf CR. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006;66(22):10983–10994. doi: 10.1158/0008-5472.CAN-06-2298. [DOI] [PubMed] [Google Scholar]
- 42.So HS, Kim HJ, Lee JH, et al. Flunarizine induces Nrf2-mediated transcriptional activation of heme oxygenase-1 in protection of auditory cells from cisplatin. Cell Death Differ. 2006;13(10):1763–1775. doi: 10.1038/sj.cdd.4401863. [DOI] [PubMed] [Google Scholar]
- 43.Mohr A, Fahrenkamp D, Rinis N, Muller-Newen G. Dominant-negative activity of the STAT3-Y705F mutant depends on the N-terminal domain. Cell Commun Signal. 2013;11(1):83. doi: 10.1186/1478-811X-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Y, Chen A. Curcumin protects hepatic stellate cells against leptin-induced activation in vitro by accumulating intracellular lipids. Endocrinology. 2010;151(9):4168–4177. doi: 10.1210/en.2010-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Y, Chen A. Curcumin prevents leptin raising glucose levels in hepatic stellate cells by blocking translocation of glucose transporter-4 and increasing glucokinase. Br J Pharmacol. 2010;161(5):1137–1149. doi: 10.1111/j.1476-5381.2010.00956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sterenczak KA, Nolte I, Murua Escobar H. RAGE splicing variants in mammals. Methods Mol Biol. 2013;963:265–276. doi: 10.1007/978-1-62703-230-8_16. [DOI] [PubMed] [Google Scholar]
- 47.Ahima RS, Osei SY. Leptin signaling. Physiol Behav. 2004;81(2):223–241. doi: 10.1016/j.physbeh.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 48.Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393(Pt 1):7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7(3–4):385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 51.Fraser JA, Kansagra P, Kotecki C, Saunders RD, McLellan LI. The modifier subunit of Drosophila glutamate-cysteine ligase regulates catalytic activity by covalent and noncovalent interactions and influences glutathione homeostasis in vivo. J Biol Chem. 2003;278(47):46369–46377. doi: 10.1074/jbc.M308035200. [DOI] [PubMed] [Google Scholar]
- 52.Fridovich I. The biology of oxygen radicals. Science. 1978;201(4359):875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 53.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 54.Seelig GF, Simondsen RP, Meister A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J Biol Chem. 1984;259(15):9345–9347. [PubMed] [Google Scholar]
- 55.Sekura R, Meister A. gamma-Glutamylcysteine synthetase. Further purification, “half of the sites” reactivity, subunits, and specificity. J Biol Chem. 1977;252(8):2599–2605. [PubMed] [Google Scholar]
- 56.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem. 1997;272(11):7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- 57.Moinova HR, Mulcahy RT. An electrophile responsive element (EpRE) regulates beta-naphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene. Constitutive expression is mediated by an adjacent AP-1 site. J Biol Chem. 1998;273(24):14683–14689. doi: 10.1074/jbc.273.24.14683. [DOI] [PubMed] [Google Scholar]
- 58.Guan SP, Tee W, Ng DS, et al. Andrographolide protects against cigarette smoke-induced oxidative lung injury via augmentation of Nrf2 activity. Br J Pharmacol. 2013;168(7):1707–1718. doi: 10.1111/bph.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sebekova K, Kupcova V, Schinzel R, Heidland A. Markedly elevated levels of plasma advanced glycation end products in patients with liver cirrhosis - amelioration by liver transplantation. J Hepatol. 2002;36(1):66–71. doi: 10.1016/s0168-8278(01)00232-x. [DOI] [PubMed] [Google Scholar]
- 60.Makita Z, Vlassara H, Cerami A, Bucala R. Immunochemical detection of advanced glycosylation end products in vivo. J Biol Chem. 1992;267(8):5133–5138. [PubMed] [Google Scholar]
- 61.Vlassara H, Uribarri J, Cai W, Striker G. Advanced glycation end product homeostasis: exogenous oxidants and innate defenses. Ann N Y Acad Sci. 2008;1126:46–52. doi: 10.1196/annals.1433.055. [DOI] [PubMed] [Google Scholar]
- 62.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251(2):87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 63.Lohwasser C, Neureiter D, Popov Y, Bauer M, Schuppan D. Role of the receptor for advanced glycation end products in hepatic fibrosis. World J Gastroenterol. 2009;15(46):5789–5798. doi: 10.3748/wjg.15.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ekong U, Zeng S, Dun H, et al. Blockade of the receptor for advanced glycation end products attenuates acetaminophen-induced hepatotoxicity in mice. J Gastroenterol Hepatol. 2006;21(4):682–688. doi: 10.1111/j.1440-1746.2006.04225.x. [DOI] [PubMed] [Google Scholar]
- 65.Yamagishi S, Nakamura K, Matsui T, Ueda S, Fukami K, Okuda S. Agents that block advanced glycation end product (AGE)-RAGE (receptor for AGEs)-oxidative stress system: a novel therapeutic strategy for diabetic vascular complications. Expert Opin Investig Drugs. 2008;17(7):983–996. doi: 10.1517/13543784.17.7.983. [DOI] [PubMed] [Google Scholar]
- 66.Fei H, Okano HJ, Li C, et al. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A. 1997;94(13):7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernotiene E, Palmer G, Gabay C. The role of leptin in innate and adaptive immune responses. Arthritis Res Ther. 2006;8(5):217. doi: 10.1186/ar2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21 (Suppl 3):S84–87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 69.Yamagishi S, Matsui T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev. 2010;3(2):101–108. doi: 10.4161/oxim.3.2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol. 2013;85(6):705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 71.Folch J, Pedros I, Patraca I, et al. Neuroprotective and anti-ageing role of leptin. J Mol Endocrinol. 2012;49(3):R149–156. doi: 10.1530/JME-12-0151. [DOI] [PubMed] [Google Scholar]
- 72.Liu XM, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol Heart Circ Physiol. 2011;300(1):H84–93. doi: 10.1152/ajpheart.00749.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HSC were transiently transfected with the plasmid p8xARE-luc. After recovery, cells were treated with or without leptin (100 ng/ml) in the presence of curcumin at indicated concentrations in serum-depleted DMEM for 24 h. Luciferase activity assays were conducted. Luciferase activities were expressed as relative units after normalization with β-galactosidase activities (means ± SD, n≥6). *P<0.05 vs. the untreated control cells (the corresponding 1st column). ‡P<0.05 vs. the cells treated with leptin only (the 2nd column). The inset denoted the plasmid p8xARE-luc in use and the application of leptin plus curcumin (Cur) to the system.