

Abstract
Recent studies have demonstrated that bone marrow-derived fibroblasts contribute significantly to the pathogenesis of renal fibrosis. However, the signaling mechanisms underlying the activation of bone marrow-derived fibroblasts in the kidney are incompletely understood. Since TGF-β1/Smad3 signaling has been shown to play an important role in the pathogenesis of kidney fibrosis, we investigated the role of Smad3 in the activation of bone marrow-derived fibroblasts in the kidney following obstructive injury using Smad3 knockout mice and Smad3 null monocytes. Compared with wild-type mice, Smad3-knockout mice accumulated significantly fewer bone marrow-derived fibroblasts in the kidney after obstructive injury. Furthermore, Smad3 knockout mice exhibited less myofibroblast activation and expressed less α-SMA in the obstructed kidney. Consistent with these findings, genetic deletion of Smad3 reduced total collagen deposition and suppressed expression of extracellular matrix proteins. Moreover, wild-type mice engrafted with Smad3−/− bone marrow cells displayed fewer bone marrow-derived fibroblasts in the kidney with obstructive injury and showed less severe renal fibrosis compared with wild-type mice engrafted with Smad3+/+ bone marrow cells. In cultured monocytes, TGF-β1 induced phosphorylation of Smad3 and Smad3 deficiency abolished TGF-β1-induced expression of α-SMA and extracellular matrix proteins. Taken together, our results demonstrate that Smad3 signaling plays an essential role in the activation of bone marrow-derived fibroblasts in the kidney during the pathogenesis of renal fibrosis.
Keywords: Cytokine, Bone marrow-derived cells, Fibroblasts, Renal Fibrosis, Extracellular matrix, Chronic kidney disease
Renal fibrosis is a hallmark of chronic kidney disease regardless of underlying etiologies1, 2. Furthermore, interstitial fibrosis is a key structural component of obstructive nephropathy, which is the major cause of chronic kidney disease in children3. Renal interstitial fibrosis is characterized by fibroblast activation and excessive production and deposition of extracellular matrix (ECM), which causes the destruction and collapse of renal parenchyma and progressive loss of kidney function. Because activated fibroblasts are the principal cells responsible for ECM production, their activation is regarded as a key event in the pathogenesis of renal fibrosis4, 5. Recent evidence indicates that these cells may originate from bone marrow-derived fibroblast progenitor cells6–11.
Bone marrow-derived fibroblasts termed fibrocytes are derived from a subpopulation of monocytes via monocyte-to-fibroblast transition12–16. These cells express mesenchymal markers such as collagen I, vimentin, and discoid domain receptor 2 (DDR2) and hematopoietic markers such as CD45 and CD11b13, 17–19. These cells in culture display an adherent, spindle-shape morphology and express α-SMA, consistent with the concept that they can differentiate into myofibroblasts17–19. We and others have shown that bone marrow-derived fibroblasts contribute significantly in the pathogenesis of renal fibrosis5, 8, 14. However, the molecular mechanisms underlying the activation of these cells in the kidney are incompletely understood.
TGF-β1 plays a key role in the pathogenesis of renal fibrosis through activation of a cascade of intracellular signaling pathways20–23. Evidence suggests that activation of the Smad signaling cascade is important in the regulation of ECM protein expression and tissue fibrosis24–28. Although Smad3 is essential for the development of renal fibrosis, the cellular and molecular mechanisms underlying its profibrotic actions are not fully understood.
In this study, we examined the role of Smad3 in the activation of bone marrow-derived fibroblasts in the kidney in a well-established model of tubulointerstitial fibrosis induced by unilateral ureteral obstruction (UUO) using Smad3 knockout (KO) mice and Smad3−/− monocytes. Our results demonstrate that genetic disruption of Smad3 prevents the development of renal fibrosis by suppressing the activation of CD45+ myeloid fibroblasts.
Materials and Methods
Animals
Animal experiments were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine (IACUC permit #: AN-5011). The investigation conforms with the recommendations in the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All efforts were made to minimize suffering. The Smad3 KO mice on a background of C57BL6/J were kindly provided by Dr. Xiao-Fan Wang29. Smad3 KO mice and WT littermates at 8–12 weeks old were subjected to UUO surgery as described11, 12, 30, 31. Mice were allowed to recover from anesthesia and were housed in standard rodent cages with ad libitum access to water and food until sacrificed.
Renal Morphology
Mice were euthanized and perfused by injections of PBS into the left ventricle of the heart to remove blood. One portion of the renal tissue was fixed in 10% buffered formalin and embedded in paraffin, cut at 5 μm thickness, and stained with hematoxylin and eosin for initial evaluation, or picrosirius red to identify collagen fibers. The picrosirius red-stained sections were scanned using a microscope equipped with a digital camera (Nikon, Melville, NY) and quantitative evaluation was performed using NIS-Elements Br 3.0 software as described11. The collagen-stained area was calculated as a percentage of the total area.
Quantitative Real-Time RT-PCR
Quantitative analysis of the target mRNA expression was performed with real-time reverse transcription – polymerase chain reaction (RT-PCR) by the relative standard curve method. Total RNA was extracted from snap-frozen kidney tissues with TRIzol Reagent (Invitrogen). Aliquots (1 μg) of total RNA were reverse-transcribed and amplified in triplicate using IQ SYBR green supermix reagent (Bio-Rad, Herculus, CA) with a real-time PCR machine (Bio-Rad, Herculus, CA), according to the manufacturer’s instructions. The specificity of real-time PCR was confirmed by melting-curve analysis. The expression levels of the target genes were normalized to the GAPDH level in each sample. The following are the primer sequences: Collagen I: Forward 5′-TGCCGCGACCTCAAGATGTG-3′ and reverse 5′-CACAAGGGTGCTGTAGGTGA-3′; Fibronectin: Forward 5′-CTTCTCCGTGGAGTTTTACCG-3′ and reverse 5′-GCTGTCAAATTGAATGGTGGTG-3′; α-SMA: Forward 5′-ACTGGGACGACATGGAAAAG-3′ and reverse 5′-CATCTCCAGAGTCCAGCACA-3′; GAPDH: Forward 5′-TGCTGAGTATGTCGTGGAGTCTA-3′ and reverse 5′-AGTGGGAGTTGCTGTTGAAATC-3′.
Immunofluorescence
Renal tissues were embedded in OCT compound, snap-frozen on dry ice, cut at 5 μm thickness, and mounted on microscope slides. After fixation, nonspecific binding was blocked with serum-free protein block (DAKO). Sectionss were then incubated with rabbit anti-phospho-Smad3 antibody (Rockland) followed by Alexa-488 conjugated donkey anti-rabbit antibody (Invitrogen), rabbit anti-collagen I antibody (Rockland) followed by Alexa-488 conjugated donkey anti-rabbit antibody (Invitrogen), rabbit anti-fibronectin antibody (Sigma) followed by Alexa-488 conjugated donkey anti-rabbit antibody (Invitrogen), or rabbit anti-α-SMA antibody (Abcam) followed by Alexa-488 conjugated donkey anti-rabbit antibody (Invitrogen). For double immunofluorescence, renal tissues were fixed and stained with primary antibodies followed by appropriate secondary antibodies sequentially. Slides were mounted with mounting medium containing DAPI. Fluorescence intensity was visualized using a microscope equipped with a digital camera (Nikon, Melville, NY). Quantitative evaluation of sections stained for α-SMA was performed using NIS-Elements Br 3.0 software. The fluorescence positive area was calculated as a percentage of the total area.
Cell Isolation and Flow Cytometry
Renal cell isolation and flow cytometry were performed as described11. Briefly, kidneys were minced and incubated at 37°C for 30 minutes in PBS containing 0.25 mg/ml Liberase TM (Roche) and 10 U/ml Dnase (Roche). Cells were filtered through a 40μm strainer, washed, centrifuged and resuspended in FACS buffer. Cells (5 × 105) were first incubated with FITC-anti-CD45 (BD Biosciences, San Jose, CA), and goat-anti-DDR2 (Santa Cruz Biotech, Santa Cruz, CA) followed by followed by anti-goat-APC (BD Biosciences, San Jose, CA). Cells incubated with irrelevant isotype-matched antibodies (BD Biosciences, San Jose, CA) and unstained cells were used as controls. The cutoffs were set according to results of controls. The fluorescence intensities were measured using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed using BD FACSDiva software.
Western Blot Analysis
Protein was extracted using the RIPA buffer containing a cocktail of proteinase inhibitors (Thermo Fisher Scientific Inc., Rockford, IL) and quantified with Bio-Rad protein assay. Equal amounts of protein were separated on SDS–polyacrylamide gels in a Tris/glycine buffer system, transferred onto nitrocellulose membranes, and blotted according to standard procedures with primary antibodies (phospho-Smad3, collagen I, fibronectin, and α-SMA). Membranes were then stripped and reblotted with anti-GAPDH antibody (Millipore, Billerica, CA). The specific bands of target proteins were analyzed using an Odyssey IR scanner and band intensities were quantified using NIH Image/J.
Bone Marrow Transplantation
Bone marrow transplantation was performed as described previously11. Briefly, bone marrow cells (5X106) from WT or Smad3 KO mice were transferred to lethally irradiated WT C57BL/6 mice. After transplantation, mice were allowed to recuperate for 2 months prior to induction of kidney injury by UUO.
Monocyte Isolation and Culture
Monocytes from spleen of WT and Smad3 KO mice were isolated as described32. Monocytes were cultured in RPMI 1640 medium containing 10% FBS, 1% penicillin and streptomycin in an atmosphere of 5% CO2 and 95% air at 37_C. For TGF-β1 treatment, cells were starved for 24 hrs by incubation with RPMI 1640 containing 1% FBS and then exposed to vehicle or TGF-β1 at 10ng/ml for 24 hrs.
Statistical Analysis
All data were expressed as mean ± SEM. Multiple group comparisons were performed by one-way ANOVA followed by the Bonferroni procedure for comparison of means. Comparisons between 2 groups were analyzed by the two-tailed student t test. P < 0.05 was considered statistically significant.
Results
Smad3 is Activated in CD45+ Myeloid Fibroblasts during Renal Fibrosis
We first characterized the activation of Smad3 in the kidney in a mouse model of tubulointerstitial fibrosis induced by UUO. Kidney sections were stained with phospho-Smad3 antibody and examined with a fluorescence microscope. Phospho-Smad3 positive signal was primarily detected in the interstitial cells of obstructed kidney (Figure 1A). To determine if Smad3 was activated in fibroblasts, renal sections were stained for phospho-Smad3 and DDR2, a fibroblast-specific marker, and examined with a fluorescence microscope. DDR2 positive fibroblasts were stained positive for phospho-Smad3 (Figure 1B). To examine if Smad3 was activated in CD45+ myeloid fibroblasts, freshly isolated renal cells were stained for CD45, collagen I, and phospho-Smad3. CD45+ and collagen I+ cells were stained positive for phospho-Smad3, indicating that Smad3 in bone marrow-derived fibroblasts are activated (Figure 1C). Western blot analysis confirmed that Smad3 was activated in the kidney in response to obstructive injury (Figure 1, D and E).
Figure 1. Smad3 is activated in myeloid fibroblasts in the kidney after obstructive injury.

A. Representative photomicrographs of kidney sections stained for phospho-Smad3 (green) and counter stained with DAPI (blue) (Original magnification: X400). B. Representative photomicrographs of kidney sections stained for phospho-Smad3 (green) and DDR2 (red) and counter stained with DAPI (blue) (Original magnification: X600). C. Representative photomicrographs of isolated renal fibroblasts stained for CD45 (red), phospho-Smad3 (green), collagen I (blue). D. Representative Western blots show activation of Smad3 in UUO kidneys of WT mice. E. Quantitative analysis of Smad3 phosphorylation in the control and UUO kidneys of WT mice. ** P < 0.01 vs WT controls. n=6.
Smad3 Deficiency Impairs Myeloid Fibroblast Accumulation
To examine if Smad3 plays a role in the accumulation of bone marrow-derived fibroblasts in the obstructed kidneys, WT and Smad3-KO mice were subjected to obstructive injury for 7 days. Kidney sections were stained for CD45 and procollagen I. Our results showed that the accumulation of CD45+ and procollagen I+ fibroblasts was markedly increased in injured kidneys of WT mice; whereas the accumulation of CD45+ and procollagen I+ fibroblasts was significantly reduced in injured kidneys of Smad3-KO mice (Figure 2, A–B). Consistent with immunofluorescence staining, flow cytometric analysis of freshly-isolated renal cells stained for CD45 and DDR2 demonstrated that Smad3 deficiency inhibited CD45+ and DDR2+ fibroblast accumulation in the kidney compared with WT mice (Figure 2, C–D). These data indicate that Smad3 plays a role in the accumulation of bone marrow-derived fibroblasts in the kidney in response to obstructive injury.
Figure 2. Smad3 deficiency suppresses the accumulation of bone marrow-derived fibroblasts in the kidney in response to obstructive injury.

A. Representative photomicrographs of kidney sections from WT and Smad3-KO mice 1 week after UUO stained for CD45 (red), procollagen I (green), and DAPI (blue). B. Quantitative analysis of CD45+ and procollagen I+ fibroblasts in the kidney of WT and Smad3-KO mice 1 week after UUO. ** P < 0.01 vs WT controls, + P < 0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n=4 per group. C. Representative cytometric diagrams showing the effect of Smad3 deficiency on the accumulation of CD45 and DDR2 dual positive fibroblasts in the kidney in response to UUO. D. Quantitative analysis of CD45 and DDR2 dual positive fibroblasts in the kidney in response to UUO. ** P < 0.01 vs WT control, + P < 0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n=3–4 per group.
Smad3 Deficiency Inhibits Myofibroblast Activation
Since TGF-β1 signaling has been shown to promote maturation of bone marrow-derived fibroblasts in vitro19, 33, we determined if Smad3 deficiency influences the development of myofibroblasts in the kidney. WT and Smad3 KO mice were subjected to UUO for 14 days. Kidney sections were stained with an antibody against α-SMA, a marker of myofibroblasts, and examined with a fluorescence microscope. The results revealed that genetic deletion of Smad3 resulted in a significant reduction in the number of myofibroblasts in obstructed kidneys compared with WT mice (Figure 3, A and B). Consistent with these findings, real-time RT-PCR showed that Smad3 deficiency significantly reduced the mRNA expression of α-SMA compared to the level in WT mice with UUO (Figure 3C). These findings were confirmed at the protein levels by Western blot analysis, which showed that Smad3 deficiency significantly reduced the protein expression levels of α-SMA in obstructed kidneys compared with WT mice (Figure 3, DE). These results indicate that Smad3 signaling plays an important role in myofibroblast activation in the kidney in response to obstructive injury.
Figure 3. Smad3 deficiency inhibits myofibroblast activation and α-SMA expression in obstructive nephropathy.

A. Representative photomicrographs of α-SMA immunofluorescence staining in the kidney of WT and Smad3-KO mice 2 weeks after UUO. B. Quantitative analysis of α-SMA protein expression in the kidney of WT and Smad3-KO mice 2 weeks after UUO. ** P <0.01 vs WT controls, + P <0.05 vs KO UUO, and # P <0.05 vs WT UUO. n=5 per group. C. The mRNA levels of α-SMA in the kidney of WT and Smad3-KO mice as determined by real-time RT-PCR. * P < 0.05 vs WT controls, + P <0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n=4 per group. D. Representative Western blots show the levels of α-SMA protein expression in the kidney of WT and Smad3-KO mice. E. Quantitative analysis of α-SMA protein expression in the kidney of WT and Smad3-KO mice. ** P <0.01 vs WT controls, + P <0.05 vs KO UUO, and # P <0.05 vs WT UUO. n=5 per group.
Smad3 Deficiency Suppresses Renal Fibrosis
Since Smad3 regulates the accumulation of bone marrow-derived fibroblasts in the kidney in response to obstructive injury, we then examined the effect of Smad3 deficiency on the development of renal fibrosis. WT and Smad3 KO mice were subjected to UUO for 14 days. WT mice developed significant collagen deposition in obstructed kidneys as demonstrated by picrosirius red staining. In contrast, Smad3 KO mice displayed much less collagen deposition in the kidney in response to obstructive injury (Figure 4, A and B). These data indicate that Smad3 plays a critical role in the pathogenesis of renal fibrosis.
Figure 4. Smad3 deficiency suppresses renal fibrosis and collagen deposition in the kidney.

A. Representative photomicrographs show kidney sections stained with picrosirius red for assessment of total collagen deposition. B. Quantitative analysis of renal interstitial collagen in different groups as indicated. ** P < 0.01 vs WT controls, + P < 0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n= 6 per group.
We next investigated the effect of genetic disruption of Smad3 on the expression and accumulation of collagen I and fibronectin, two major components of ECM. There was a marked increase in the mRNA expression levels of collagen I and fibronectin in obstructed kidneys of WT mice, whereas genetic disruption of Smad3 significantly suppressed the mRNA expression levels of these matrix components in obstructed kidneys (Figure 5, A and B). We then examined ECM protein expression in the kidney in response to obstructive injury. Immunofluorescence staining revealed that genetic disruption of Smad3 inhibited collagen I and fibronectin protein expression in the obstructed kidneys compared with WT mice (Figure 5, C and D). These results were confirmed by Western blot analysis (Figure 5, E–H). These data indicate that genetic deletion of Smad3 prevents renal fibrosis by inhibiting production and deposition of ECM proteins.
Figure 5. Smad3 deficiency inhibits collagen I and fibronectin expression in the kidney.

A. The mRNA levels of collagen I in the kidney of WT and Smad3-KO mice as determined by real-time RT-PCR. * P < 0.05 vs WT controls, + P < 0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n=4 per group. B. The mRNA levels of fibronectin in the kidney of WT and Smad3-KO mice as determined by real-time RT-PCR. * P < 0.05 vs WT controls, + P < 0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n=4 per group. C. Representative photomicrographs of collagen I immunofluorescence staining in the kidney of WT and Smad3-KO mice at day 14 after UUO (original magnification X400). D. Representative photomicrographs of fibronectin immunofluorescence staining in the kidney of WT and Smad3-KO mice at day 14 after UUO (original magnification X400). E. Representative Western blots show the protein levels of collagen I in the kidney of WT and Smad3-KO mice. F. Quantitative analysis of collagen I protein expression in the kidney of WT and Smad3-KO mice. * P < 0.05 vs WT controls, + P < 0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n=4 per group. G. Representative Western blots show the protein levels of fibronectin in the kidney of WT and Smad3-KO mice. H. Quantitative analysis of fibronectin protein expression in the kidney of WT and Smad3-KO mice. * P < 0.05 vs WT controls, + P < 0.05 vs KO UUO, and # P < 0.05 vs WT UUO. n=4 per group.
Smad3 Deficiency in Bone Marrow-derived Cells Reduces Renal Fibrosis
To determine the role of Smad3 in bone marrow-derived cells in the development of renal fibrosis, we performed bone marrow transplant with Smad3+/+ or Smad3−/− bone marrow cells. Compared with WT mice transplanted with Smad3+/+ bone marrow cells, WT mice transplanted with Smad3−/− bone marrow cells accumulated fewer bone marrow-derived fibroblasts (Figure 6 A and B) and showed a lesser degree of renal fibrosis (Figure 6 C and D). These data indicate that myeloid Smad3 signaling is important for the activation of bone marrow-derived fibroblasts and the development of renal fibrosis.
Figure 6. Smad3 deficiency in bone marrow-derived cells inhibits myeloid fibroblast accumulation and fibrosis.

A. Representative photomicrographs of kidney sections from WT mice transplanted with Smad3+/+ bone marrow cells and WT mice transplanted with Smad3−/− bone marrow cells 1 week after UUO stained for CD45 (red), procollagen I (green), and DAPI (blue). B. Quantitative analysis of CD45+ and procollagen I+ fibroblasts in the kidney. ** P < 0.01 vs WT→WT controls, + P < 0.05 vs KO→WT UUO, and # P < 0.05 vs WT→WT UUO. n=5 per group. C. Representative photomicrographs show kidney sections stained with picrosirius red for assessment of total collagen deposition. D. Quantitative analysis of renal interstitial collagen content in different groups as indicated. ** P < 0.01 vs WT→WT controls, + P < 0.05 vs KO→WT UUO, and # P < 0.05 vs WT→WT UUO. n=5 per group.
Smad3 Deficiency Inhibits Monocyte-to-Fibroblast Transition in vitro
To determine if Smad3 signaling regulates monocyte-to-fibroblast transition in vitro, we treated monocytes from spleens of WT and Smad3 KO mice with TGF-β1. Smad3−/− monocytes express less fibronectin and α-SMA at basal conditions compared with Smad3+/+ monocytes. Treatment of Smad3+/+ monocytes with TGF-β1 induced phosphorylation of Smad3 as well as expression of collagen I, fibronectin, and α-SMA. In contrast, treatment of Smad3−/− monocytes with TGF-β1 did not induce expression of collagen I, fibronectin and α-SMA (Figure 7). These data support our in vivo observations that Smad3 signaling plays an important role in activation of bone marrow-derived fibroblasts and pathogenesis of renal fibrosis.
Figure 7. Smad3 deficiency inhibits TGF-β1-induced monocyte-to-fibroblast transition.

A. Representative Western blots show Smad3 is activated in monocytes treated with TGF-β1. B. The mRNA levels of collagen I as determined by real-time RT-PCR. * P < 0.05 vs WT controls, + P < 0.05 vs KO-TGF-β1, and ## P < 0.01 vs WT-TGF-β1. n=4 per group. C. The mRNA levels of fibronectin as determined by real-time RT-PCR. * P < 0.05 vs WT controls, + P < 0.05 vs KO-TGF-β1, and # P < 0.05 vs WT-TGF-β1. n=4 per group. D. The mRNA levels of α-SMA as determined by real-time RT-PCR. ** P < 0.01 vs WT controls, + P < 0.05 vs KO-TGF-β1, and ## P < 0.01 vs WT-TGF-β1. n=4 per group. E. Representative Western blots show the protein levels of collagen I, fibronectin, and α-SMA.
Discussion
Activated fibroblasts are responsible for wound healing and organ fibrosis. Recent studies have shown that bone marrow-derived fibroblasts play a significant role in the pathogenesis of renal fibrosis8, 11, 14, 34. The signaling mechanisms underlying the accumulation and activation of bone marrow-derived fibroblasts in the kidney are incompletely understood. We have previously shown that CXCL16 plays an obligatory role in the recruitment of myeloid fibroblasts into the kidney and the development of renal fibrosis in response to obstructive injury11. In the present study, we demonstrate that Smad3 signaling is pathologically important in the activation of myeloid fibroblasts and the development of renal fibrosis because genetic disruption of Smad3 inhibits the activation of bone marrow-derived fibroblasts in the kidney in response to obstructive injury in vivo and suppresses monocyte to fibroblast transition in vitro. These data indicate Smad3 signaling plays an important role in the transformation of bone marrow-derived fibroblasts in the kidney.
TGF-β1 is a multifunctional cytokine that regulates ECM production by signaling through the type I and type II serine/threonine kinase receptors35. TGF-β1 binds to receptor II and results in phosphorylation of receptor I. The activated receptor I then directly signals to downstream intracellular substrates, the receptor-associated Smads (R-Smads), Smad2 and Smad3 through phosphorylation. Activated R-Smads heteroligomerize with the common Smad (Co-Smad), Smad4, and translocate into the nucleus to regulate target gene expression36. A large body of evidence has shown that TGF-β1 plays a central role in the pathogenesis of renal fibrosis20–22. Smad3 is a major downstream signaling molecule of TGF-β1 in mediating the pathogenesis of fibrosis27, 37. In our study, we show that Smad3 signaling in myeloid fibroblasts is activated in response to obstructive injury in vivo. Genetic disruption of Smad3 suppressed the accumulation of bone marrow-derived fibroblasts in the kidney. These results indicate that Smad3 signaling plays an important role in the activation of bone marrow-derived fibroblasts.
Myofibroblasts are the active form of fibroblasts that are generally considered to be the main source of increased ECM deposition in renal fibrosis4, 5. Furthermore, experimental and clinical studies have shown that the number of interstitial myofibroblasts correlates closely with the severity of tubulointerstitial fibrosis and the progression of kidney disease38–40. In the present study, we demonstrate that myofibroblasts identified as α-SMA positive cells accumulate in the kidney of WT mice following obstructive injury, and their accumulation is significantly reduced in the obstructed kidney of Smad3-KO mice. These results strongly indicate that Smad3 signaling activates bone marrow-derived myofibroblasts in the kidney.
A key feature of renal fibrosis is the dramatic increase and deposition of extracellular matrix proteins including collagens and fibronectin. Morphometric analysis of picrosirius red staining of kidney sections at day 14 after obstructive injury demonstrates the presence of interstitial collagen deposition. This collagen deposition is significantly attenuated in the obstructed kidney of Smad3-KO mice. In keeping with these findings, we further illustrate that the expression of two major ECM proteins, collagen I and fibronectin, are markedly increased in the obstructed kidney of WT mice, whereas these responses are significantly attenuated in the injured kidney of Smad3-KO mice.
Although studies have shown activation of Smad3 signaling plays an important role in the pathogenesis of renal fibrosis, the cell types in which Smad3 is activated has not been clearly defined. One study claims that activation of Smad3 in tubular epithelial cells mediates renal fibrosis27. Another study reports that Smad2/3 is activated in cells located in renal interstitial space41. Using a phospho-Smad3 specific antibody, our results show that Smad3 is activated in bone marrow-derived fibroblasts in the kidney in response to obstructive injury. Furthermore, the results of bone marrow chimeric experiments demonstrate that Smad3 signaling in bone marrow-derived fibroblasts contribute significantly to the pathogenesis of renal fibrosis following obstructive injury. The importance of Smad3 signaling in the activation of bone marrow-derived fibroblasts was further supported by our in vitro studies.
In summary, our study defines a novel mechanism by which Smad3 signaling regulates the pathogenesis of renal fibrosis. In response to injury, activated Smad3 signaling transforms CD45+ myeloid fibroblasts into myofibroblasts, which contribute to the pathogenesis of renal fibrosis (Figure 8). These data suggest that Smad3 signaling in bone marrow-derived fibroblasts is essential for the development of renal fibrosis.
Figure 8. Schematic illustration of the signaling events leading to activation of myeloid fibroblasts.
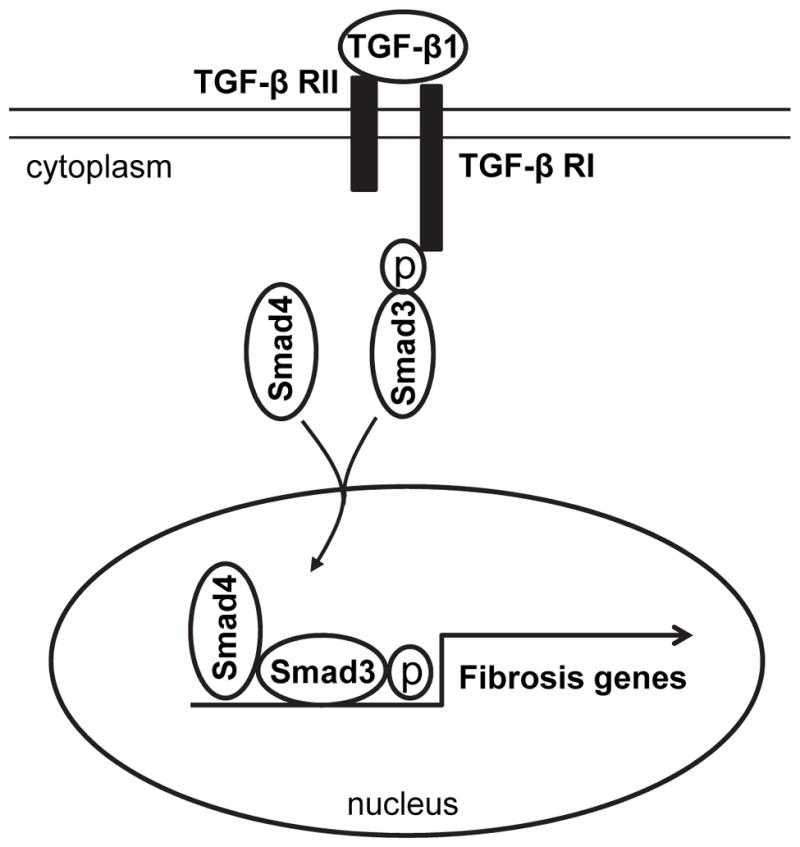
TGF-β1 binding to its receptor results in Smad3 phosphorylation. The phosphorylated Smad3 along with Smad4 is translocated into the nucleus, where it induces expression of ECM genes and activation of myeloid fibroblasts.
Acknowledgments
Sources of Funding: This work was supported in part by the NIH grants - K08HL92958, R01DK95835 and an American Heart Association grant - 11BGIA7840054 to Y.W. The Cytometry and Cell Sorting Core at Baylor College of Medicine was supported in part by the NIH grants - AI036211, CA125123, and RR024574.
We thank Dr. William E. Mitch for helpful discussion and Dr. Xiao-Fan Wang for providing Smad3 KO mice. We also thank Joel M. Sederstrom of the flow cytometry core at Baylor College of Medicine for technical assistance.
Footnotes
Disclosure
None
References
- 1.Schainuck LI, Striker GE, Cutler RE, Benditt EP. Structural-functional correlations in renal disease. II. The correlations. Hum Pathol. 1970;1(4):631–641. doi: 10.1016/s0046-8177(70)80061-2. [DOI] [PubMed] [Google Scholar]
- 2.Nath KA. The tubulointerstitium in progressive renal disease. Kidney Int. 1998;54(3):992–994. doi: 10.1046/j.1523-1755.1998.00079.x. [DOI] [PubMed] [Google Scholar]
- 3.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15(3–4):290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- 4.Neilson EG. Mechanisms of disease: Fibroblasts--a new look at an old problem. Nat Clin Pract Nephrol. 2006;2(2):101–108. doi: 10.1038/ncpneph0093. [DOI] [PubMed] [Google Scholar]
- 5.Strutz F, Muller GA. Renal fibrosis and the origin of the renal fibroblast. Nephrol Dial Transplant. 2006;21(12):3368–3370. doi: 10.1093/ndt/gfl199. [DOI] [PubMed] [Google Scholar]
- 6.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110(3):341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Deane JA, Campanale NV, Bertram JF, Ricardo SD. The contribution of bone marrow-derived cells to the development of renal interstitial fibrosis. Stem Cells. 2007;25(3):697–706. doi: 10.1634/stemcells.2006-0133. [DOI] [PubMed] [Google Scholar]
- 8.Sakai N, Wada T, Yokoyama H, et al. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci U S A. 2006;103(38):14098–14103. doi: 10.1073/pnas.0511200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimm PC, Nickerson P, Jeffery J, et al. Neointimal and tubulointerstitial infiltration by recipient mesenchymal cells in chronic renal-allograft rejection. N Engl J Med. 2001;345(2):93–97. doi: 10.1056/NEJM200107123450203. [DOI] [PubMed] [Google Scholar]
- 10.Broekema M, Harmsen MC, van Luyn MJ, et al. Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol. 2007;18(1):165–175. doi: 10.1681/ASN.2005070730. [DOI] [PubMed] [Google Scholar]
- 11.Chen G, Lin SC, Chen J, et al. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol. 2011;22(10):1876–1886. doi: 10.1681/ASN.2010080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Lin SC, Chen G, et al. Adiponectin Promotes Monocyte-to-Fibroblast Transition in Renal Fibrosis. J Am Soc Nephrol. 2013 doi: 10.1681/ASN.2013030217. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1(1):71–81. [PMC free article] [PubMed] [Google Scholar]
- 14.Niedermeier M, Reich B, Rodriguez Gomez M, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A. 2009;106(42):17892–17897. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. Journal of leukocyte biology. 2008;83(6):1323–1333. doi: 10.1189/jlb.1107782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia Y, Entman ML, Wang Y. Critical Role of CXCL16 in Hypertensive Kidney Injury and Fibrosis. Hypertension. 2013;62(6):1129–1137. doi: 10.1161/HYPERTENSIONAHA.113.01837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metz CN. Fibrocytes: a unique cell population implicated in wound healing. Cell Mol Life Sci. 2003;60(7):1342–1350. doi: 10.1007/s00018-003-2328-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36(4):598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166(12):7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 20.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331(19):1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 21.Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature. 1990;346(6282):371–374. doi: 10.1038/346371a0. [DOI] [PubMed] [Google Scholar]
- 22.Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. Journal of the American Society of Nephrology : JASN. 2002;13(10):2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 23.Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7(7):1056–1067. doi: 10.7150/ijbs.7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Latella G, Vetuschi A, Sferra R, et al. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. 2009;29(7):997–1009. doi: 10.1111/j.1478-3231.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 25.Huang XR, Chung AC, Yang F, et al. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension. 2010;55(5):1165–1171. doi: 10.1161/HYPERTENSIONAHA.109.147611. [DOI] [PubMed] [Google Scholar]
- 26.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276(20):17058–17062. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 27.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112(10):1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J, Shi W, Wang YL, et al. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282(3):L585–593. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 29.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol. 1999;19(4):2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Chen J, Yan J, et al. Effect of interleukin 6 deficiency on renal interstitial fibrosis. PLoS One. 2012;7(12):e52415. doi: 10.1371/journal.pone.0052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia Y, Entman ML, Wang Y. CCR2 Regulates the Uptake of Bone Marrow-Derived Fibroblasts in Renal Fibrosis. PLoS One. 2013;8(10):e77493. doi: 10.1371/journal.pone.0077493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crawford JR, Pilling D, Gomer RH. Improved serum-free culture conditions for spleen-derived murine fibrocytes. J Immunol Methods. 2010;363(1):9–20. doi: 10.1016/j.jim.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong KM, Belperio JA, Keane MP, Burdick MD, Strieter RM. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282(31):22910–22920. doi: 10.1074/jbc.M703597200. [DOI] [PubMed] [Google Scholar]
- 34.Lebleu VS, Taduri G, O'Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19(8):1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 36.Miyazono K. Positive and negative regulation of TGF-beta signaling. J Cell Sci. 2000;113 ( Pt 7):1101–1109. doi: 10.1242/jcs.113.7.1101. [DOI] [PubMed] [Google Scholar]
- 37.Roberts AB, Piek E, Bottinger EP, Ashcroft G, Mitchell JB, Flanders KC. Is Smad3 a major player in signal transduction pathways leading to fibrogenesis? Chest. 2001;120(1 Suppl):43S–47S. doi: 10.1378/chest.120.1_suppl.s43-a. [DOI] [PubMed] [Google Scholar]
- 38.Zhang G, Moorhead PJ, el Nahas AM. Myofibroblasts and the progression of experimental glomerulonephritis. Exp Nephrol. 1995;3(5):308–318. [PubMed] [Google Scholar]
- 39.Roberts IS, Burrows C, Shanks JH, Venning M, McWilliam LJ. Interstitial myofibroblasts: predictors of progression in membranous nephropathy. J Clin Pathol. 1997;50(2):123–127. doi: 10.1136/jcp.50.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Essawy M, Soylemezoglu O, Muchaneta-Kubara EC, Shortland J, Brown CB, el Nahas AM. Myofibroblasts and the progression of diabetic nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 1997;12(1):43–50. doi: 10.1093/ndt/12.1.43. [DOI] [PubMed] [Google Scholar]
- 41.Inazaki K, Kanamaru Y, Kojima Y, et al. Smad3 deficiency attenuates renal fibrosis, inflammation,and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66(2):597–604. doi: 10.1111/j.1523-1755.2004.00779.x. [DOI] [PubMed] [Google Scholar]