

Abstract
Background and purpose
Obstructive sleep apnea (OSA), a condition associated with chronic intermittent hypoxia (CIH), carries an increased risk of stroke. However, CIH has been reported to either increase or decrease brain injury in models of focal cerebral ischemia. The factors determining the differential effects of CIH on ischemic injury and their mechanisms remain unclear. Here, we tested the hypothesis that the intensity of the hypoxic challenge determines the protective or destructive nature of CIH by modulating mitochondrial resistance to injury.
Methods
Male C57Bl/6J mice were exposed to CIH with 10% or 6% O2 for up to 35 days and subjected to transient middle cerebral artery occlusion (MCAO). Motor deficits and infarct volume were assessed 3 days later. Intra-ischemic CBF was measured by laser-Doppler flowmetry and resting CBF by arterial spin labeling MRI. Ca2+-induced mitochondrial depolarization and reactive oxygen species (ROS) production were evaluated in isolated brain mitochondria.
Results
We found that 10% CIH is neuroprotective, while 6% CIH exacerbates tissue damage. No differences in resting or intra-ischemic CBF were observed between 6% and 10% CIH. However, 10% CIH reduced, while 6% CIH increases mitochondrial ROS production and susceptibility to Ca2+-induced depolarizations.
Conclusions
The influence of CIH on the ischemic brain is dichotomous and can be attributed in part to changes in the mitochondrial susceptibility to injury. The findings highlight a previously unappreciated complexity in the effect of CIH on the brain, which needs to be considered in evaluating the neurological impact of conditions associated with cyclic hypoxia.
Keywords: sleep apnea, cerebral blood flow, free radicals, mitochondria
Introduction
Obstructive sleep apnea (OSA) is a common form of sleep disordered breathing, affecting approximately 4% of men and 2% of women1. It is characterized by cyclical periods of airflow cessation (apneas), increased intrathoracic pressure, intermittent hypoxia, hypercapnia and sleep deprivation2. OSA is well established as an independent risk factor for stroke, increasing stroke risk by 2-3 fold3-5, an effect that seems to be related to the number of apnea-hypopnea episodes/hour of sleep5. Yet, the mechanisms by which OSA increases susceptibility to cerebral ischemia remain to be defined.
Chronic intermittent hypoxia (CIH) reproduces selected features of OSA and is commonly used as model of the human disease6. In CIH, as in OSA, there is an impairment in cerebrovascular regulation, consisting of attenuation of endothelial dependent responses and of the increases in cerebral blood flow (CBF) induced by neural activity7-10. While these deleterious cerebrovascular effects may reduce cerebrovascular reserves and contribute to the increase in the risk of stroke in OSA patients4,5, there is experimental evidence that CIH can also reduce cerebral ischemic brain injury11. Furthermore, mortality is reduced in older patients with OSA12, possibly owing to a “preconditioning” effect of CIH13. This raises the intriguing possibility that the influence of CIH on the ischemic brain may in fact be dichotomous and that the fate of the ischemic tissue may depend on factors other than cerebrovascular reserves.
The intensity of the hypoxic episodes and the period of CIH exposure are likely to influence reaction of the brain tissue to ischemic injury. Indeed, studies in the heart have indicated that the duration of CIH exposure can determine the outcome of myocardial ischemia14-16, an effect in part related to changes in the susceptibility of mitochondria to damage17,18. However, it is unclear how the duration of CIH and the intensity of the episodic hypoxia influence the outcome of cerebral ischemia and whether mitochondria may play a role in determining the fate of the ischemic tissue.
The key aim of this study was to test the hypothesis that intensity and duration of CIH are critical factors determining the impact of CIH on the post-ischemic brain. We found that CIH with cyclic hypoxia induced by 10% O2 for 35 days is neuroprotective in a mouse model of middle cerebral artery occlusion (MCAO), while CIH with 6% O2 exacerbates the injury. These effects could not be attributed to differences in cerebrovascular reserves, but to changes in mitochondrial function modulating mitochondrial depolarization and production of reactive oxygen species (ROS). Our findings support the hypothesis that the effects of CIH on the ischemic brain can be either protective or destructive, and that CIH-induced changes in mitochondrial resistance to injury have a key role in determining the fate of the ischemic tissue.
Methods
Mice
All procedures were approved by the institutional animal care and use committee of Weill Cornell Medical College. Experiments were performed in male C57Bl/6J mice (aged 7-13 weeks) (Jackson Laboratory).
Chronic intermittent hypoxia (CIH)
CIH was used to reproduce the cyclic hypoxia experienced by patients with OSA. Although all OSA models have limitations19, CIH is frequently used to investigate the cerebral and extracerebral effect of cyclic hypoxia6. CIH was induced using a custom system designed by Dr George J. Delagrammatikas and described previously7,20. Briefly, mice were randomly assigned to CIH (10% or 6% O2) or sham (room air) groups. In CIH groups, oxygen levels within the animals’ cage were changed from normal (21±1%) to low (10±1% or 6±1%) O2 for 90 seconds at 90-second intervals, resulting in 20 hypoxic episodes per hour. The cages of sham treated mice were infused at the same rate with room air. Hypoxia/sham cycling was induced throughout the light (sleep) phase (8:00 am to 4:00 pm; 8 hrs)20,21. During the remaining 16 hours of the day (4:00 pm to 8:00am), both CIH and sham cages were infused with room air. CIH and sham protocols were repeated for 14, 21 or 35 days, during which time mice had free access to food and water. The length of the CIH period was determined based on previous studies demonstrating that CIH for 35 days was sufficient to induce alterations in NMDA receptor trafficking and function20, or cerebrovascular regulation7. Systolic blood pressure (SBP) was measured using a non-invasive tail cuff system (Model MC4000, Hatteras Instruments), as previously described20.
Transient focal cerebral ischemia
Transient middle cerebral artery occlusion (MCAO) was induced using an intraluminal filament, as described22-25. Briefly, mice were anesthetized with isoflurane (1.5 - 2%) and rectal temperature maintained at 37° C. A heat-blunted suture was inserted into the right external carotid artery and advanced along the internal carotid artery until it obstructed the MCA. The common carotid artery was simultaneously ligated for the duration of the ischemic period (30 - 35 min). In all studies, cerebral blood flow (CBF) was monitored using laser Doppler flowmetry (Periflux System 5010, Perimed) in the ischemic territory (2mm posterior, 5mm lateral to bregma), as determined in preliminary studies26. Three days after MCAO functional impairment was assessed using the hanging-wire test27, and infarct volume was quantified in cresyl violet stained sections and corrected for swelling as previously described24. Blood glucose and hematocrit were determined at the time of euthanasia.
Real-Time PCR
Total RNA was isolated from a 1 mm thick coronal slice collected at the center of the middle cerebral artery territory in non-ischemic mice23,28. Quantitative determination of gene expression was performed on a Chromo 4 detector (Peltier Thermal Cycler, MJ Research) using a two-step cycling protocol, as described. Primers have been previously described23,28. Relative expression levels were calculated according to Livak and Schmittgen29. Quantities of all targets in test samples were normalized to the mouse hypoxanthine-guanine phosphoribosyltransferase (HPRT) housekeeping gene and values were correlated to sham-treated samples28.
Mitochondria isolation and measurements of Ca2+ induced mitochondrial depolarization
Brain mitochondria were isolated and purified from the forebrain of CIH and sham naïve mice that were not subjected to MCAO, as described previously30. Ca2+ induced mitochondrial depolarization was assessed using the fluorescent mitochondrial membrane potential indicator Safranin O (Molecular Probes)30,31. Briefly, 100 μg of purified mitochondria were suspended in KCl buffer containing glutamate (5mM), malate (5mM) and ADP (0.2 mM). Safranin O (5 μm) was added and fluorescence intensity was measured spectrophotometrically. Boluses of 50 nmoles CaCl2 were added sequentially until mitochondrial Ca2+ uptake declined, as indicated by the mitochondrial depolarization resulting from the mitochondrial permeability transition induced by Ca2+ overload. The amount of Ca2+ needed to cause mitochondrial depolarization was estimated from standard curves.
Measurement of mitochondrial reactive oxygen species
In isolated and purified brain mitochondria from CIH and sham mice (see above), ROS emission was measured by Amplex Red (Invitrogen) fluorescence, as described32. Briefly, 100 μg mitochondria were added to 1 mL incubation buffer. Standard curves were used to calculate H2O2 emission rates after sequential addition of substrate (5 mM glutamate, 2 mM malate), the complex I inhibitor rotenone (1 μM), and the complex III inhibitor antimycin A (1.8 μM).
Statistical Analysis
Mice were randomly assigned to the experimental groups, and analyses were performed by an investigator blinded to the treatment protocol. Data are expressed as mean±SE. Intergroup differences were analyzed using a Student's unpaired t-test or one-way ANOVA with Bonferroni's post-hoc analysis, as appropriate. Differences were considered statistically significant for p<0.05. Group sizes were calculated based on power analysis. We used a power of 0.8 and significance levels of 0.05.
Results
Influence of CIH on physiological parameters and HIF-1 α target genes
Exposure to CIH with 10% O2 (10% CIH) for 14 – 35 days had no effect on body weight, plasma glucose or hematocrit (Table I). Exposure to 6% CIH transiently increased hematocrit and reduced plasma glucose at 21 days, and decreased body weight both at 21 and 35 days (Table I). SBP tended to increase, but the effect did not reach statistical significance (Table I). Next, we assessed messenger RNA expression of the HIF-1α target genes EPO, VEGF-A and Glut-1, which are induced by CIH33. Both 10% and 6% CIH increased forebrain EPO mRNA expression at 35 days (Figure I). The induction was more pronounced with 6% than with 10% CIH, in accordance with the intensity of the hypoxic challenge. Increases (<2 fold) in VEGF-A at 10% CIH and Glut-1 at 6% CIH were also observed (Figure I).
Opposing effects of CIH on ischemic brain injury
Exposure to 6% or 10% CIH for 14 days had no effect on the size of the infarct (Fig 1A,B). Exposure to 6% CIH for 21 days exacerbated ischemic brain injury, increasing infarct size by ≈50%, while 10% CIH was without effect (Fig 2A,B). Surprisingly, exposure to 10% CIH for 35 days reduced infarct volume by ≈50%, whereas 6% CIH increased infarct volume and functional impairment at the hanging wire test (Fig. 3A,B). The reduction in infarct volume with 10% CIH was not associated with motor improvement at the hanging wire test, perhaps reflecting the ability of this test to detect more readily worsening than improvement in this model.
Figure 1. Influence of 14 days CIH on focal cerebral ischemic injury.
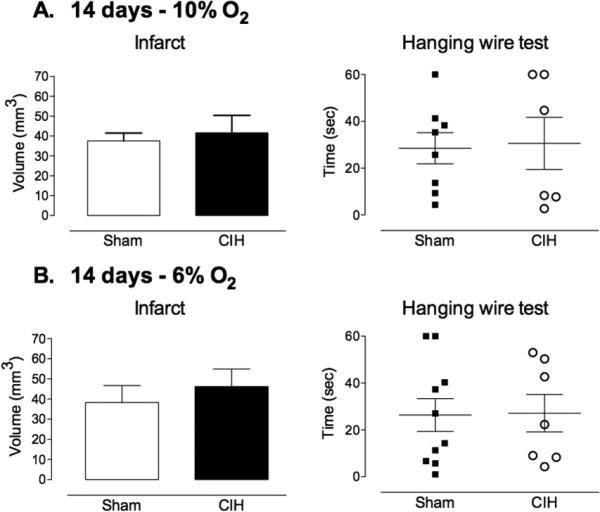
A. Exposure to 10% CIH for 14 days prior to induction of ischemia had no effect on infarct volume or functional impairment (hanging-wire test) evaluated at 72 hrs (n=6-8/group). B. Similarly, exposure to 6% CIH for 14 days did not alter infarct volume or functional impairment evaluated at 72 hrs (n=7-10/group).
Figure 2. Influence of 21 days CIH on focal cerebral ischemic injury.
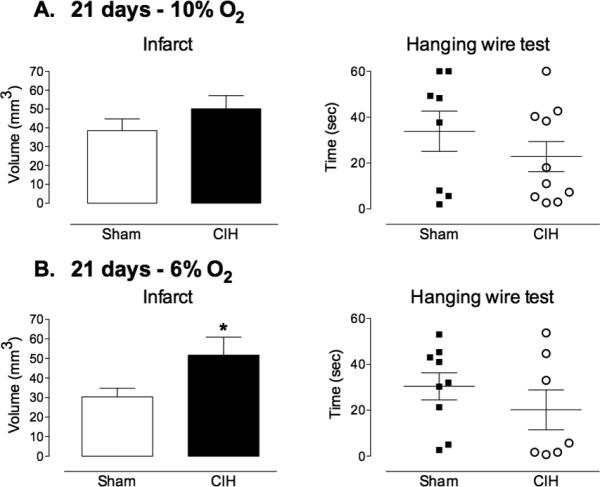
A. Exposure to 10% CIH for 21 days prior to induction of ischemia had no effect on infarct volume or functional impairment (hanging-wire test) evaluated at 72 hrs (n=8-10/group). B. Exposure to 6% CIH significantly increased infarct volume compared to sham treated mice, while functional impairment was not affected (n=7-9/group; *p<0.05, t-test).
Figure 3. Influence of 35 days CIH on focal cerebral ischemic injury.
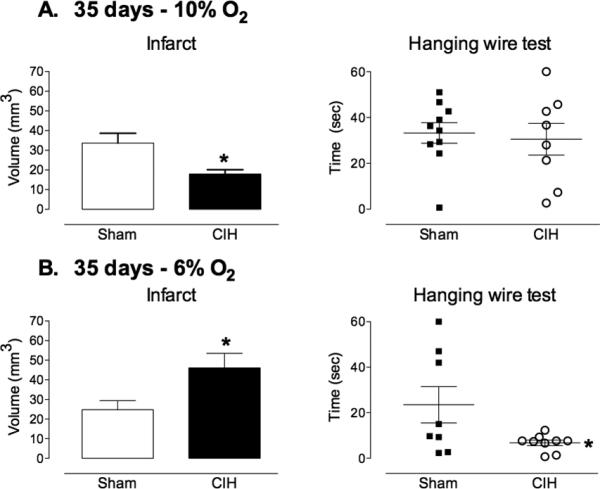
A. Exposure to 10% CIH for 35 days prior to induction of ischemia resulted in a significant reduction in infarct volume (n=8-10/group; *p<0.05, t-test). Functional impairment (hanging-wire test), was not affected (n=8-10/group). B. Exposure to 6% CIH increased both infarct volume and functional impairment 72 hrs after induction of cerebral ischemia (n=8-10/group; *p<0.05, t-test).
CIH does not affect resting or intraischemic CBF
Since CIH alters cerebrovascular reactivity and may reduce cerebrovascular reserves7,10, we examined whether the differences in tissue outcome between 6 and 10% CIH could be related to changes in intraischemic CBF. However, no differences in the reduction in CBF produced by MCA occlusion were observed among the groups (Figure IIA; p>0.05). Since LDF cannot measure resting CBF quantitatively, we used ASL-MRI to examine if the worsening of injury with 6% CIH could be related to reductions in resting CBF, which could result in more severe ischemia after MCAO. However, resting CBF did not differ between mice exposed to sham treatment or CIH (CIH 10%: Sham 146±2, CIH: 144±10; CIH 6%: Sham 170±6, CIH 181±5 ml/100g/min) at 35 days (Figure IIB; p>0.05).
Effects of CIH on pro-inflammatory gene expression in brain
Next, we examined if CIH could influence the outcome of cerebral ischemia by modulating inflammatory gene expression. Exposure to 6% CIH for 35 days increased expression of regulated and normal T cell expressed and secreted/CCL5 (RANTES), monocyte chemotactic protein-1 (MCP-1), inducible nitric oxide synthase (INOS), intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) (Figure 4). There was a trend for pro-inflammatory gene expression to be downregulated in mice exposed to 10% CIH, particularly INOS, which was significantly reduced (Figure 4). These changes in inflammatory gene expression were not associated with evidence of tissue injury both with 6% or 10% CIH, as assessed by fluoro jade-B (Figure IIIA,B). As a positive control, fluoro jade-B was able to detect brain damage after MCAO (Figure IIIC).
Figure 4. CIH induced changes in pro-inflammatory gene expression in non-ischemic mice.
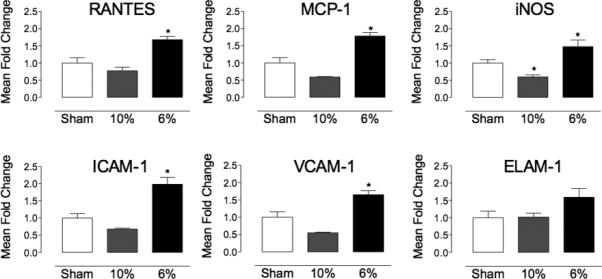
mRNA expression of pro-inflammatory genes RANTES, MCP-1, iNOS, ICAM-1, VCAM-1 and ELAM-1 tended to be decreased in non-ischemic mice exposed to 10% CIH (35 days; n=5-15/group; *p<0.05 from sham, ANOVA), while expression was increased following exposure to 6% CIH (35 days; n=5-15 /group; *p<0.05 from sham, ANOVA).
Opposing effects of CIH on mitochondrial susceptibility to Ca2+ induced depolarization
Mitochondria are critical modulators of ischemic brain injury and are influenced by intermittent hypoxia17,18,34,35. Therefore, we tested the hypothesis that CIH-induced changes in mitochondrial resistance to injury play a role in the opposing effects on post-ischemic brain damage. Mitochondria were isolated from non-ischemic mice subjected to sham treatment or 10% or 6% CIH for 35 days, and their susceptibility to Ca2+-induced loss of membrane potential (ΔΨ) was tested. Sham treated mice maintained ΔΨ in response to up to 200 nmoles CaCl2. Beyond this point, administration of an additional aliquot of CaCl2 (40 nmol) resulted in a complete loss of ΔΨ, consistent with mitochondrial permeability transition pore (mPTP) opening (Figure 5A & B). In contrast, mitochondria isolated from mice exposed to 10% O2 CIH had an increased Ca2+ capacity, maintaining ΔΨ in response to up to 240 nmoles CaCl2 (Figure 5A), whereas mitochondrial Ca2+ capacity of mice exposed to 6% O2 CIH was reduced, with loss of ΔΨ occurring in response to 120 nmoles CaCl2 (Figure 5B).
Figure 5. Effect of CIH on mitochondrial Ca2+-induced membrane depolarization in non-ischemic mice.
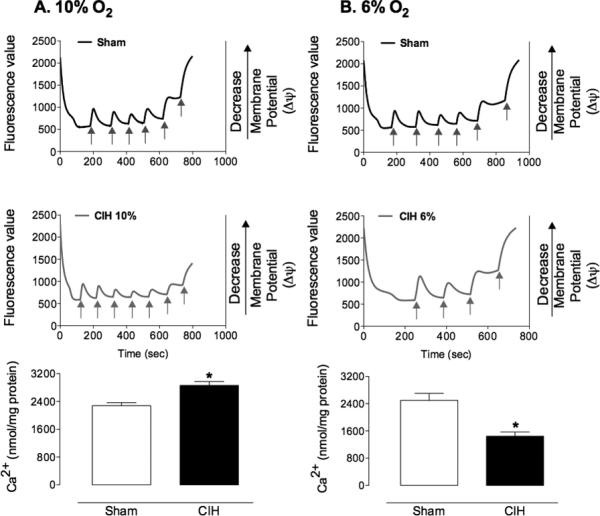
A. Representative trace and group data illustrating exposure to 10% CIH decreased susceptibility of mitochondria to Ca2+ induced loss of membrane potential (n=9/group; *p<0.05, t-test). B. In contrast, exposure to 6% CIH increased susceptibility to Ca2+ induced membrane depolarization (n=9/group; *p<0.05, t-test). Additions of Ca2+ are indicated by arrows.
Effect of CIH on mitochondrial ROS production
ROS promote mPTP opening and subsequent loss of membrane potential34. In order to gain an insight into the mechanism by which CIH alters susceptibility of mitochondria to Ca2+ induced depolarization, we evaluated mitochondrial ROS production with substrate (malate, glutamate), rotenone (complex I inhibitor) and antimycin A (complex III inhibitor), in mice exposed to 10% and 6% CIH for 35 days. Consistent with a reduced susceptibility to Ca2+ induced membrane depolarization and mPTP transition, exposure to 10% CIH attenuated mitochondrial ROS production, as compared to sham-treated mice (Figure 6A). In contrast, ROS production was similar in mice exposed to either sham treatment or 6% CIH (Figure 6B). While there was a tendency for ROS production in response to substrate to increase in 6% CIH mice relative to control, this effect did not reach statistical significance (p>0.05).
Figure 6. Effect of CIH on mitochondrial ROS production in non-ischemic mice.

A. Representative Amplex Red fluorescence traces (top panel) and group data (bottom panel) showing that exposure to 10% CIH decreased the rate of H2O2 emission from isolated mitochondria, following sequential additions of substrate (succinate), rotenone, and antimycin A, relative to sham treated controls (n=5-6/group; *p<0.05, t-test). B. Representative trace (top panel) and group data (bottom panel) showing that exposure to 6% CIH did not attenuate mitochondrial H2O2 production relative to sham treated controls (n=5/group,). The additions of mitochondria (mito), substrate (succinate), rotenone and antimycin A are indicated by arrows.
Discussion
We have demonstrated that CIH can either ameliorate or worsen ischemic brain injury depending on the intensity of the hypoxic challenge. Thus, 10% CIH induces neuroprotection while 6% CIH exacerbates ischemic brain injury. Differences in the effects of 10% and 6% CIH on outcome were independent of alterations in CBF, as both basal cerebral perfusion, measured using ASL-MRI, and the degree of blood flow reduction in response to MCAO were similar in 10% and 6% CIH mice. However, the expression of key pro-inflammatory mediators was altered by CIH, increasing with 6% CIH and showing a tendency to decrease with 10% CIH, in agreement with their effects on outcome. Furthermore, the ability of cerebral mitochondria to maintain membrane potential in response to Ca2+ overload was enhanced by 10% CIH and reduced by 6% CIH, an effect associated with differences in mitochondrial ROS production. Collectively, our findings unveil a dichotomous effect of CIH on the post-ischemic brain that is dependent on the severity of cyclic hypoxic episodes.
CIH exposure exerts opposing effects on the ischemic brain
Decreasing the intensity from 10% to 6% CIH resulted in a switch from beneficial to detrimental effects of CIH on the ischemic brain. These opposing effects of 10% and 6% CIH were first observed after 21 days exposure, when 6% CIH increased susceptibility to ischemic brain injury. By 35 days, however, the dichotomy was fully developed, with 10% CIH decreasing and 6% CIH exacerbating the injury. These effects could not be attributed to changes in SBP, blood glucose, hematocrit or CBF, because changes in these parameters did not correlate with the post-ischemic outcome. Interestingly, while the pathogenic effects of CIH developed more rapidly, neuroprotective effects of CIH on the ischemic brain took more than 21 days to develop. This observation is consistent with the idea that exposure to CIH induces delayed cerebral ischemic tolerance33. Indeed, others have shown that hypoxic episodes can induce short-term36-39 or long lasting11 preconditioning in the brain. However, our results indicate that the effects of CIH are not uniformly beneficial, and that their protective or destructive character depends on the intensity and duration of the recurring hypoxic challenge. Clinical data suggest that older patients with moderate OSA may have reduced mortality compared to the general population12, attesting to a potential preconditioning effect of mild CIH13. Therefore, it would be important to test if the dichotomous effect of CIH also occurs in older animals.
CIH induced vascular dysfunction does not influence the outcome of cerebral ischemia
Post-ischemic cerebral perfusion has profound effects on the outcome of MCAO40. Thus, alterations in CBF regulation reduce intra-ischemic CBF and exacerbate focal ischemic brain injury41-43, whereas counteracting post-ischemic cerebrovascular dysregulation ameliorates the damage26. Since 10% CIH alters critical vasomotor responses of the cerebral circulation7, we anticipated that intra-ischemic CBF would be reduced, owing to the impaired vasodilatatory capacity of cerebral blood vessels and failure of collateral flow. Contrary to this prediction, however, we found that 10% CIH does not exacerbate the CBF reduction induced by MCAO, and results in less damage, due to its beneficial parenchymal effects. Collectively, these observations indicate that the “preconditioning” effect of CIH may protect the post-ischemic brain from the deleterious effects of cerebrovascular dysfunction.
CIH influences inflammation and the tolerance of mitochondria to Ca2+-induced membrane depolarization
Pro-inflammatory markers were slightly upregulated in the brain of mice exposed to 6% CIH in the absence of changes in tissue injury, and tended to be reduced in mice exposed to 10% CIH. This supports the idea that inflammation contributes to the CIH-induced changes in susceptibility ischemic brain injury and is in agreement with previous studies reporting alterations in mediators and markers of inflammation in the brain following CIH11,44 and in the blood of OSA patients45,46. However, the significance of these relatively minor changes in inflammatory gene expression remain to be established and a more detailed analysis of the cellular and molecular features of post-ischemic inflammation would be needed to assess the impact of CIH-induced immunomodulation on the outcome of cerebral ischemia.
A more striking and novel finding of the present study is that CIH exerts a profound influence on cerebral mitochondria. Mitochondria play a central role in cell death and survival47 and are critical for buffering excessive post-ischemic intracellular Ca2+ levels34. Here, we found that cerebral mitochondria isolated from mice exposed to 10% CIH had a significant reduction in susceptibility to Ca2+ induced membrane depolarization, consistent with a protection from injury, while 6% CIH increased susceptibility to Ca2+ induced depolarization. Although changes in mitochondrial function have been reported following CIH33, we found, for the first time, that the effects of CIH on mitochondrial Ca2+ buffering capacity can be either protective or harmful depending on the intensity of the cyclic hypoxia.
Induction of mPTP and loss of mitochondrial membrane potential occurs due to excessive accumulation of Ca2+ and results in impaired ATP production and cell death. This loss of mitochondrial membrane potential can be promoted by oxidative stress34. Indeed, we observed a reduction in mitochondrial ROS production in mice exposed to 10% CIH, in which susceptibility to Ca2+ induced membrane depolarization is attenuated. This is consistent with changes in mitochondrial ROS production in response to CIH, altering the stability of the mPTP and the susceptibility of the brain to ischemic injury. In agreement with this hypothesis, 6% CIH did not increase mitochondrial ROS production relative to control, whereas mitochondrial ROS levels were higher in 6% than in 10% CIH. This supports the idea that mitochondrial ROS play an important role in the effects of CIH on cerebral mitochondria and subsequent injury development. Interestingly, we previously reported that cerebrovascular ROS production is increased by 10% CIH, an effect mediated by a NOX2-containing NADPH oxidase7. In contrast, in the present study we observed that mitochondrial ROS are reduced by the same CIH regimen. While cell-type specific effects cannot be ruled out, these observations raise the intriguing possibility that CIH has differential effects on ROS production depending on the enzymatic system and/or cellular compartment generating the radicals. Further studies targeting specifically mitochondrial and extramitochondrial sources of ROS would be required to address this issue.
Conclusions
This study demonstrates that CIH can have both beneficial and deleterious effects on the ischemic brain. Neuroprotection was observed following exposure to 10% CIH, while injury exacerbation was seen with 6% CIH, suggesting that the intensity of the hypoxic insult is critical in determining the effect of CIH on tissue outcome. At the mechanistic level, this study provides novel evidence for a key role of mitochondria in the effects of CIH on the ischemic brain, with alterations in the susceptibility to Ca2+-induced membrane depolarization and mitochondrial ROS production observed. While further studies are required to define the mechanisms of these mitochondrial effects, the dichotomous nature of the impact of CIH on the post-ischemic brain unveils a novel and intriguing aspect of the pathobiology of CIH. This may be of great relevance to clinical conditions associated with cyclic changes in oxygenation levels.
Supplementary Material
Acknowledgements
None
Sources of funding
This work was funded by NIH grants R01-NS73666 and P01-HL96571. KAJ is supported by a National Health and Medical Research Council of Australia CJ Martin Fellowship and American Australian Association Sir Keith Murdoch Fellowship. HUV acknowledges financial support from the Nancy M. & Samuel C. Fleming Research Scholar Award in Intercampus Collaborations. We are grateful for the support of the Feil Family Foundation.
Footnotes
Conflict of interest/disclosures
The authors have no conflicts of interest or disclosures to declare.
References
- 1.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. The New England journal of medicine. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 2.Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol. Rev. 2010;90:47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durgan DJ, Bryan RM. Cerebrovascular consequences of obstructive sleep apnea. J Am Heart Assoc. 2012;1:e000091. doi: 10.1161/JAHA.111.000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. The New England journal of medicine. 2005;353:2034–2041. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 5.Redline S, Yenokyan G, Gottlieb DJ, Shahar E, O'Connor GT, Resnick HE, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am. J. Respir. Crit. Care Med. 2010;182:269–277. doi: 10.1164/rccm.200911-1746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veasey S. Insight from animal models into the cognitive consequences of adult sleep-disordered breathing. ILAR J. 2009;50:307–311. doi: 10.1093/ilar.50.3.307. [DOI] [PubMed] [Google Scholar]
- 7.Capone C, Faraco G, Coleman C, Young CN, Pickel VM, Anrather J, et al. Endothelin 1-dependent neurovascular dysfunction in chronic intermittent hypoxia. Hypertension. 2012;60:106–113. doi: 10.1161/HYPERTENSIONAHA.112.193672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Culebras A. Sleep and Stroke. Semin Neurol. 2009;29:438–445. doi: 10.1055/s-0029-1237121. [DOI] [PubMed] [Google Scholar]
- 9.Phillips SA, Olson EB, Morgan BJ, Lombard JH. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. American journal of physiology. 2004;286:H388–93. doi: 10.1152/ajpheart.00683.2003. [DOI] [PubMed] [Google Scholar]
- 10.Crossland RF, Durgan DJ, Lloyd EE, Phillips SC, Reddy AK, Marrelli SP, et al. A new rodent model for obstructive sleep apnea: effects on ATP-mediated dilations in cerebral arteries. AJP: Regulatory, Integrative and Comparative Physiology. 2013;305:R334–42. doi: 10.1152/ajpregu.00244.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stowe AM, Altay T, Freie AB, Gidday JM. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol. 2011;69:975–985. doi: 10.1002/ana.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavie P, Lavie L, Herer P. All-cause mortality in males with sleep apnoea syndrome: declining mortality rates with age. Eur. Respir. J. 2005;25:514–520. doi: 10.1183/09031936.05.00051504. [DOI] [PubMed] [Google Scholar]
- 13.Lavie L, Lavie P. Ischemic preconditioning as a possible explanation for the age decline relative mortality in sleep apnea. Med. Hypotheses. 2006;66:1069–1073. doi: 10.1016/j.mehy.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 14.Park A-M, Suzuki YJ. Effects of intermittent hypoxia on oxidative stress-induced myocardial damage in mice. J. Appl. Physiol. 2007;102:1806–1814. doi: 10.1152/japplphysiol.01291.2006. [DOI] [PubMed] [Google Scholar]
- 15.Joyeux-Faure M, Stanke-Labesque F, Lefebvre B, Béguin P, Godin-Ribuot D, Ribuot C, et al. Chronic intermittent hypoxia increases infarction in the isolated rat heart. J. Appl. Physiol. 2005;98:1691–1696. doi: 10.1152/japplphysiol.01146.2004. [DOI] [PubMed] [Google Scholar]
- 16.Milano G, Abruzzo PM, Bolotta A, Marini M, Terraneo L, Ravara B, et al. Impact of the phosphatidylinositide 3-kinase signaling pathway on the cardioprotection induced by intermittent hypoxia. PLoS One. 2013;8:e76659. doi: 10.1371/journal.pone.0076659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z-H, Cai X-L, Wu L, Yu Z, Liu J-L, Zhou Z-N, et al. Mitochondrial energy metabolism plays a critical role in the cardioprotection afforded by intermittent hypobaric hypoxia. Exp. Physiol. 2012;97:1105–1118. doi: 10.1113/expphysiol.2012.065102. [DOI] [PubMed] [Google Scholar]
- 18.Kolár F, Neckar J, Ostadal B. MCC-134, a blocker of mitochondrial and opener of sarcolemmal ATP-sensitive K+ channels, abrogates cardioprotective effects of chronic hypoxia. Physiol Res. 2005;54:467–471. [PubMed] [Google Scholar]
- 19.Davis EM, O'Donnell CP. Rodent models of sleep apnea. Respir Physiol Neurobiol. 2013;188:355–361. doi: 10.1016/j.resp.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman CG, Wang G, Park L, Anrather J, Delagrammatikas GJ, Chan J, et al. Chronic intermittent hypoxia induces NMDA receptor-dependent plasticity and suppresses nitric oxide signaling in the mouse hypothalamic paraventricular nucleus. JNeurosci. 2010;30:12103–12112. doi: 10.1523/JNEUROSCI.3367-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boland LL, Shahar E, Iber C, Knopman DS, Kuo TF, Nieto FJ. Measures of cognitive function in persons with varying degrees of sleep-disordered breathing: the Sleep Heart Health Study. J Sleep Res. 2002;11:265–272. doi: 10.1046/j.1365-2869.2002.00308.x. [DOI] [PubMed] [Google Scholar]
- 22.Jackman K, Kunz A, Iadecola C. Modeling focal cerebral ischemia in vivo. Methods Mol. Biol. 2011;793:195–209. doi: 10.1007/978-1-61779-328-8_13. [DOI] [PubMed] [Google Scholar]
- 23.Jackman K, Kahles T, Lane D, Garcia-Bonilla L, Abe T, Capone C, et al. Progranulin deficiency promotes post-ischemic blood-brain barrier disruption. J Neurosci. 2013;33:19579–19589. doi: 10.1523/JNEUROSCI.4318-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hochrainer K, Jackman K, Anrather J, Iadecola C. Reperfusion rather than ischemia drives the formation of ubiquitin aggregates after middle cerebral artery occlusion. Stroke. 2012;43:2229–2235. doi: 10.1161/STROKEAHA.112.650416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abe T, Zhou P, Jackman K, Capone C, Casolla B, Hochrainer K, et al. Lipoprotein Receptor-Related Protein-6 Protects the Brain From Ischemic Injury. Stroke. 2013;44:2284–2291. doi: 10.1161/STROKEAHA.113.001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, et al. Neurovascular Protection by Ischemic Tolerance: Role of Nitric Oxide and Reactive Oxygen Species. Journal of Neuroscience. 2007;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abe T, Kunz A, Shimamura M, Zhou P, Anrather J, Iadecola C. The neuroprotective effect of prostaglandin E2 EP1 receptor inhibition has a wide therapeutic window, is sustained in time and is not sexually dimorphic. Journal of Cerebral Blood Flow & Metabolism. 2008;29:66–72. doi: 10.1038/jcbfm.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunz A, Abe T, Hochrainer K, Shimamura M, Anrather J, Racchumi G, et al. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. Journal of Neuroscience. 2008;28:1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Zhou P, Qian L, Zhou T, Iadecola C. Mitochondria are involved in the neurogenic neuroprotection conferred by stimulation of cerebellar fastigial nucleus. Journal of Neurochemistry. 2005;95:221–229. doi: 10.1111/j.1471-4159.2005.03358.x. [DOI] [PubMed] [Google Scholar]
- 31.Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starkov AA. Measurement of mitochondrial ROS production. Methods Mol. Biol. 2010;648:245–255. doi: 10.1007/978-1-60761-756-3_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochimica et biophysica acta. 2010;1802:80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Douglas RM, Ryu J, Kanaan A, del Carmen Rivero M, Dugan LL, Haddad GG, et al. Neuronal death during combined intermittent hypoxia/hypercapnia is due to mitochondrial dysfunction. AJP: Cell Physiology. 2010;298:C1594–C1602. doi: 10.1152/ajpcell.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin AMY, Dung S-W, Chen C-F, Chen W-H, Ho L-T. Hypoxic preconditioning prevents cortical infarction by transient focal ischemia-reperfusion. Ann N Y Acad Sci. 2003;993:168–78. doi: 10.1111/j.1749-6632.2003.tb07527.x. [DOI] [PubMed] [Google Scholar]
- 37.Bernaudin M, Nedelec A-S, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. Journal of Cerebral Blood Flow & Metabolism. 2002;22:393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Gong SJ, Chen LY, Zhang M, Gong JX, Ma YX, Zhang JM, et al. Intermittent hypobaric hypoxia preconditioning induced brain ischemic tolerance by up-regulating glial glutamate transporter-1 in rats. Neurochem Res. 2012;37:527–537. doi: 10.1007/s11064-011-0639-3. [DOI] [PubMed] [Google Scholar]
- 39.Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport. 2001;12:1663–1669. doi: 10.1097/00001756-200106130-00030. [DOI] [PubMed] [Google Scholar]
- 40.Jackman K, Iadecola C. Neurovascular regulation in the ischemic brain. Antioxidants & Redox Signaling. 2013 doi: 10.1089/ars.2013.5669. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. Journal of Neuroscience. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iadecola C, Sugimoto K, Niwa K, Kazama K, Ross ME. Increased susceptibility to ischemic brain injury in cyclooxygenase-1-deficient mice. J. Cereb. Blood Flow Metab. 2001;21:1436–1441. doi: 10.1097/00004647-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. Journal of Cerebral Blood Flow & Metabolism. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 44.Aviles-Reyes RX, Angelo MF, Villarreal A, Rios H, Lazarowski A, Ramos AJ. Intermittent hypoxia during sleep induces reactive gliosis and limited neuronal death in rats: implications for sleep apnea. Journal of Neurochemistry. 2010;112:854–869. doi: 10.1111/j.1471-4159.2009.06535.x. [DOI] [PubMed] [Google Scholar]
- 45.Ohga E, Tomita T, Wada H, Yamamoto H, Nagase T, Ouchi Y. Effects of obstructive sleep apnea on circulating ICAM-1, IL-8, and MCP-1. J. Appl. Physiol. 2003;94:179–184. doi: 10.1152/japplphysiol.00177.2002. [DOI] [PubMed] [Google Scholar]
- 46.Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, et al. Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation. 2003;107:1129–1134. doi: 10.1161/01.cir.0000052627.99976.18. [DOI] [PubMed] [Google Scholar]
- 47.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen A-L. Mitochondrial calcium and the permeability transition in cell death. Biochimica et biophysica acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.